
Antenatal and Postnatal Sequelae of Oxidative Stress in Preterm Infants: A Narrative Review Targeting Pathophysiological Mechanisms


 , , ,
, , , {kind=link}
{kind=link}
Abstract
1. Introduction
2. OS and Pregnancy Disorders
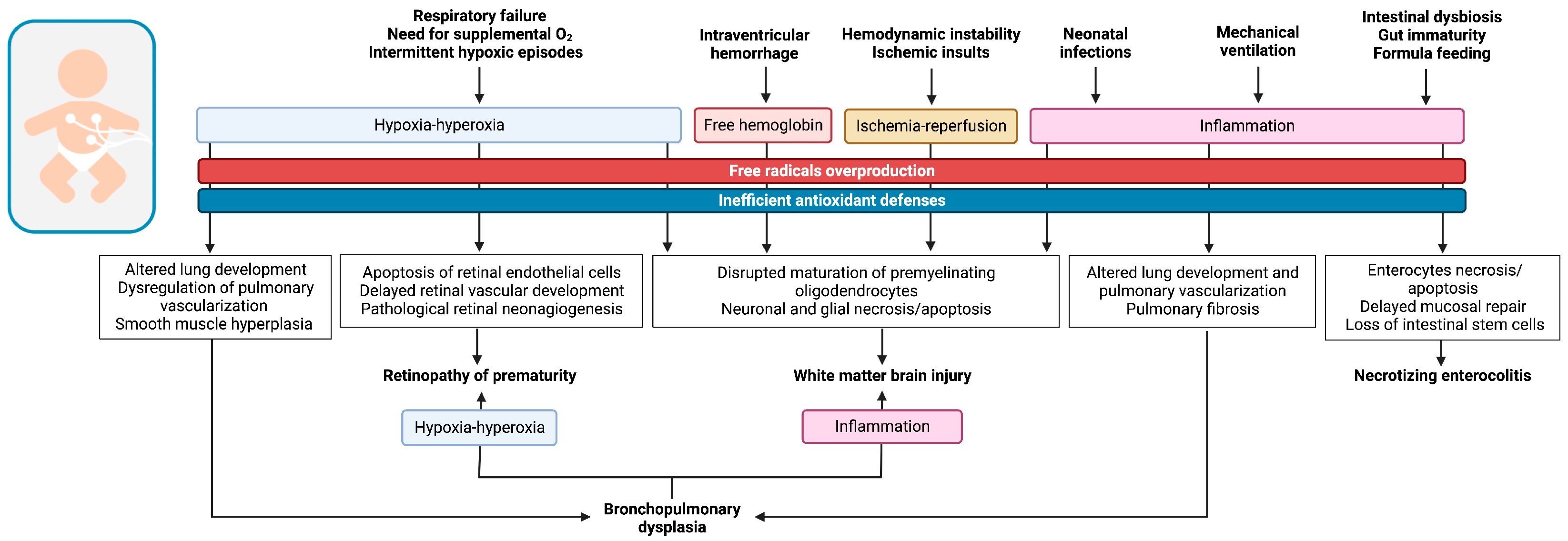
3. OS and Prematurity-Related Diseases
3.1. Respiratory Diseases of Preterm Infants
3.2. Retinopathy of Prematurity
3.3. Prematurity-Related Brain Injury
3.4. Necrotizing Enterocolitis
3.5. Neonatal Infections
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ozsurekci, Y.; Aykac, K. Oxidative Stress Related Diseases in Newborns. Oxidative Med. Cell. Longev. 2016, 2016, 2768365. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Arrigo, T.; Barberi, I.; Reiter, R.J.; Gitto, E. Oxidative Stress-Mediated Aging during the Fetal and Perinatal Periods. Oxidative Med. Cell. Longev. 2014, 2014, 358375. [Google Scholar] [CrossRef]
- Davis, J.M.; Auten, R.L. Maturation of the antioxidant system and the effects on preterm birth. Semin. Fetal Neonatal Med. 2010, 15, 191–195. [Google Scholar] [CrossRef]
- Folkerth, R.D.; Haynes, R.L.; Borenstein, N.S.; Belliveau, R.A.; Trachtenberg, F.; Rosenberg, P.A.; Volpe, J.J.; Kinney, H.C. Developmental Lag in Superoxide Dismutases Relative to Other Antioxidant Enzymes in Premyelinated Human Telencephalic White Matter. J. Neuropathol. Exp. Neurol. 2004, 63, 990–999. [Google Scholar] [CrossRef]
- Thibeault, D.W. The precarious antioxidant defenses of the preterm infant. Am. J. Perinatol. 2000, 17, 167–182. [Google Scholar] [CrossRef]
- Ferrante, G.; Montante, C.; Notarbartolo, V.; Giuffrè, M. Antioxidants: Role the in prevention and treatment of bronchopulmonary dysplasia. Paediatr. Respir. Rev. 2022, 42, 53–58. [Google Scholar] [CrossRef]
- Martini, S.; Castellini, L.; Parladori, R.; Paoletti, V.; Aceti, A.; Corvaglia, L. Free Radicals and Neonatal Brain Injury: From Underlying Pathophysiology to Antioxidant Treatment Perspectives. Antioxidants 2021, 10, 2012. [Google Scholar] [CrossRef]
- Lembo, C.; Buonocore, G.; Perrone, S. Oxidative Stress in Preterm Newborns. Antioxidants 2021, 10, 1672. [Google Scholar] [CrossRef]
- Falsaperla, R.; Lombardo, F.; Filosco, F.; Romano, C.; Saporito, M.A.N.; Puglisi, F.; Piro, E.; Ruggieri, M.; Pavone, P. Oxidative Stress in Preterm Infants: Overview of Current Evidence and Future Prospects. Pharmaceuticals 2020, 13, 145. [Google Scholar] [CrossRef]
- Sultana, Z.; Maiti, K.; Aitken, R.J.; Morris, J.; Dedman, L.; Smith, R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am. J. Reprod. Immunol. 2017, 77, e12653. [Google Scholar] [CrossRef]
- Pijnenborg, R.; Dixon, G.; Robertson, W.; Brosens, I. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta 1980, 1, 3–19. [Google Scholar] [CrossRef]
- Watson, A.L.; Skepper, J.N.; Jauniaux, E.; Burton, G.J. Susceptibility of Human Placental Syncytiotrophoblastic Mitochondria to Oxygen-Mediated Damage in Relation to Gestational Age. J. Clin. Endocrinol. Metab. 1998, 83, 1697–1705. [Google Scholar] [CrossRef]
- Genbacev, O.; Zhou, Y.; Ludlow, J.W.; Fisher, S.J. Regulation of Human Placental Development by Oxygen Tension. Science 1997, 277, 1669–1672. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.-P.; Skepper, J.N.; Burton, G.J. Onset of Maternal Arterial Blood Flow and Placental Oxidative Stress: A Possible Factor in Human Early Pregnancy Failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Jauniaux, E.; Hempstock, J.; Greenwold, N.; Burton, G.J. Trophoblastic Oxidative Stress in Relation to Temporal and Regional Differences in Maternal Placental Blood Flow in Normal and Abnormal Early Pregnancies. Am. J. Pathol. 2003, 162, 115–125. [Google Scholar] [CrossRef]
- Tsonis, O.; Balogun, S.; Adjei, J.O.; Mogekwu, O.; Iliodromiti, S. Management of recurrent miscarriages: An overview of current evidence. Curr. Opin. Obstet. Gynecol. 2021, 33, 370–377. [Google Scholar] [CrossRef]
- Simşek, M.; Naziroğlu, M.; Simşek, H.; Cay, M.; Aksakal, M.; Kumru, S. Blood plasma levels of lipoperoxides, glutathione peroxidase, beta carotene, vitamin A and E in women with habitual abortion. Cell Biochem. Funct. 1998, 16, 227–231. [Google Scholar] [CrossRef]
- Biri, A.; Bozkurt, N.; Turp, A.; Kavutcu, M.; Himmetoglu, Ö.; Durak, I. Role of Oxidative Stress in Intrauterine Growth Restriction. Gynecol. Obstet. Investig. 2007, 64, 187–192. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 204: Fetal Growth Restriction. Obstet. Gynecol. 2019, 133, e97–e109. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.C.; Cluver, C.A.; Kingdom, J.; Tong, S. Pre-eclampsia. Lancet 2021, 398, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Jaleel, A.; Tamimi, W.; Al Kadri, H.M.F. Role of oxidative stress in the pathogenesis of preeclampsia. Arch. Gynecol. Obstet. 2010, 282, 469–474. [Google Scholar] [CrossRef]
