
Electronegative LDL Is Associated with Plaque Vulnerability in Patients with Ischemic Stroke and Carotid Atherosclerosis

 , , , , , , , ,
, , , , , , , ,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Study Population
2.3. Carotid Plaque Imaging
2.3.1. Carotid Ultrasound Protocol
2.3.2. Carotid 18F-FDG PET/CT Protocol
2.4. Collection of Blood Samples
2.5. Serum Determinations
2.6. LDL(−) Quantification
2.7. Effect of Lipoproteins on Cells
2.7.1. ChE Capacity of HDL
2.7.2. Inflammatory Effect of LDL(−) on Cells
2.8. Outcomes
2.9. Statistical Analysis
3. Results
3.1. Study Population
3.2. Lipid Profile and Lipoprotein-Related Molecules
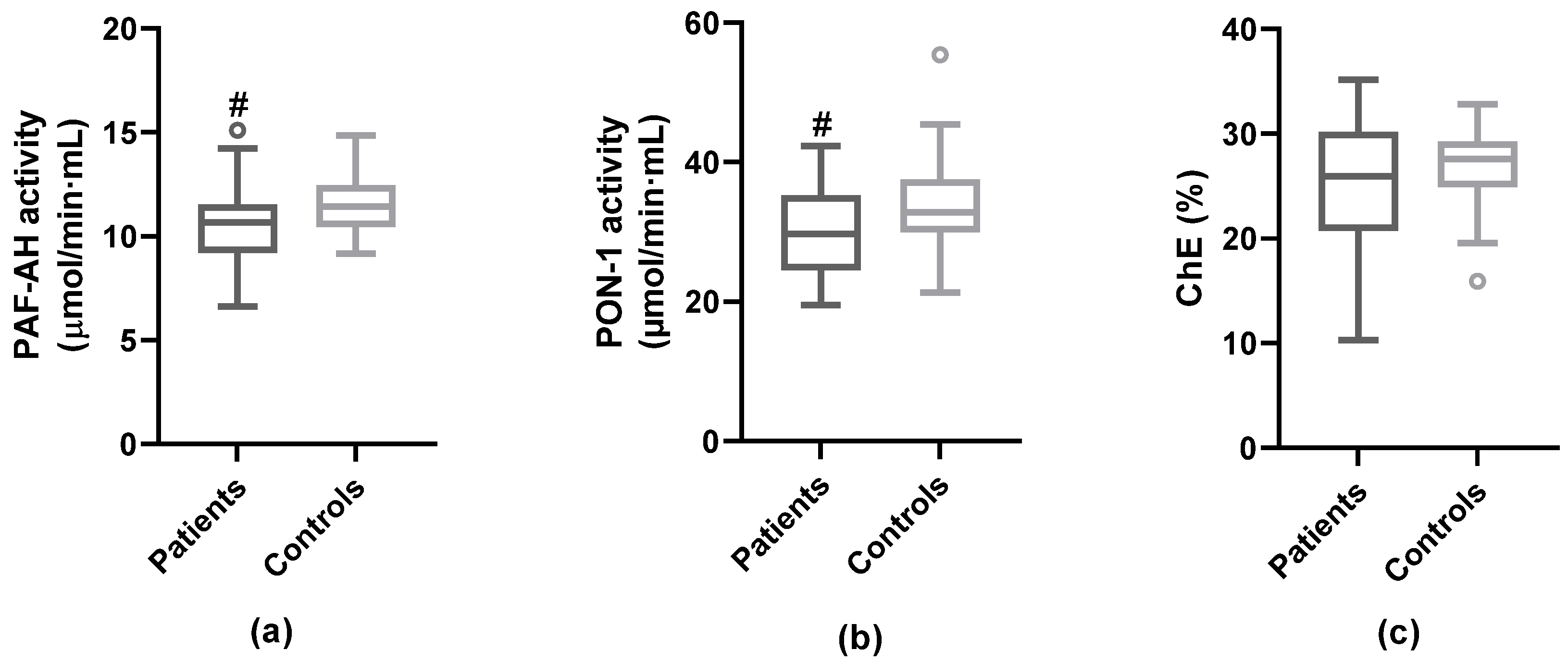
3.3. Anti-Atherogenic Properties of HDL
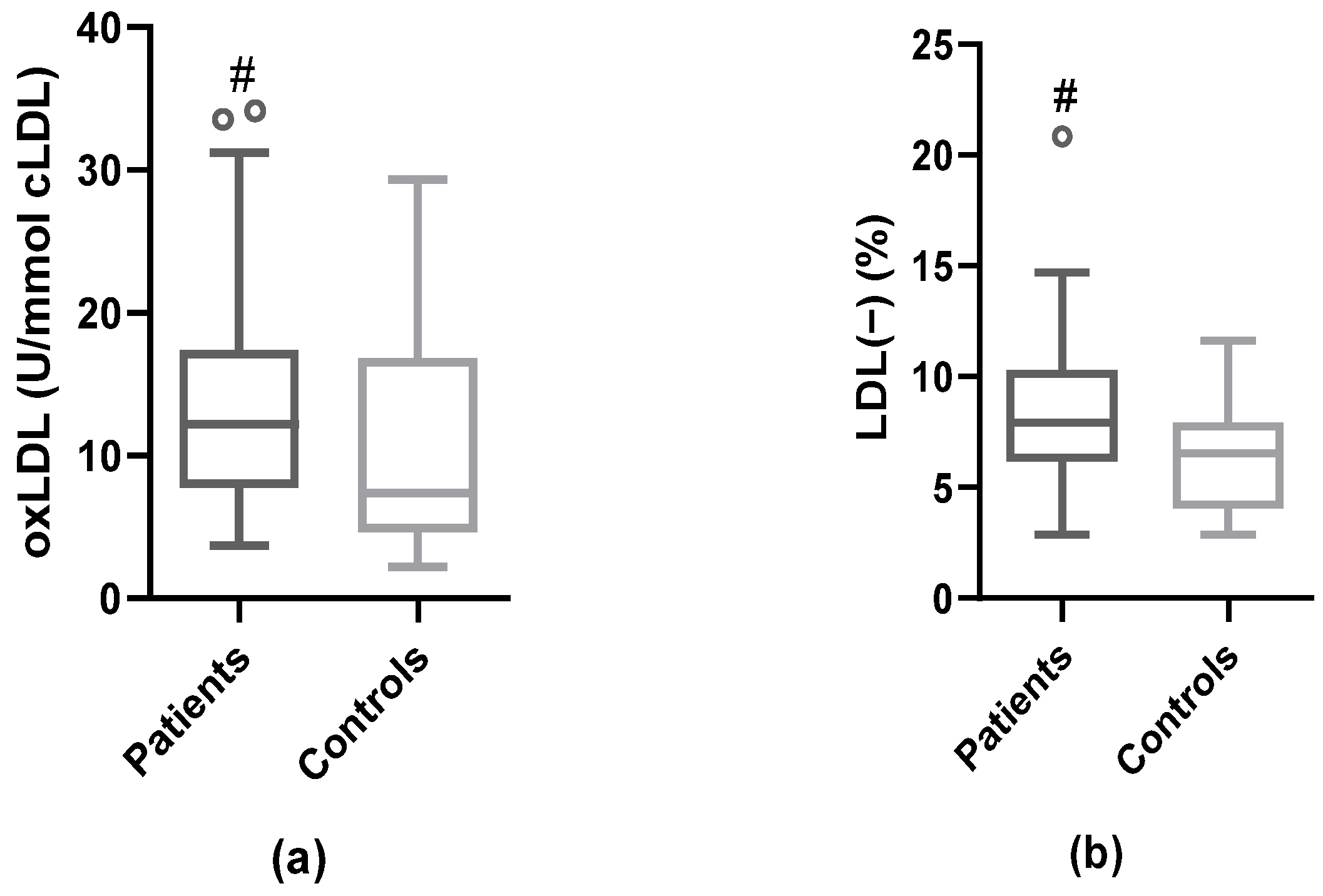
3.4. Modified LDL: oxLDL and LDL(−)
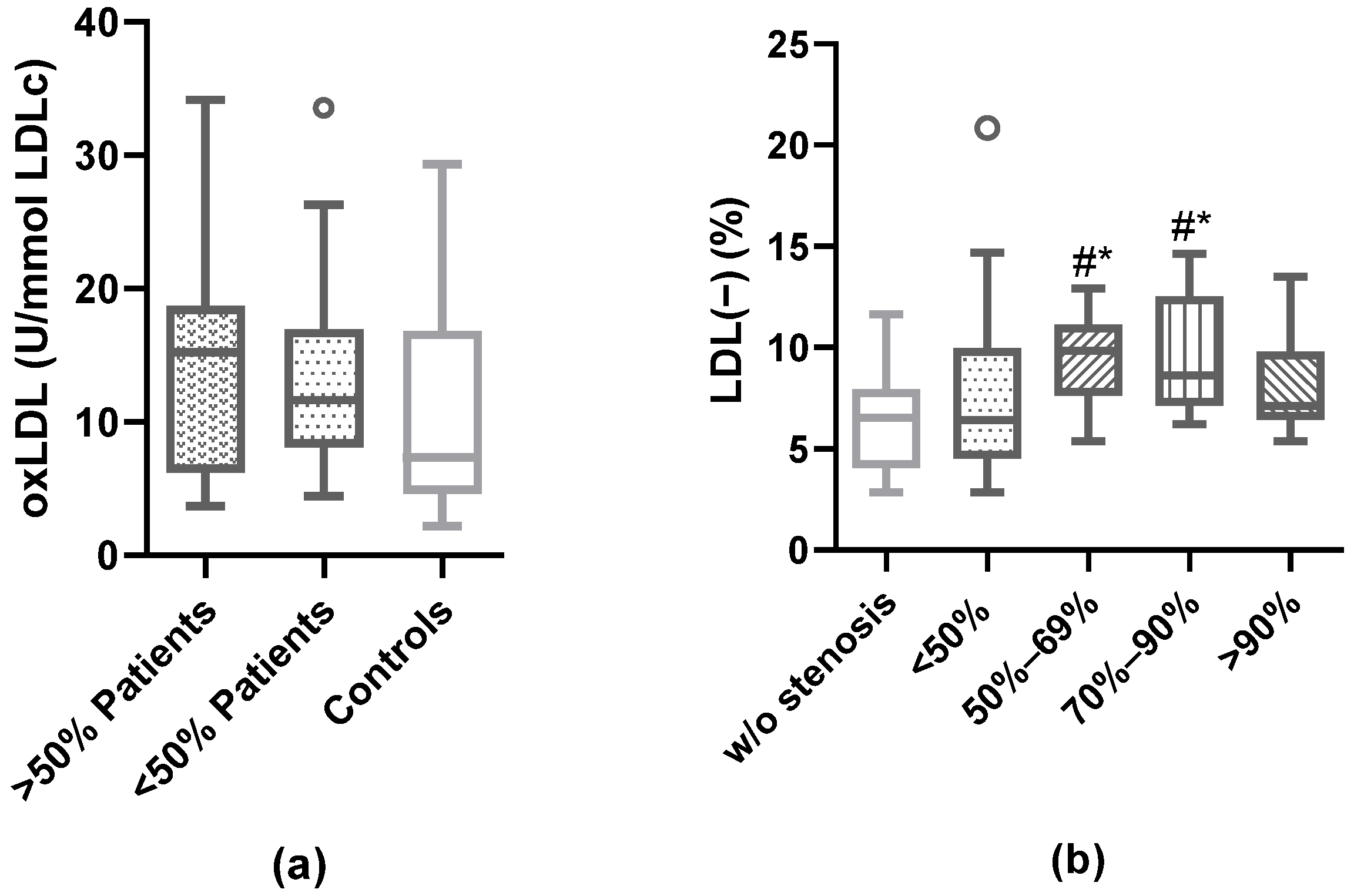
3.5. Association of Plaque Characteristics with Modified LDL
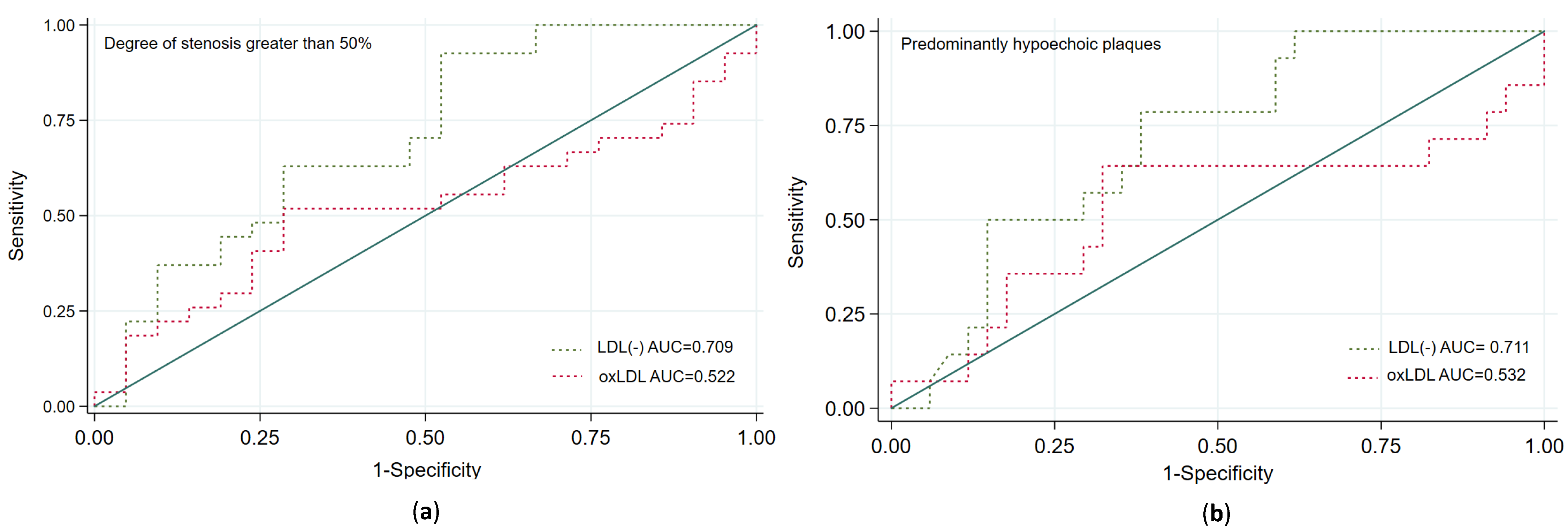
3.6. Prediction of Carotid Plaque Vulnerability According to the Plasma Proportion of LDL(−)
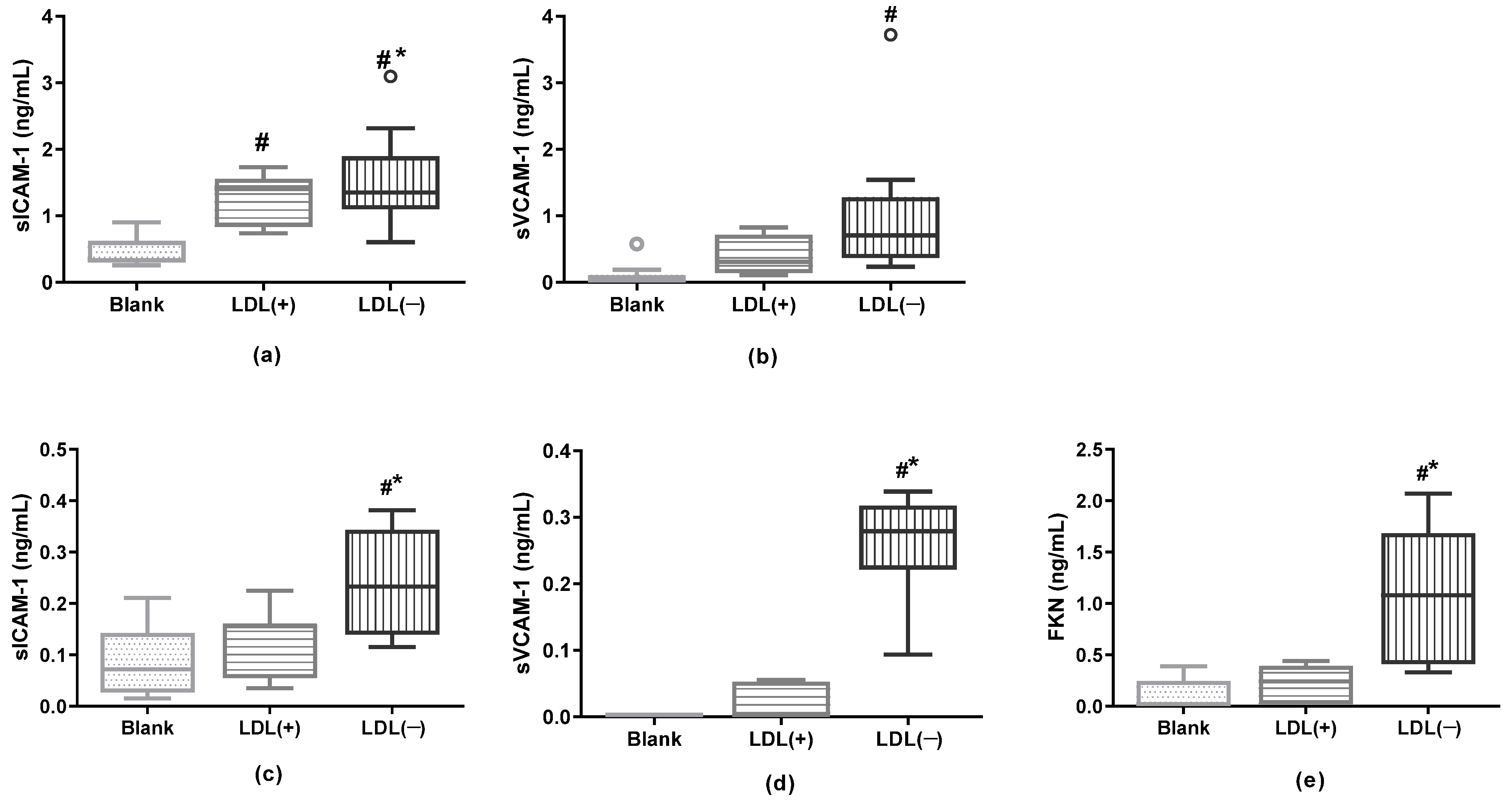
3.7. LDL(−) as an Inductor of Inflammation in Cultured Cells
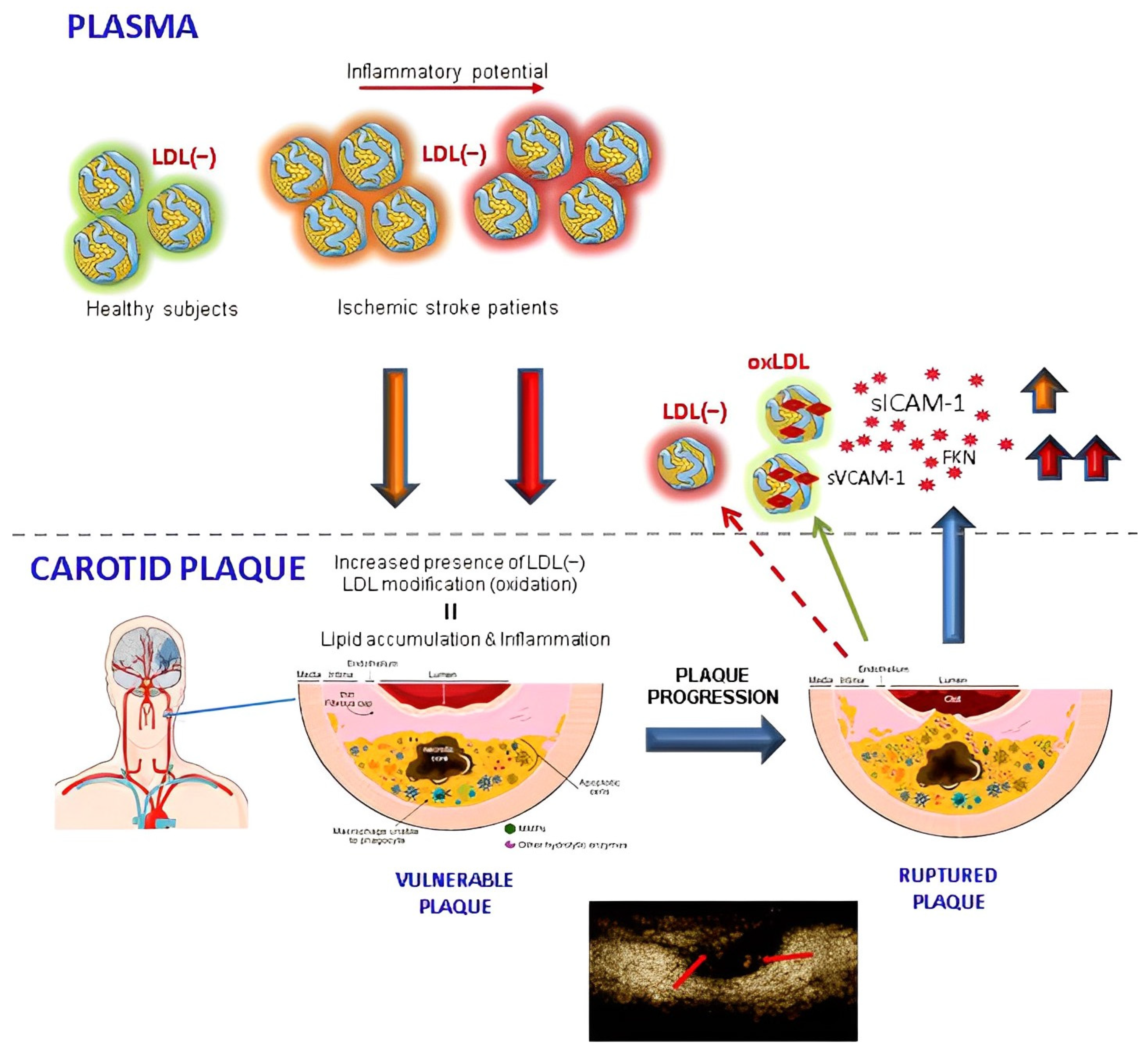
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonati, L.H.; Kakkos, S.; Berkefeld, J.; de Borst, G.J.; Bulbulia, R.; Halliday, A.; van Herzeele, I.; Koncar, I.; McCabe, D.J.; Lal, A.; et al. European Stroke Organisation guideline on endarterectomy and stenting for carotid artery stenosis. Eur. Stroke J. 2021, 6, I–XLVII. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M. ACST: Which subgroups will benefit most from carotid endarterectomy? Lancet 2004, 364, 1122–1123, author reply 1125–1126. [Google Scholar] [CrossRef] [PubMed]
- Camps-Renom, P.; Prats-Sánchez, L.; Casoni, F.; González-de-Echávarri, J.M.; Marrero-González, P.; Castrillón, I.; Marín, R.; Jiménez-Xarrié, E.; Delgado-Mederos, R.; Martínez-Domeño, A.; et al. Plaque neovascularization detected with contrast-enhanced ultrasound predicts ischaemic stroke recurrence in patients with carotid atherosclerosis. Eur. J. Neurol. 2020, 27, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.J.; Camps-Renom, P.; Giannotti, N.; Martí-Fàbregas, J.; Murphy, S.; McNulty, J.; Barry, M.; Barry, P.; Calvet, D.; Coutts, S.B.; et al. Carotid Plaque Inflammation Imaged by 18F-Fluorodeoxyglucose Positron Emission Tomography and Risk of Early Recurrent Stroke. Stroke 2019, 50, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Puig, N.; Camps-Renom, P.; Camacho, M.; Aguilera-Simón, A.; Jiménez-Altayó, F.; Fernández-León, A.; Marín, R.; Martí-Fàbregas, J.; Sánchez-Quesada, J.L.; Jiménez-Xarrié, E.; et al. Plasma sICAM-1 as a Biomarker of Carotid Plaque Inflammation in Patients with a Recent Ischemic Stroke. Transl. Stroke Res. 2022, 13, 745–756. [Google Scholar] [CrossRef]