- Sankaralingam, S.; Arenas, I.A.; Lalu, M.M.; Davidge, S.T. Preeclampsia: Current understanding of the molecular basis of vascular dysfunction. Expert Rev. Mol. Med. 2006, 8, 1–20. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 190: Gestational Diabetes Mellitus. Obstet. Gynecol. 2018, 131, e49–e64. [Google Scholar] [CrossRef]
- Phoswa, W.N.; Khaliq, O.P. The Role of Oxidative Stress in Hypertensive Disorders of Pregnancy (Preeclampsia, Gestational Hypertension) and Metabolic Disorder of Pregnancy (Gestational Diabetes Mellitus). Oxidative Med. Cell. Longev. 2021, 2021, 5581570. [Google Scholar] [CrossRef]
- Arribas, L.; Almansa, I.; Miranda, M.; Muriach, M.; Romero, F.J.; Villar, V.M. Serum Malondialdehyde Concentration and Glutathione Peroxidase Activity in a Longitudinal Study of Gestational Diabetes. PLoS ONE 2016, 11, e0155353. [Google Scholar] [CrossRef]
- Biri, A.; Onan, A.; Devrim, E.; Babacan, F.; Kavutcu, M.; Durak, I. Oxidant Status in Maternal and Cord Plasma and Placental Tissue in Gestational Diabetes. Placenta 2006, 27, 327–332. [Google Scholar] [CrossRef]
- Coughlan, M.; Vervaart, P.; Permezel, M.; Georgiou, H.; Rice, G. Altered Placental Oxidative Stress Status in Gestational Diabetes Mellitus. Placenta 2004, 25, 78–84. [Google Scholar] [CrossRef]
- Rudge, M.V.C.; Costa, E.; Barbisan, L.F.; Damasceno, D.C.; Bueno, A.; Saito, F.H.; Calderon, I.; Rodrigues, M.M.P. Evaluation of cell proliferation and apoptosis in placentas of rats with severe diabetes. Braz. Arch. Biol. Technol. 2012, 55, 243–250. [Google Scholar] [CrossRef]
- Correa, A.; Gilboa, S.M.; Besser, L.M.; Botto, L.D.; Moore, C.A.; Hobbs, C.A.; Cleves, M.A.; Riehle-Colarusso, T.J.; Waller, D.K.; Reece, E.A. Diabetes mellitus and birth defects. Am. J. Obstet. Gynecol. 2008, 199, 237.e1–237.e9. [Google Scholar] [CrossRef]
- Tinker, S.C.; Gilboa, S.M.; Moore, C.A.; Waller, D.K.; Simeone, R.M.; Kim, S.Y.; Jamieson, D.J.; Botto, L.D.; Reefhuis, J. Specific birth defects in pregnancies of women with diabetes: National Birth Defects Prevention Study, 1997–2011. Am. J. Obstet. Gynecol. 2020, 222, 176.e1–176.e11. [Google Scholar] [CrossRef]
- Weksler-Zangen, S.; Yaffe, P.; Ornoy, A. Reduced SOD activity and increased neural tube defects in embryos of the sensitive but not of the resistant Cohen diabetic rats cultured under diabetic conditions. Birth Defects Res. 2003, 67, 429–437. [Google Scholar] [CrossRef]
- Moore, T.A.; Ahmad, I.M.; Zimmerman, M.C. Oxidative Stress and Preterm Birth: An Integrative Review. Biol. Res. Nurs. 2018, 20, 497–512. [Google Scholar] [CrossRef]
- Woods, J. Reactive Oxygen Species and Preterm Premature Rupture of Membranes—A Review. Placenta 2001, 22, S38–S44. [Google Scholar] [CrossRef]
- Kumar, N.; Nandula, P.; Menden, H.; Jarzembowski, J.; Sampath, V. Placental TLR/NLR expression signatures are altered with gestational age and inflammation. J. Matern. Fetal Neonatal Med. 2017, 30, 1588–1595. [Google Scholar] [CrossRef]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef]
- Menon, R.; Fortunato, S.J.; Milne, G.L.; Brou, L.M.; Carnevale, C.M.; Sanchez, S.C.M.; Hubbard, L.B.; Lappas, M.; Drobek, C.O.B.; Taylor, R.N. Amniotic Fluid Eicosanoids in Preterm and Term Births: Effects of Risk Factors for Spontaneous Preterm Labor. Obstet. Gynecol. 2011, 118, 121–134. [Google Scholar] [CrossRef]
- Dutta, E.H.; Behnia, F.; Boldogh, I.; Saade, G.R.; Taylor, B.D.; Kacerovský, M.; Menon, R. Oxidative stress damage-associated molecular signaling pathways differentiate spontaneous preterm birth and preterm premature rupture of the membranes. Mol. Hum. Reprod. 2016, 22, 143–157. [Google Scholar] [CrossRef]
- Indrio, F.; Martini, S.; Francavilla, R.; Corvaglia, L.; Cristofori, F.; Mastrolia, S.A.; Neu, J.; Rautava, S.; Spena, G.R.; Raimondi, F.; et al. Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-term Health Development. Front. Pediatr. 2017, 5, 178. [Google Scholar] [CrossRef] [PubMed]
- Moraes-Souza, R.Q.; Vesentini, G.; Paula, V.G.; Sinzato, Y.K.; Soares, T.S.; Gelaleti, R.B.; Volpato, G.T.; Damasceno, D.C. Oxidative Stress Profile of Mothers and Their Offspring after Maternal Consumption of High-Fat Diet in Rodents: A Systematic Review and Meta-Analysis. Oxidative Med. Cell. Longev. 2021, 2021, 9073859. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, S.M.; Braz, G.R.F.; Freitas, C.D.M.; de Santana, D.F.; Sellitti, D.F.; Fernandes, M.P.; Lagranha, C.J. Oxidative injuries induced by maternal low-protein diet in female brainstem. Nutr. Neurosci. 2018, 21, 580–588. [Google Scholar] [CrossRef]
- Tain, Y.-L.; Hsu, C.-N. Maternal High-Fat Diet and Offspring Hypertension. Int. J. Mol. Sci. 2022, 23, 8179. [Google Scholar] [CrossRef] [PubMed]
- Edlow, A.G. Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat. Diagn. 2017, 37, 95–110. [Google Scholar] [CrossRef]