- Kamtchum-Tatuene, J.; Saba, L.; Heldner, M.R.; Poorthuis, M.H.F.; de Borst, G.J.; Rundek, T.; Kakkos, S.K.; Chaturvedi, S.; Topakian, R.; Polak, J.F.; et al. Interleukin-6 Predicts Carotid Plaque Severity, Vulnerability, and Progression. Circ. Res. 2022, 131, e22–e33. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Hindy, G.; Engström, G.; Larsson, S.C.; Traylor, M.; Markus, H.S.; Melander, O.; Orho-Melander, M. Stroke Genetics Network (SiGN) Role of Blood Lipids in the Development of Ischemic Stroke and its Subtypes: A Mendelian Randomization Study. Stroke 2018, 49, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Labreuche, J.; Elbaz, A.; Touboul, P.-J.; Driss, F.; Jaillard, A.; Bruckert, E. GENIC Investigators Blood Lipids in Brain Infarction Subtypes. Cerebrovasc. Dis. 2006, 22, 101–108. [Google Scholar] [CrossRef]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Pan, Y.; Wang, M.; Meng, X.; Jiang, Y.; Li, Z.; Li, H.; Wang, Y.; Wang, Y. Inverse Association between High-Density Lipoprotein Cholesterol and Adverse Outcomes among Acute Ischemic Stroke Patients with Diabetes Mellitus. Biomedicines 2021, 9, 1947. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Itabe, H.; Uno, M.; Kitazato, K.T.; Horiguchi, H.; Shinno, K.; Nagahiro, S. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Lehti, S.; Nguyen, S.D.; Belevich, I.; Vihinen, H.; Heikkilä, H.M.; Soliymani, R.; Käkelä, R.; Saksi, J.; Jauhiainen, M.; Grabowski, G.A.; et al. Extracellular Lipids Accumulate in Human Carotid Arteries as Distinct Three-Dimensional Structures and Have Proinflammatory Properties. Am. J. Pathol. 2018, 188, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kubo, T.; Okumoto, Y.; Ishibashi, K.; Komukai, K.; Tanimoto, T.; Ino, Y.; Kitabata, H.; Hirata, K.; Imanishi, T.; et al. Circulating malondialdehyde-modified low-density lipoprotein levels are associated with the presence of thin-cap fibroatheromas determined by optical coherence tomography in coronary artery disease. Eur. Heart J. Cardiovasc. Imaging 2013, 14, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yang, Y.; Su, Z.; Yue, W.; Hao, H.; Ren, L.; Wang, Y.; Cao, Y.; Wang, Y. Association of Oxidized Low-Density Lipoprotein With Prognosis of Stroke and Stroke Subtypes. Stroke 2017, 48, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Dai, L.; Zhang, N.; Lin, J.; Chen, G.; Zuo, Y.; Li, H.; Wang, Y.; Meng, X.; Wang, Y. Oxidized low-density lipoprotein (LDL) and LDL cholesterol are associated with outcomes of minor stroke and TIA. Atherosclerosis 2020, 297, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-C.; Chang, P.