- Sinzato, Y.K.; Paula, V.G.; Gallego, F.Q.; Moraes-Souza, R.Q.; Corrente, J.E.; Volpato, G.T.; Damasceno, D.C. Maternal Diabetes and Postnatal High-Fat Diet on Pregnant Offspring. Front. Cell Dev. Biol. 2022, 10, 818621. [Google Scholar] [CrossRef]
- Pedroza, A.; Ferreira, D.S.; Santana, D.F.; da Silva, P.T.; de Aguiar Júnior, F.C.A.; Sellitti, D.F.; Lagranha, C.J. A maternal low-protein diet and neonatal overnutrition result in similar changes to glomerular morphology and renal cortical oxidative stress measures in male Wistar rats. Appl. Physiol. Nutr. Metab. 2019, 44, 164–171. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Chen, J.; Jones, R.H.; Smith, N.H.; Ozanne, S.E. Poor maternal nutrition leads to alterations in oxidative stress, antioxidant defense capacity, and markers of fibrosis in rat islets: Potential underlying mechanisms for development of the diabetic phenotype in later life. FASEB J. 2010, 24, 2762–2771. [Google Scholar] [CrossRef]
- Sikalidis, A.K.; Maykish, A. The Gut Microbiome and Type 2 Diabetes Mellitus: Discussing A Complex Relationship. Biomedicines 2020, 8, 8. [Google Scholar] [CrossRef]
- Hu, C.; Yan, Y.; Ji, F.; Zhou, H. Maternal Obesity Increases Oxidative Stress in Placenta and It Is Associated with Intestinal Microbiota. Front. Cell. Infect. Microbiol. 2021, 11, 671347. [Google Scholar] [CrossRef]
- Mazenc, A.; Mervant, L.; Maslo, C.; Lencina, C.; Bézirard, V.; Levêque, M.; Ahn, I.; Alquier-Bacquié, V.; Naud, N.; Héliès-Toussaint, C.; et al. Maternal heme-enriched diet promotes a gut pro-oxidative status associated with microbiota alteration, gut leakiness and glucose intolerance in mice offspring. Redox Biol. 2022, 53, 102333. [Google Scholar] [CrossRef]
- Wang, Y.W.; Yu, H.R.; Tiao, M.M.; Tain, Y.L.; Lin, I.C.; Sheen, J.M.; Lin, Y.J.; Chang, K.A.; Chen, C.C.; Tsai, C.C.; et al. Maternal Obesity Related to High Fat Diet Induces Placenta Remodeling and Gut Microbiome Shaping That Are Responsible for Fetal Liver Lipid Dysmetabolism. Front. Nutr. 2021, 8, 736944. [Google Scholar] [CrossRef]
- Gao, Y.; Nanan, R.; Macia, L.; Tan, J.; Sominsky, L.; Quinn, T.P.; O’Hely, M.; Ponsonby, A.-L.; Tang, M.L.; Collier, F.; et al. The maternal gut microbiome during pregnancy and offspring allergy and asthma. J. Allergy Clin. Immunol. 2021, 148, 669–678. [Google Scholar] [CrossRef]
- Sweet, D.G.; Carnielli, V.; Greisen, G.; Hallman, M.; Ozek, E.; Pas, A.T.; Plavka, R.; Roehr, C.C.; Saugstad, O.D.; Simeoni, U.; et al. European Consensus Guidelines on the Management of Respiratory Distress Syndrome–2019 Update. Neonatology 2019, 115, 432–450. [Google Scholar] [CrossRef]
- Gilfillan, M.; Bhandari, A.; Bhandari, V. Diagnosis and management of bronchopulmonary dysplasia. BMJ 2021, 375, n1974. [Google Scholar] [CrossRef]
- Cannavò, L.; Perrone, S.; Viola, V.; Marseglia, L.; Di Rosa, G.; Gitto, E. Oxidative Stress and Respiratory Diseases in Preterm Newborns. Int. J. Mol. Sci. 2021, 22, 12504. [Google Scholar] [CrossRef]
- Choi, Y.; Rekers, L.; Dong, Y.; Holzfurtner, L.; Goetz, M.J.; Shahzad, T.; Zimmer, K.-P.; Behnke, J.; Behnke, J.; Bellusci, S.; et al. Oxygen Toxicity to the Immature Lung—Part I: Pathomechanistic Understanding and Preclinical Perspectives. Int. J. Mol. Sci. 2021, 22, 11006. [Google Scholar] [CrossRef]
- Saugstad, O.D.; Oei, J.-L.; Lakshminrusimha, S.; Vento, M. Oxygen therapy of the newborn from molecular understanding to clinical practice. Pediatr. Res. 2019, 85, 20–29. [Google Scholar] [CrossRef]
- Jensen, E.A.; Whyte, R.K.; Schmidt, B.; Bassler, D.; Vain, N.E.; Roberts, R.S.; Shah, P.; Brown, L.; Wenger, L.; Frye, S.; et al. Association between Intermittent Hypoxemia and Severe Bronchopulmonary Dysplasia in Preterm Infants. Am. J. Respir. Crit. Care Med. 2021, 204, 1192–1199. [Google Scholar] [CrossRef]
- Bik-Multanowski, M.; Revhaug, C.; Grabowska, A.; Dobosz, A.; Madetko-Talowska, A.; Zasada, M.; Saugstad, O.D. Hyperoxia induces epigenetic changes in newborn mice lungs. Free. Radic. Biol. Med. 2018, 121, 51–56. [Google Scholar] [CrossRef]
- Damianos, A.; Kulandavelu, S.; Chen, P.; Nwajei, P.; Batlahally, S.; Sharma, M.; Alvarez-Cubela, S.; Domínguez-Bendala, J.; Zambrano, R.; Huang, J.; et al. Neonatal intermittent hypoxia persistently impairs lung vascular development and induces longterm lung mitochondrial DNA damage. J. Appl. Physiol. 2022, 133, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Gronbach, J.; Shahzad, T.; Radajewski, S.; Chao, C.-M.; Bellusci, S.; Morty, R.E.; Reicherzer, T.; Ehrhardt, H. The Potentials and Caveats of Mesenchymal Stromal Cell-Based Therapies in the Preterm Infant. Stem Cells Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, G.; Carota, G.; Volti, G.L.; Giuffrè, M. Biomarkers of Oxidative Stress for Neonatal Lung Disease. Front. Pediatr. 2021, 9, 618867. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Granese, R.; Falsaperla, R.; Reiter, R.J.; Corsello, G.; Gitto, E. Role of oxidative stress in neonatal respiratory distress syndrome. Free. Radic. Biol. Med. 2019, 142, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Negi, R.; Pande, D.; Karki, K.; Kumar, A.; Khanna, R.S.; Khanna, H.D. A novel approach to study oxidative stress in neonatal respiratory distress syndrome. BBA Clin. 2015, 3, 65–69. [Google Scholar] [CrossRef]
- Dizdar, E.A.; Uras, N.; Oguz, S.; Erdeve, O.; Sari, F.N.; Aydemir, C.; Dilmen, U. Total antioxidant capacity and total oxidant status after surfactant treatment in preterm infants with respiratory distress syndrome. Ann. Clin. Biochem. 2011, 48, 462–467. [Google Scholar] [CrossRef]
- Elkabany, Z.A.; El-Farrash, R.A.; Shinkar, D.M.; Ismail, E.A.; Nada, A.S.; Farag, A.S.; Elsayed, M.A.; Salama, D.H.; Macken, E.L.; Gaballah, S.A. Oxidative stress markers in neonatal respiratory distress syndrome: Advanced oxidation protein products and 8-hydroxy-2-deoxyguanosine in relation to disease severity. Pediatr. Res. 2020, 87, 74–80. [Google Scholar] [CrossRef]
- Carvalho, C.G.; Procianoy, R.S.; Neto, E.C.; Silveira, R.C. Preterm Neonates with Respiratory Distress Syndrome: Ventilator-Induced Lung Injury and Oxidative Stress. J. Immunol. Res. 2018, 2018, 696375. [Google Scholar] [CrossRef]
- Jensen, E.A.; Dysart, K.; Gantz, M.G.; McDonald, S.; Bamat, N.A.; Keszler, M.; Kirpalani, H.; Laughon, M.M.; Poindexter, B.B.; Duncan, A.F.; et al. The Diagnosis of Bronchopulmonary Dysplasia in Very Preterm Infants. An Evidence-based Approach. Am. J. Respir. Crit. Care Med. 2019, 200, 751–759. [Google Scholar] [CrossRef]
- Higgins, R.D.; Jobe, A.H.; Koso-Thomas, M.; Bancalari, E.; Viscardi, R.M.; Ha rtert, T.V.; Ryan, R.M.; Kallapur, S.G.; Steinhorn, R.H.; Konduri, G.G.; et al. Bronchopulmonary Dysplasia: Executive Summary of a Workshop. J. Pediatr. 2018, 197, 300–308. [Google Scholar] [CrossRef]
- Sahni, M.; Bhandari, V. Patho-mechanisms of the origins of bronchopulmonary dysplasia. Mol. Cell. Pediatr. 2021, 8, 21. [Google Scholar] [CrossRef]
- Yucel, O.E.; Eraydin, B.; Niyaz, L.; Terzi, O. Incidence and risk factors for retinopathy of prematurity in premature, extremely low birth weight and extremely low gestational age infants. BMC Ophthalmol. 2022, 22, 367. [Google Scholar] [CrossRef]
- Dammann, O.; Hartnett, M.E.; Stahl, A. Retinopathy of prematurity. Dev. Med. Child Neurol. 2022. [CrossRef]
- Hellström, A.; Smith, L.E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457. [Google Scholar] [CrossRef]
- Graziosi, A.; Perrotta, M.; Russo, D.; Gasparroni, G.; D’Egidio, C.; Marinelli, B.; Di Marzio, G.; Falconio, G.; Mastropasqua, L.; Volti, G.L.; et al. Oxidative Stress Markers and the Retinopathy of Prematurity. J. Clin. Med. 2020, 9, 2711. [Google Scholar] [CrossRef]
- Stone, W.L.; Shah, D.; Hollinger, S.M. Retinopathy of prematurity an oxidative stress neonatal disease. Front. Biosci. 2016, 21, 165–177. [Google Scholar] [CrossRef]
- Saito, Y.; Geisen, P.; Uppal, A.; Hartnett, M.E. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol. Vis. 2007, 13, 840–853. [Google Scholar]
- Uno, K.; Prow, T.W.; Bhutto, I.A.; Yerrapureddy, A.; McLeod, D.S.; Yamamoto, M.; Reddy, S.P.; Lutty, G.A. Role of Nrf2 in retinal vascular development and the vaso-obliterative phase of oxygen-induced retinopathy. Exp. Eye Res. 2010, 90, 493–500. [Google Scholar] [CrossRef]
- Hartnett, M.E.; Penn, J.S. Mechanisms and Management of Retinopathy of Prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef]
- Hartnett, M.E. Pathophysiology and Mechanisms of Severe Retinopathy of Prematurity. Ophthalmology 2015, 122, 200–210. [Google Scholar] [CrossRef]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter Via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Fevereiro-Martins, M.D.R.; Marques-Neves, C.A.M.; Guimarães, H.; Bicho, M.D.P. Retinopathy of prematurity: A review of pathophysiology and signaling pathways. Surv. Ophthalmol. 2023, 68, 175–210. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, M.H.; Martinez-Bermudez, A.K.; Gobeil, F., Jr.; Marrache, A.M.; Hou, X.; Speranza, G.; Abran, D.; Quiniou, C.; Lachapelle, P.; Roberts, J., II; et al. Role of thromboxane in retinal microvascular degeneration in oxygen-induced retinopathy. J. Appl. Physiol. 2001, 90, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Gobeil, F.; Peri, K.; Speranza, G.; Marrache, A.M.; Lachapelle, P.; Roberts, J.; Varma, D.R.; Chemtob, S. Augmented Vasoconstriction and Thromboxane Formation by 15-F 2t -Isoprostane (8-Iso-Prostaglandin F 2α ) in Immature Pig Periventricular Brain Microvessels. Stroke 2000, 31, 516–524; discussion 525. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.J.; Beharry, K.D.; Brock, R.S.; Abad-Santos, P.; Abad-Santos, M.; Modanlou, H.D. Effects of Brief, Clustered Versus Dispersed Hypoxic Episodes on Systemic and Ocular Growth Factors in a Rat Model of Oxygen-Induced Retinopathy. Pediatr. Res. 2008, 64, 50–55. [Google Scholar] [CrossRef]