-Y.; Lu, S.-C. L5-LDL from ST-elevation myocardial infarction patients induces IL-1β production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H265–H274. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.-Y.; Chen, F.-Y.; Hsu, J.-F.; Fu, R.-H.; Chang, C.-M.; Chang, C.-T.; Liu, C.-H.; Wu, J.-R.; Lee, A.-S.; Chan, H.-C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef]
- Estruch, M.; Sánchez-Quesada, J.L.; Ordóñez Llanos, J.; Benítez, S. Electronegative LDL: A circulating modified LDL with a role in inflammation. Mediators Inflamm. 2013, 2013, 181324. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Chen, C.-H.; Chen, Y.-M.; Hsieh, T.-Y.; Li, J.-P.; Shen, M.-Y.; Lan, J.-L.; Chen, D.-Y. Association between Negatively Charged Low-Density Lipoprotein L5 and Subclinical Atherosclerosis in Rheumatoid Arthritis Patients. J. Clin. Med. 2019, 8, E177. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Berliner, J.A.; Subbanagounder, G.; Hama, S.; Lusis, A.J.; Castellani, L.W.; Reddy, S.; Shih, D.; Shi, W.; Watson, A.D.; et al. HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Mackness, M.I.; Durrington, P.N.; Mackness, B. The role of paraoxonase 1 activity in cardiovascular disease: Potential for therapeutic intervention. Am. J. Cardiovasc. Drugs Drugs Devices Interv. 2004, 4, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chapman, M.J. Antiatherogenic small, dense HDL—Guardian angel of the arterial wall? Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 144–153. [Google Scholar] [CrossRef]
- Bartlett, E.S.; Walters, T.D.; Symons, S.P.; Fox, A.J. Quantification of carotid stenosis on CT angiography. AJNR Am. J. Neuroradiol. 2006, 27, 13–19. [Google Scholar]
- von Reutern, G.-M.; Goertler, M.-W.; Bornstein, N.M.; Del Sette, M.; Evans, D.H.; Hetzel, A.; Kaps, M.; Perren, F.; Razumovky, A.; von Reutern, M.; et al. Grading carotid stenosis using ultrasonic methods. Stroke 2012, 43, 916–921. [Google Scholar] [CrossRef]
- Adams, H.P.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef]
- Norris, S.L.; Grothaus, L.C.; Buchner, D.M.; Pratt, M. Effectiveness of physician-based assessment and counseling for exercise in a staff model HMO. Prev. Med. 2000, 30, 513–523. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; García-Arellano, A.; Toledo, E.; Salas-Salvadó, J.; Buil-Cosiales, P.; Corella, D.; Covas, M.I.; Schröder, H.; Arós, F.; Gómez-Gracia, E.; et al. A 14-item Mediterranean diet assessment tool and obesity indexes among high-risk subjects: The PREDIMED trial. PLoS ONE 2012, 7, e43134. [Google Scholar] [CrossRef]