- McColm, J.R.; Geisen, P.; Hartnett, M.E. VEGF isoforms and their expression after a single episode of hypoxia or repeated fluctuations between hyperoxia and hypoxia: Relevance to clinical ROP. Mol. Vis. 2004, 10, 512–520. [Google Scholar]
- Di Fiore, J.M.; Kaffashi, F.; Loparo, K.; Sattar, A.; Schluchter, M.; Foglyano, R.; Martin, R.J.; Wilson, C.G. The relationship between patterns of intermittent hypoxia and retinopathy of prematurity in preterm infants. Pediatr. Res. 2012, 72, 606–612. [Google Scholar] [CrossRef]
- Di Fiore, J.M.; Bloom, J.N.; Orge, F.; Schutt, A.; Schluchter, M.; Cheruvu, V.K.; Walsh, M.; Finer, N.; Martin, R.J. A Higher Incidence of Intermittent Hypoxemic Episodes Is Associated with Severe Retinopathy of Prematurity. J. Pediatr. 2010, 157, 69–73. [Google Scholar] [CrossRef]
- Aranda, J.V.; Cai, C.L.; Ahmad, T.; Bronshtein, V.; Sadeh, J.; Valencia, G.B.; Lazzaro, D.R.; Beharry, K.D. Pharmacologic synergism of ocular ketorolac and systemic caffeine citrate in rat oxygen-induced retinopathy. Pediatr. Res. 2016, 80, 554–565. [Google Scholar] [CrossRef]
- Panfoli, I.; Candiano, G.; Malova, M.; De Angelis, L.C.; Cardiello, V.; Buonocore, G.; Ramenghi, L.A. Oxidative Stress as a Primary Risk Factor for Brain Damage in Preterm Newborns. Front. Pediatr. 2018, 6, 369. [Google Scholar] [CrossRef]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative stress disrupts oligodendrocyte maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Coviello, C.; Perrone, S.; Buonocore, G.; Negro, S.; Longini, M.; Dani, C.; de Vries, L.S.; Groenendaal, F.; Vijlbrief, D.C.; Benders, M.J.N.L.; et al. Isoprostanes as Biomarker for White Matter Injury in Extremely Preterm Infants. Front. Pediatr. 2021, 8, 618622. [Google Scholar] [CrossRef]
- Ophelders, D.R.M.G.; Gussenhoven, R.; Klein, L.; Jellema, R.K.; Westerlaken, R.J.; Hütten, M.C.; Vermeulen, J.; Wassink, G.; Gunn, A.J.; Wolfs, T.G. Preterm brain injury, antenatal triggers, and therapeutics: Timing is key. Cells 2020, 9, 1871. [Google Scholar] [CrossRef]
- Brekke, E.; Berger, H.R.; Widerøe, M.; Sonnewald, U.; Morken, T.S. Glucose and Intermediary Metabolism and Astrocyte–Neuron Interactions Following Neonatal Hypoxia–Ischemia in Rat. Neurochem. Res. 2017, 42, 115–132. [Google Scholar] [CrossRef]
- Hope, P.L.; Cady, E.B.; Delpy, D.T.; Ives, N.K.; Gardiner, R.M.; Reynolds, E.O.R. Brain Metabolism and Intracellular pH During Ischaemia: Effects of Systemic Glucose and Bicarbonate Administration Studied by31P and1H Nuclear Magnetic Resonance Spectroscopy In Vivo in the Lamb. J. Neurochem. 1988, 50, 1394–1402. [Google Scholar] [CrossRef]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age 1997, 20, 127–140. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]
- Laptook, A.R. Birth Asphyxia and Hypoxic-Ischemic Brain Injury in the Preterm Infant. Clin. Perinatol. 2016, 43, 529–545. [Google Scholar] [CrossRef]
- Di Fiore, J.M.; Vento, M. Intermittent hypoxemia and oxidative stress in preterm infants. Respir. Physiol. Neurobiol. 2019, 266, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, J.M.; Raffay, T.M. The relationship between intermittent hypoxemia events and neural outcomes in neonates. Exp. Neurol. 2021, 342, 113753. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A. White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Liu, Y.; Yuan, X.; Yang, L.; Zhang, R.; Zhang, X.; Wang, X.; Xu, F.; Zhu, C. Early application of caffeine improves white matter development in very preterm infants. Respir. Physiol. Neurobiol. 2020, 281, 103495. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J. The Redox Activity of Hemoglobins: From Physiologic Functions to Pathologic Mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef]
- Pandya, C.D.; Vekaria, H.; Joseph, B.; Slone, S.A.; Gensel, J.C.; Sullivan, P.G.; Miller, B.A. Hemoglobin induces oxidative stress and mitochondrial dysfunction in oligodendrocyte progenitor cells. Transl. Res. 2021, 231, 13–23. [Google Scholar] [CrossRef]
- Ballabh, P.; de Vries, L.S. White matter injury in infants with intraventricular haemorrhage: Mechanisms and therapies. Nat. Rev. Neurol. 2021, 17, 199–214. [Google Scholar] [CrossRef]
- Wan, J.; Ren, H.; Wang, J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 2019, 4, 93–95. [Google Scholar] [CrossRef]
- Romantsik, O.; Bruschettini, M.; Ley, D. Intraventricular Hemorrhage and White Matter Injury in Preclinical and Clinical Studies. Neoreviews 2019, 20, e636–e652. [Google Scholar] [CrossRef]
- Adler, I.; Batton, D.; Betz, B.; Bezinque, S.; Ecklund, K.; Junewick, J.; McCauley, R.; Miller, C.; Seibert, J.; Specter, B.; et al. Mechanisms of injury to white matter adjacent to a large intraventricular hemorrhage in the preterm brain. J. Clin. Ultrasound 2010, 38, 254–258. [Google Scholar] [CrossRef]
- Zia, M.T.; Csiszar, A.; Labinskyy, N.; Hu, F.; Vinukonda, G.; LaGamma, E.F.; Ungvari, Z.; Ballabh, P.; Kim, Y.M.; Guzik, T.J.; et al. Oxidative-Nitrosative Stress in a Rabbit Pup Model of Germinal Matrix Hemorrhage: Role of NAD(P)H oxidase. Stroke 2009, 40, 2191–2198. [Google Scholar] [CrossRef]
- Goulding, D.S.; Vogel, R.C.; Gensel, J.C.; Morganti, J.M.; Stromberg, A.J.; Miller, B.A. Acute brain inflammation, white matter oxidative stress, and myelin deficiency in a model of neonatal intraventricular hemorrhage. J. Neurosurg. Pediatr. 2020, 26, 613–623. [Google Scholar] [CrossRef]
- Jaganjac, M.; Cipak, A.; Schaur, R.J.; Zarkovic, N. Pathophysiology of neutrophil-mediated extracellular redox reactions. Front. Biosci. 2016, 21, 839–855. [Google Scholar] [CrossRef]
- Lu, H.-Y.; Zhang, Q.; Wang, Q.-X.; Lu, J.-Y. Contribution of Histologic Chorioamnionitis and Fetal Inflammatory Response Syndrome to Increased Risk of Brain Injury in Infants With Preterm Premature Rupture of Membranes. Pediatr. Neurol. 2016, 61, 94–98.e1. [Google Scholar] [CrossRef]
- Anblagan, D.; Pataky, R.; Evans, M.J.; Telford, E.J.; Serag, A.; Sparrow, S.; Piyasena, C.; Semple, S.I.; Wilkinson, A.G.; Bastin, M.E.; et al. Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 2016, 6, 37932. [Google Scholar] [CrossRef]
- Procianoy, R.S.; Silveira, R.C. Association between high cytokine levels with white matter injury in preterm infants with sepsis. Pediatr. Crit. Care Med. 2012, 13, 183–187. [Google Scholar] [CrossRef]
- Gagliardi, L.; Bellu’, R.; Zanini, R.; Dammann, O. Bronchopulmonary dysplasia and brain white matter damage in the preterm infant: A complex relationship. Paediatr. Périnat. Epidemiol. 2009, 23, 582–590. [Google Scholar] [CrossRef]
- Folkerth, R.D.; Keefe, R.J.; Haynes, R.L.; Trachtenberg, F.L.; Volpe, J.J.; Kinney, H.C. Interferon-γ Expression in Periventricular Leukomalacia in the Human Brain. Brain Pathol. 2004, 14, 265–274. [Google Scholar] [CrossRef]
- Kadhim, H.; Tabarki, B.; Verellen, G.; De Prez, C.; Rona, A.-M.; Sebire, G. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology 2001, 56, 1278–1284. [Google Scholar] [CrossRef]
- Yoon, B.H.; Romero, R.; Kim, C.J.; Koo, J.N.; Choe, G.; Syn, H.C.; Chi, J.-G. High expression of tumor necrosis factor-α and interleukin-6 in periventricular leukomalacia. Am. J. Obstet. Gynecol. 1997, 177, 406–411. [Google Scholar] [CrossRef]
- Haynes, R.L.; Folkerth, R.D.; Keefe, R.J.; Sung, I.; Swzeda, L.I.; Rosenberg, P.; Volpe, J.J.; Kinney, H.C. Nitrosative and Oxidative Injury to Premyelinating Oligodendrocytes in Periventricular Leukomalacia. J. Neuropathol. Exp. Neurol. 2003, 62, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Walker, W.A. Necrotizing Enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, S.C.; Ching, Y.; Yu, D.; Carpenter, J.; Kenny, M.; Weldon, C.; Lillehei, C.; Valim, C.; Horbar, J.D.; Jaksic, T. Mortality of necrotizing enterocolitis expressed by birth weight categories. J. Pediatr. Surg. 2009, 44, 1072–1076; discussion 1075–1076. [Google Scholar] [CrossRef] [PubMed]
- Hackam, D.J.; Sodhi, C.P. Bench to bedside—New insights into the pathogenesis of necrotizing enterocolitis. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, C.; Dilli, D.; Uras, N.; Ulu, H.O.; Oguz, S.S.; Erdeve, O.; Dilmen, U. Total oxidant status and oxidative stress are increased in infants with necrotizing enterocolitis. J. Pediatr. Surg. 2011, 46, 2096–2100. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.L.; Negro, S.; Cornacchione, S.; Longini, M.; Proietti, F.; Soubasi, V.; Benders, M.J.; Van Bel, F.; Buonocore, G. May oxidative stress biomarkers in cord blood predict the occurrence of necrotizing enterocolitis in preterm infants? J. Matern. Fetal Neonatal Med. 2012, 25 (Suppl. 1), 128–131. [Google Scholar] [CrossRef]
- Yazji, I.; Sodhi, C.P.; Lee, E.K.; Good, M.; Egan, C.E.; Afrazi, A.; Neal, M.D.; Jia, H.; Lin, J.; Ma, C.; et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS–NO–nitrite signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 9451–9456. [Google Scholar] [CrossRef]
- Ferretti, E.; Tremblay, E.; Thibault, M.-P.; Grynspan, D.; Burghardt, K.M.; Bettolli, M.; Babakissa, C.; Levy, E.; Beaulieu, J.-F. The nitric oxide synthase 2 pathway is targeted by both pro- and anti-inflammatory treatments in the immature human intestine. Nitric Oxide 2017, 66, 53–61. [Google Scholar] [CrossRef]
- Whitehouse, J.S.; Xu, H.; Shi, Y.; Noll, L.; Kaul, S.; Jones, D.W.; Pritchard, K.A.; Oldham, K.T.; Gourlay, D.M. Mesenteric Nitric Oxide and Superoxide Production in Experimental Necrotizing Enterocolitis. J. Surg. Res. 2010, 161, 1–8. [Google Scholar] [CrossRef]
- Grishin, A.; Bowling, J.; Bell, B.; Wang, J.; Ford, H.R. Roles of nitric oxide and intestinal microbiota in the pathogenesis of necrotizing enterocolitis. J. Pediatr. Surg. 2016, 51, 13–17. [Google Scholar] [CrossRef]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Hassan Talukder, M.A.; Chen, Y.-R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef]