- Kernan, W.N.; Ovbiagele, B.; Black, H.R.; Bravata, D.M.; Chimowitz, M.I.; Ezekowitz, M.D.; Fang, M.C.; Fisher, M.; Furie, K.L.; Heck, D.V.; et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 2160–2236. [Google Scholar] [CrossRef] [PubMed]
- Geroulakos, G.; Ramaswami, G.; Nicolaides, A.; James, K.; Labropoulos, N.; Belcaro, G.; Holloway, M. Characterization of symptomatic and asymptomatic carotid plaques using high-resolution real-time ultrasonography. Br. J. Surg. 1993, 80, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Deyama, J.; Nakamura, T.; Takishima, I.; Fujioka, D.; Kawabata, K.; Obata, J.; Watanabe, K.; Watanabe, Y.; Saito, Y.; Mishina, H.; et al. Contrast-enhanced ultrasound imaging of carotid plaque neovascularization is useful for identifying high-risk patients with coronary artery disease. Circ. J. Off. J. Jpn. Circ. Soc. 2013, 77, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Benítez, S.; Sánchez-Quesada, J.L.; Ribas, V.; Jorba, O.; Blanco-Vaca, F.; González-Sastre, F.; Ordóñez-Llanos, J. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation 2003, 108, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Sánchez-Quesada, J.L.; Antón, R.; Camacho, M.; Julve, J.; Escolà-Gil, J.C.; Vila, L.; Ordóñez-Llanos, J.; Blanco-Vaca, F. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: A new mechanism linking HDL protein composition and antiatherogenic potential. Circ. Res. 2004, 95, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quesada, J.L.; Vinagre, I.; De Juan-Franco, E.; Sánchez-Hernández, J.; Bonet-Marques, R.; Blanco-Vaca, F.; Ordóñez-Llanos, J.; Pérez, A. Impact of the LDL subfraction phenotype on Lp-PLA2 distribution, LDL modification and HDL composition in type 2 diabetes. Cardiovasc. Diabetol. 2013, 12, 112. [Google Scholar] [CrossRef]
- Benítez, S.; Sánchez-Quesada, J.L.; Lucero, L.; Arcelus, R.; Ribas, V.; Jorba, O.; Castellví, A.; Alonso, E.; Blanco-Vaca, F.; Ordóñez-Llanos, J. Changes in low-density lipoprotein electronegativity and oxidizability after aerobic exercise are related to the increase in associated non-esterified fatty acids. Atherosclerosis 2002, 160, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Cedó, L.; Fernández-Castillejo, S.; Rubió, L.; Metso, J.; Santos, D.; Muñoz-Aguayo, D.; Rivas-Urbina, A.; Tondo, M.; Méndez-Lara, K.A.; Farràs, M.; et al. Phenol-Enriched Virgin Olive Oil Promotes Macrophage-Specific Reverse Cholesterol Transport In Vivo. Biomedicines 2020, 8, E266. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama-Levy, S.; Van Lenten, B.J.; Fonarow, G.C.; Cardinez, C.J.; Castellani, L.W.; Brennan, M.-L.; Lusis, A.J.; Fogelman, A.M. Mildly oxidized LDL induces an increased apolipoprotein J/paraoxonase ratio. J. Clin. Investig. 1997, 99, 2005–2019. [Google Scholar] [CrossRef]
- Cubedo, J.; Padró, T.; Vilahur, G.; Crea, F.; Storey, R.F.; Lopez Sendon, J.L.; Kaski, J.C.; Sionis, A.; Sans-Rosello, J.; Fernández-Peregrina, E.; et al. Glycosylated apolipoprotein J in cardiac ischaemia: Molecular processing and circulating levels in patients with acute ischaemic events. Eur. Heart J. 2022, 43, 153–163. [Google Scholar] [CrossRef]
- Zhang, B.; Matsunaga, A.; Rainwater, D.L.; Miura, S.-I.; Noda, K.; Nishikawa, H.; Uehara, Y.; Shirai, K.; Ogawa, M.; Saku, K. Effects of rosuvastatin on electronegative LDL as characterized by capillary isotachophoresis: The ROSARY Study. J. Lipid Res. 2009, 50, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Benítez, S.; Ordóñez-Llanos, J.; Franco, M.; Marín, C.; Paz, E.; López-Miranda, J.; Otal, C.; Pérez-Jiménez, F.; Sánchez-Quesada, J.L. Effect of simvastatin in familial hypercholesterolemia on the affinity of electronegative low-density lipoprotein subfractions to the low-density lipoprotein receptor. Am. J. Cardiol. 2004, 93, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-S.; Ke, L.-Y.; Chan, H.-C.; Chan, H.-C.; Chen, C.-C.; Cheng, K.-H.; Lee, H.-C.; Kuo, H.-F.; Chang, C.-T.; Chang, K.-C.; et al. Four Statin Benefit Groups Defined by The 2013 ACC/AHA New Cholesterol Guideline are Characterized by Increased Plasma Level of Electronegative Low-Density Lipoprotein. Acta Cardiol. Sin. 2016, 32, 667–675. [Google Scholar] [CrossRef]
- Puig, N.; Montolio, L.; Camps-Renom, P.; Navarra, L.; Jiménez-Altayó, F.; Jiménez-Xarrié, E.; Sánchez-Quesada, J.L.; Benitez, S. Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages. Cells 2020, 9, 583. [Google Scholar] [CrossRef] [PubMed]
- De Castellarnau, C.; Sánchez-Quesada, J.L.; Benítez, S.; Rosa, R.; Caveda, L.; Vila, L.; Ordóñez-Llanos, J. Electronegative LDL from normolipemic subjects induces IL-8 and monocyte chemotactic protein secretion by human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2281–2287. [Google Scholar] [CrossRef]