- Sullivan, S.; Schanler, R.J.; Kim, J.H.; Patel, A.L.; Trawöger, R.; Kiechl-Kohlendorfer, U.; Chan, G.M.; Blanco, C.L.; Abrams, S.; Cotten, C.M.; et al. An Exclusively Human Milk-Based Diet Is Associated with a Lower Rate of Necrotizing Enterocolitis than a Diet of Human Milk and Bovine Milk-Based Products. J. Pediatr. 2010, 156, 562–567.e1. [Google Scholar] [CrossRef]
- Shoji, H.; Shimizu, T.; Shinohara, K.; Oguchi, S.; Shiga, S.; Yamashiro, Y. Suppressive effects of breast milk on oxidative DNA damage in very low birthweight infants. Arch. Dis. Child. Fetal Neonatal Ed. 2004, 89, F136–F138. [Google Scholar] [CrossRef]
- Friel, J.K.; Martin, S.M.; Langdon, M.; Herzberg, G.R.; Buettner, G.R. Milk from Mothers of Both Premature and Full-Term Infants Provides Better Antioxidant Protection than Does Infant Formula. Pediatr. Res. 2002, 51, 612–618. [Google Scholar] [CrossRef]
- Friel, J.K.; Diehl-Jones, B.; Cockell, K.A.; Chiu, A.; Rabanni, R.; Davies, S.S.; Roberts, L.J. Evidence of Oxidative Stress in Relation to Feeding Type During Early Life in Premature Infants. Pediatr. Res. 2011, 69, 160–164. [Google Scholar] [CrossRef]
- Good, M.; Sodhi, C.P.; Yamaguchi, Y.; Jia, H.; Lu, P.; Fulton, W.B.; Martin, L.Y.; Prindle, T.; Nino, D.F.; Zhou, Q.; et al. The human milk oligosaccharide 2′-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. Br. J. Nutr. 2016, 116, 1175–1187. [Google Scholar] [CrossRef]
- Kim, M.; Christley, S.; Alverdy, J.C.; Liu, D.; An, G. Immature Oxidative Stress Management as a Unifying Principle in the Pathogenesis of Necrotizing Enterocolitis: Insights from an Agent-Based Model. Surg. Infect. 2012, 13, 18–32. [Google Scholar] [CrossRef]
- Carlisle, E.M.; Morowitz, M.J. The intestinal microbiome and necrotizing enterocolitis. Curr. Opin. Pediatr. 2013, 25, 382–387. [Google Scholar] [CrossRef]
- Stewart, C.J.; Embleton, N.D.; Marrs, E.C.L.; Smith, D.P.; Nelson, A.; Abdulkadir, B.; Skeath, T.; Petrosino, J.F.; Perry, J.D.; Berrington, J.E.; et al. Temporal bacterial and metabolic development of the preterm gut reveals specific signatures in health and disease. Microbiome 2016, 4, 67. [Google Scholar] [CrossRef]
- Pammi, M.; Cope, J.; Tarr, P.I.; Warner, B.B.; Morrow, A.L.; Mai, V.; Gregory, K.E.; Kroll, J.S.; McMurtry, V.; Ferris, M.J.; et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: A systematic review and meta-analysis. Microbiome 2017, 5, 31. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, Z.; Morales, M.; Wang, Y.; Khafipour, E.; Friel, J. Feeding practice influences gut microbiome composition in very low birth weight preterm infants and the association with oxidative stress: A prospective cohort study. Free. Radic. Biol. Med. 2019, 142, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Shang, Q.; Gao, Z.; Hao, F.; Guo, H.; Guo, C. Fecal microbiota transplantation (FMT) could reverse the severity of experimental necrotizing enterocolitis (NEC) via oxidative stress modulation. Free. Radic. Biol. Med. 2017, 108, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.; Michels, M.; Ávila, P.; Burger, H.; Milioli, M.V.M.; Dal-Pizzol, F. The protective effects of fecal microbiota transplantation in an experimental model of necrotizing enterocolitis. J. Pediatr. Surg. 2019, 54, 1578–1583. [Google Scholar] [CrossRef]
- Goldstein, S.L.; Somers, M.J.; Baum, M.A.; Symons, J.M.; Brophy, P.D.; Blowey, D.; Bunchman, T.E.; Baker, C.; Mottes, T.; Mcafee, N.; et al. Pediatric patients with multi-organ dysfunction syndrome receiving continuous renal replacement therapy. Kidney Int. 2005, 67, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Shankaran, S.; Laptook, A.R.; Walsh, M.C.; Hale, E.C.; Newman, N.S.; Schibler, K.; Carlo, W.A.; et al. Neonatal Outcomes of Extremely Preterm Infants from the NICHD Neonatal Research Network. Pediatrics 2010, 126, 443–456. [Google Scholar] [CrossRef]
- Spasojević, I.; Obradović, B.; Spasić, S. Bench-to-bedside review: Neonatal sepsis—Redox processes in pathogenesis. Crit. Care 2012, 16, 221. [Google Scholar] [CrossRef]
- Volante, E.; Moretti, S.; Pisani, F.; Bevilacqua, G. Early diagnosis of bacterial infection in the neonate. J. Matern. Fetal Neonatal Med. 2004, 16 (Suppl. 2), 13–16. [Google Scholar] [CrossRef]
- Segura-Cervantes, E.; Mancilla-Ramírez, J.; González-Canudas, J.; Alba, E.; Santillán-Ballesteros, R.; Morales-Barquet, D.; Sandoval-Plata, G.; Galindo-Sevilla, N. Inflammatory Response in Preterm and Very Preterm Newborns with Sepsis. Mediat. Inflamm. 2016, 2016, 6740827. [Google Scholar] [CrossRef]
- Poggi, C.; Dani, C. Sepsis and Oxidative Stress in the Newborn: From Pathogenesis to Novel Therapeutic Targets. Oxidative Med. Cell. Longev. 2018, 2018, 9390140. [Google Scholar] [CrossRef]
- Figueras-Aloy, J.; Gómez, L.; Rodríguez-Miguélez, J.M.; Jordán, Y.; Salvia, M.D.; Jiménez, W.; Carbonell-Estrany, X. Plasma nitrite/nitrate and endothelin-1 concentrations in neonatal sepsis. Acta Paediatr. 2003, 92, 582–587. [Google Scholar] [CrossRef]
- Marom, D.; Yuhas, Y.; Sirota, L.; Livni, G.; Ashkenazi, S. Nitric Oxide Levels in Preterm and Term Infants and in Premature Infants with Bacteremia. Biol. Neonate 2004, 86, 160–164. [Google Scholar] [CrossRef]
- Mittal, R.; Gonzalez-Gomez, I.; Goth, K.A.; Prasadarao, N.V. Inhibition of Inducible Nitric Oxide Controls Pathogen Load and Brain Damage by Enhancing Phagocytosis of Escherichia coli K1 in Neonatal Meningitis. Am. J. Pathol. 2010, 176, 1292–1305. [Google Scholar] [CrossRef]
- Özalkaya, E.; Karatekin, G.; Topçuoğlu, S.; Karatepe, H.; Hafızoğlu, T.; Baran, P.; Ovalı, F. Neonatology oxidative status in preterm infants with premature preterm rupture of membranes and fetal inflammatuar response syndrome. Pediatr. Neonatol. 2017, 58, 437–441. [Google Scholar] [CrossRef][Green Version]
- Bharadwaj, S.K.; Bhat, B.V.; Vickneswaran, V.; Adhisivam, B.; Bobby, Z.; Habeebullah, S. Oxidative Stress, Antioxidant Status and Neurodevelopmental Outcome in Neonates Born to Pre-eclamptic Mothers. Indian J. Pediatr. 2018, 85, 351–357. [Google Scholar] [CrossRef]
- Coutinho, F.G.; Diniz, E.M.D.A.; Kandler, I.; Cianciarullo, M.A.; Dos Santos, N.R. Assessment of oxidative damage and enzymatic antioxidant system activity on the umbilical cord blood and saliva from preterm newborns with risk factors for early-onset neonatal sepsis. Rev. Assoc. Med. Bras. 2018, 64, 888–895. [Google Scholar] [CrossRef]
- Cancelier, A.C.; Petronilho, F.; Reinke, A.; Constantino, L.; Machado, R.; Ritter, C.; Dal-Pizzol, F. Inflammatory and oxidative parameters in cord blood as diagnostic of early-onset neonatal sepsis: A case-control study. Pediatr. Crit. Care Med. 2009, 10, 467–471. [Google Scholar] [CrossRef]
- Asci, A.; Surmeli-Onay, O.; Erkekoglu, P.; Yigit, S.; Yurdakok, M.; Kocer-Gumusel, B. Oxidant and antioxidant status in neonatal proven and clinical sepsis according to selenium status. Pediatr. Int. 2015, 57, 1131–1137. [Google Scholar] [CrossRef]
- Gitto, E.; Karbownik, M.; Reiter, R.J.; Tan, D.X.; Cuzzocrea, S.; Chiurazzi, P.; Cordaro, S.; Corona, G.; Trimarchi, G.; Barberi, I. Effects of Melatonin Treatment in Septic Newborns. Pediatr. Res. 2001, 50, 756–760. [Google Scholar] [CrossRef]
- Batra, S.; Kumar, R.; Kapoor, A.K.; Ray, G. Alterations in antioxidant status during neonatal sepsis. Ann. Trop. Paediatr. 2000, 20, 27–33. [Google Scholar] [CrossRef]
- Valerio, T.A.; Cancelier, A.C.; Constantino, L.; Petronilho, F.; Ritter, C.; Dal-Pizzol, F. Inflammatory and oxidative cord blood parameters as predictors of neonatal sepsis severity. Rev. Bras. Ter. Intensiv. 2012, 24, 30–34. [Google Scholar] [CrossRef]
- Kapoor, K.; Basu, S.; Das, B.K.; Bhatia, B.D. Lipid Peroxidation and Antioxidants in Neonatal Septicemia. J. Trop. Pediatr. 2006, 52, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Singla, M.; Samal, S.; Lodha, R.; Medigeshi, G.R. Respiratory Syncytial Virus-Induced Oxidative Stress Leads to an Increase in Labile Zinc Pools in Lung Epithelial Cells. Msphere 2020, 5, e00447-20. [Google Scholar] [CrossRef] [PubMed]
- Kumova, O.K.; Galani, I.-E.; Rao, A.; Johnson, H.; Triantafyllia, V.; Matt, S.M.; Pascasio, J.; Gaskill, P.J.; Andreakos, E.; Katsikis, P.D.; et al. Severity of neonatal influenza infection is driven by type I interferon and oxidative stress. Mucosal Immunol. 2022, 15, 1309–1320. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, S.; Aceti, A.; Della Gatta, A.N.; Beghetti, I.; Marsico, C.; Pilu, G.; Corvaglia, L. Antenatal and Postnatal Sequelae of Oxidative Stress in Preterm Infants: A Narrative Review Targeting Pathophysiological Mechanisms. Antioxidants 2023, 12, 422. https://doi.org/10.3390/antiox12020422
Martini S, Aceti A, Della Gatta AN, Beghetti I, Marsico C, Pilu G, Corvaglia L. Antenatal and Postnatal Sequelae of Oxidative Stress in Preterm Infants: A Narrative Review Targeting Pathophysiological Mechanisms. Antioxidants. 2023; 12(2):422. https://doi.org/10.3390/antiox12020422
Chicago/Turabian StyleMartini, Silvia, Arianna Aceti, Anna Nunzia Della Gatta, Isadora Beghetti, Concetta Marsico, Gianluigi Pilu, and Luigi Corvaglia. 2023. "Antenatal and Postnatal Sequelae of Oxidative Stress in Preterm Infants: A Narrative Review Targeting Pathophysiological Mechanisms" Antioxidants 12, no. 2: 422. https://doi.org/10.3390/antiox12020422
APA StyleMartini, S., Aceti, A., Della Gatta, A. N., Beghetti, I., Marsico, C., Pilu, G., & Corvaglia, L. (2023). Antenatal and Postnatal Sequelae of Oxidative Stress in Preterm Infants: A Narrative Review Targeting Pathophysiological Mechanisms. Antioxidants, 12(2), 422. https://doi.org/10.3390/antiox12020422