- Bancells, C.; Benítez, S.; Jauhiainen, M.; Ordóñez-Llanos, J.; Kovanen, P.T.; Villegas, S.; Sánchez-Quesada, J.L.; Öörni, K. High binding affinity of electronegative LDL to human aortic proteoglycans depends on its aggregation level. J. Lipid Res. 2009, 50, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Ligi, D.; Benitez, S.; Croce, L.; Rivas-Urbina, A.; Puig, N.; Ordóñez-Llanos, J.; Mannello, F.; Sanchez-Quesada, J.L. Electronegative LDL induces MMP-9 and TIMP-1 release in monocytes through CD14 activation: Inhibitory effect of glycosaminoglycan sulodexide. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quesada, J.L.; Camacho, M.; Antón, R.; Benítez, S.; Vila, L.; Ordóñez-Llanos, J. Electronegative LDL of FH subjects: Chemical characterization and induction of chemokine release from human endothelial cells. Atherosclerosis 2003, 166, 261–270. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Lee, A.-S.; Lu, L.-S.; Ke, L.-Y.; Chen, W.-Y.; Dong, J.-W.; Lu, J.; Chen, Z.; Chu, C.-S.; Chan, H.-C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Cui, R.; Chen, C.-H.; Du, J. Oxidized low-density lipoprotein stimulates p53-dependent activation of proapoptotic Bax leading to apoptosis of differentiated endothelial progenitor cells. Endocrinology 2007, 148, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, J.-H.; Burns, A.R.; Chen, H.-H.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.-Y.; Sawamura, T.; Chen, C.-H. Mediation of electronegative low-density lipoprotein signaling by LOX-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-T.; Wang, G.-J.; Kuo, C.-C.; Hsieh, J.-Y.; Lee, A.-S.; Chang, C.-M.; Wang, C.-C.; Shen, M.-Y.; Huang, C.-C.; Sawamura, T.; et al. Electronegative Low-density Lipoprotein Increases Coronary Artery Disease Risk in Uremia Patients on Maintenance Hemodialysis. Medicine 2016, 95, e2265. [Google Scholar] [CrossRef] [PubMed]
- Puertas-Umbert, L.; Puig, N.; Camacho, M.; Dantas, A.P.; Marín, R.; Martí-Fàbregas, J.; Jiménez-Xarrié, E.; Benitez, S.; Camps-Renom, P.; Jiménez-Altayó, F. Serum from Stroke Patients with High-Grade Carotid Stenosis Promotes Cyclooxygenase-Dependent Endothelial Dysfunction in Non-ischemic Mice Carotid Arteries. Transl. Stroke Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gho, Y.S.; Kleinman, H.K.; Sosne, G. Angiogenic activity of human soluble intercellular adhesion molecule-1. Cancer Res. 1999, 59, 5128–5132. [Google Scholar] [PubMed]
- Koch, A.E.; Halloran, M.M.; Haskell, C.J.; Shah, M.R.; Polverini, P.J. Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature 1995, 376, 517–519. [Google Scholar] [CrossRef]
- Lee, S.-J.; Namkoong, S.; Kim, Y.-M.; Kim, C.-K.; Lee, H.; Ha, K.-S.; Chung, H.-T.; Kwon, Y.-G.; Kim, Y.-M. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2836–H2846. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| <50% Group (n = 27) | ≥50% Group (n = 37) | p | All Stroke Patients (n = 64) | Control Group (n = 27) | p | |
|---|---|---|---|---|---|---|
| Triglycerides (mM), md (IQR) | 1.11 (0.88–1.53) | 1.20 (0.94–1.63) | 0.601 | 1.17 (0.92–1.61) | 1.18 (0.90–1.45) | 0.212 |
| Total cholesterol (mM), m ± sd/md (IQR) | 3.80 ± 1.03 | 3.95 ± 1.186 | 0.588 | 3.75 (2.91–4.65) | 4.64 (4.00–5.88) | <0.001 |
| VLDLc (mM), md (IQR) | 0.22 (0.18–0.31) | 0.24 (0.19–0.33) | 0.601 | 0.23 (0.18–0.32) | 0.24 (0.18–0.29) | 0.524 |
| LDLc (mM), md (IQR) | 2.48 (1.76–3.48) | 2.31 (1.88–3.20) | 0.949 | 2.41 (1.82–3.29) | 3.09 (2.58–3.91) | <0.001 |
| HDLc (mM), m ± sd/md (IQR) | 1.07 ± 0.37 | 1.02 ± 0.28 | 0.541 | 1.03 (0.80–1.32) | 1.34 (1.15–1.59) | <0.001 |
| LDLc/HDLc ratio, md (IQR) | 2.42 (1.85–2.93) | 2.57 (1.98–3.11) | 0.510 | 2.48 (1.92–3.03) | 2.25 (1.98–2.80) | 0.507 |
| NEFA (mM), md (IQR) | 0.47 (0.30–0.69) | 0.38 (0.26–0.57) | 0.404 | 0.42 (0.27–0.61) | 0.43 (0.24–0.48) | 0.621 |
| apoB (g/L), md (IQR) | 0.68 (0.56–0.90) | 0.66 (0.60–0.80) | 0.826 | 0.68 (0.58–0.83) | 0.91 (0.78–1.02) | <0.001 |
| apoA-I (g/L), md (IQR) | 1.22 (1.14–1.50) | 1.22 (1.10–1.47) | 0.703 | 1.22 (1.10–1.47) | 1.62 (1.44–1.87) | <0.001 |
| apoA-II (g/L), m ± sd | 0.30 ± 0.07 | 0.28 ± 0.08 | 0.468 | 0.30 ± 0.08 | 0.37 ± 0.08 | <0.001 |
| apoE (g/L), m ± sd | 0.04 ± 0.02 | 0.04 ± 0.02 | 0.978 | 0.04 ± 0.02 | 0.05 ± 0.02 | 0.006 |
| apoC-III (g/L), md (IQR) | 0.05 (0.03–0.09) | 0.05 (0.01–0.11) | 0.713 | 0.05 (0.02–0.10) | 0.07 (0.03–0.11) | 0.144 |
| apoJ (mg/L), md (IQR) | 170 (153–219) | 180 (147–218) | 0.952 | 179 (148–218) | 145 (114–170) | <0.001 |
| Total PAF-AH activity (µmol/min*mL) m ± sd | 19.10 ± 3.33 | 19.07 ± 3.58 | 0.974 | 19.08 ± 3.41 | 21.49 ± 2.66 | 0.003 |
| Echolucency | Predominantly Hypoechoic (n = 19) | Predominantly Hyperechoic (n = 45) | p |
|---|---|---|---|
| LDL(−) (%), md (IQR) | 9.5 (7.1–11.7) | 7.4 (5.6–9.9) | 0.010 |
| oxLDL (U/mmol LDLc), md (IQR) | 15.7 (5.4–21.6) | 11.4 (8.2–17.1) | 0.734 |
| LDLc (mM), md (IQR) | 3.02 (1.85–3.32) | 2.33 (1.76–2.90) | 0.228 |
| Intraplaque neovascularization | Present (n = 28) | Absent (n = 9) | p |
| LDL(−) (%), md (IQR) | 7.9 (6.5–10.5) | 6.5 (3.8–7.6) | 0.047 |
| oxLDL (U/mmol LDLc), md (IQR) | 13.2 (7.0–17.1) | 12.3 (11.6–13.1) | 0.976 |
| LDLc (mM), md (IQR) | 2.35 (1.89–3.20) | 2.71 (2.01–3.70) | 0.481 |
| Diffuse intraplaque neovascularization | Present (n = 14) | Absent (n = 23) | p |
| LDL(−) (%), md (IQR) | 9.0 (7.1–11.7) | 7.0 (4.6–9.2) | 0.033 |
| oxLDL (U/mmol LDLc), md (IQR) | 15.5 (6.2–17.1) | 11.9 (8.8–14.5) | 0.569 |
| LDLc (mM), md (IQR) | 3.03 (1.90–3.44) | 2.40 (1.82–3.1) | 0.468 |
| Intraplaque inflammation | SUVmax ≥ 2.85 g/mL (n = 26) | SUVmax < 2.85 g/mL (n = 38) | p |
| LDL(−) (%), md (IQR) | 7.2 (5.4–10.9) | 8.0 (6.2–10.2) | 0.642 |
| oxLDL (U/mmol LDLc), md (IQR) | 15.9 (10.1–18.7) | 9.9 (7.0–16.9) | 0.127 |
| LDLc (mM), md (IQR) | 2.26 (1.89–3.18) | 2.52 (1.75–3.37) | 0.603 |
| OR | 95% CI | p | |
|---|---|---|---|
| (a) Plaque stenosis ≥50% | |||
| Proportion of LDL(−) (×1 increase in the logarithm) | 5.40 | 1.15–25.44 | 0.033 |
| Hypertension | 0.12 | 0.01–1.02 | 0.052 |
| (b) Predominantly hypoechoic plaque | |||
| Proportion of LDL(−) (×1 increase in the logarithm) | 7.52 | 1.26–44.83 | 0.027 |
| Prior statin therapy | 0.23 | 0.06–0.79 | 0.020 |
| (c) Diffuse intraplaque neovascularization | |||
| Proportion of LDL(−) (×1 increase in the logarithm) | 10.77 | 1.21–95.93 | 0.033 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puig, N.; Camps-Renom, P.; Solé, A.; Aguilera-Simón, A.; Jiménez-Xarrié, E.; Fernández-León, A.; Camacho, M.; Guasch-Jiménez, M.; Marin, R.; Martí-Fàbregas, J.; et al. Electronegative LDL Is Associated with Plaque Vulnerability in Patients with Ischemic Stroke and Carotid Atherosclerosis. Antioxidants 2023, 12, 438. https://doi.org/10.3390/antiox12020438
Puig N, Camps-Renom P, Solé A, Aguilera-Simón A, Jiménez-Xarrié E, Fernández-León A, Camacho M, Guasch-Jiménez M, Marin R, Martí-Fàbregas J, et al. Electronegative LDL Is Associated with Plaque Vulnerability in Patients with Ischemic Stroke and Carotid Atherosclerosis. Antioxidants. 2023; 12(2):438. https://doi.org/10.3390/antiox12020438
Chicago/Turabian StylePuig, Núria, Pol Camps-Renom, Arnau Solé, Ana Aguilera-Simón, Elena Jiménez-Xarrié, Alejandro Fernández-León, Mercedes Camacho, Marina Guasch-Jiménez, Rebeca Marin, Joan Martí-Fàbregas, and et al. 2023. "Electronegative LDL Is Associated with Plaque Vulnerability in Patients with Ischemic Stroke and Carotid Atherosclerosis" Antioxidants 12, no. 2: 438. https://doi.org/10.3390/antiox12020438
APA StylePuig, N., Camps-Renom, P., Solé, A., Aguilera-Simón, A., Jiménez-Xarrié, E., Fernández-León, A., Camacho, M., Guasch-Jiménez, M., Marin, R., Martí-Fàbregas, J., Martínez-Domeño, A., Prats-Sánchez, L., Casoni, F., Pérez, B., Jiménez-Altayó, F., Sánchez-Quesada, J. L., & Benitez, S. (2023). Electronegative LDL Is Associated with Plaque Vulnerability in Patients with Ischemic Stroke and Carotid Atherosclerosis. Antioxidants, 12(2), 438. https://doi.org/10.3390/antiox12020438