
The NADPH Oxidase Inhibitor, Mitoapocynin, Mitigates DFP-Induced Reactive Astrogliosis in a Rat Model of Organophosphate Neurotoxicity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Nanoparticle Synthesis and Characterization
2.1.2. MPO Loading and Release Kinetics
2.2. Animals
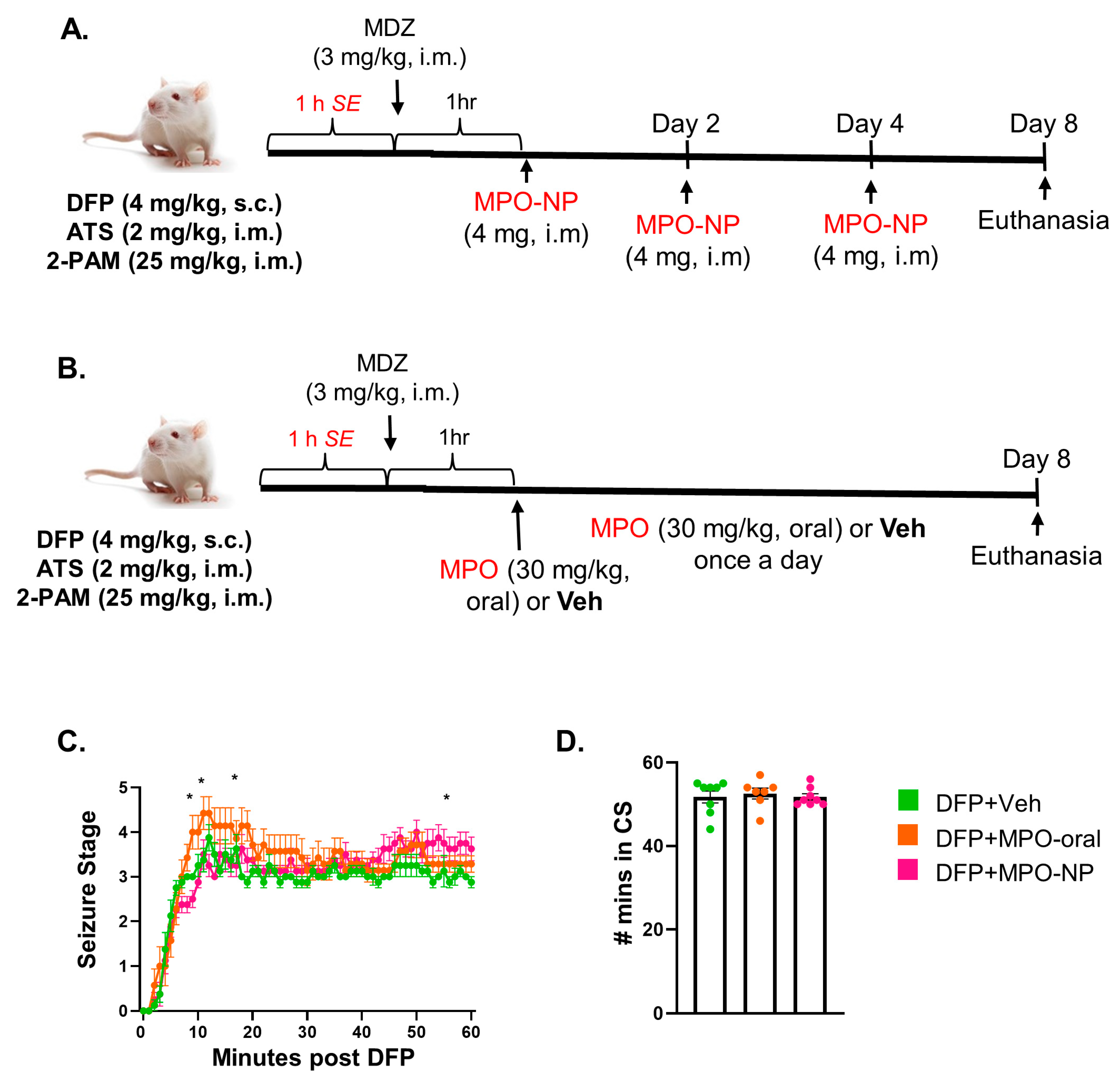
2.3. DFP Challenge, SE Monitoring, and Dosing Regimens
2.4. Euthanasia and Tissue Collection
2.5. Mitoapocynin Extraction and Quantification
2.5.1. Extraction of Mitoapocynin from Sera and Brain Tissue for LC-MS/MS
2.5.2. Mitoapocynin LC-MS/MS Quantification
2.6. Immunohistochemistry and Fluoro-Jade B Staining (FJB)
2.7. Imaging, Cell Quantification, and Morphometric Analysis
2.8. Statistical Analyses
3. Results
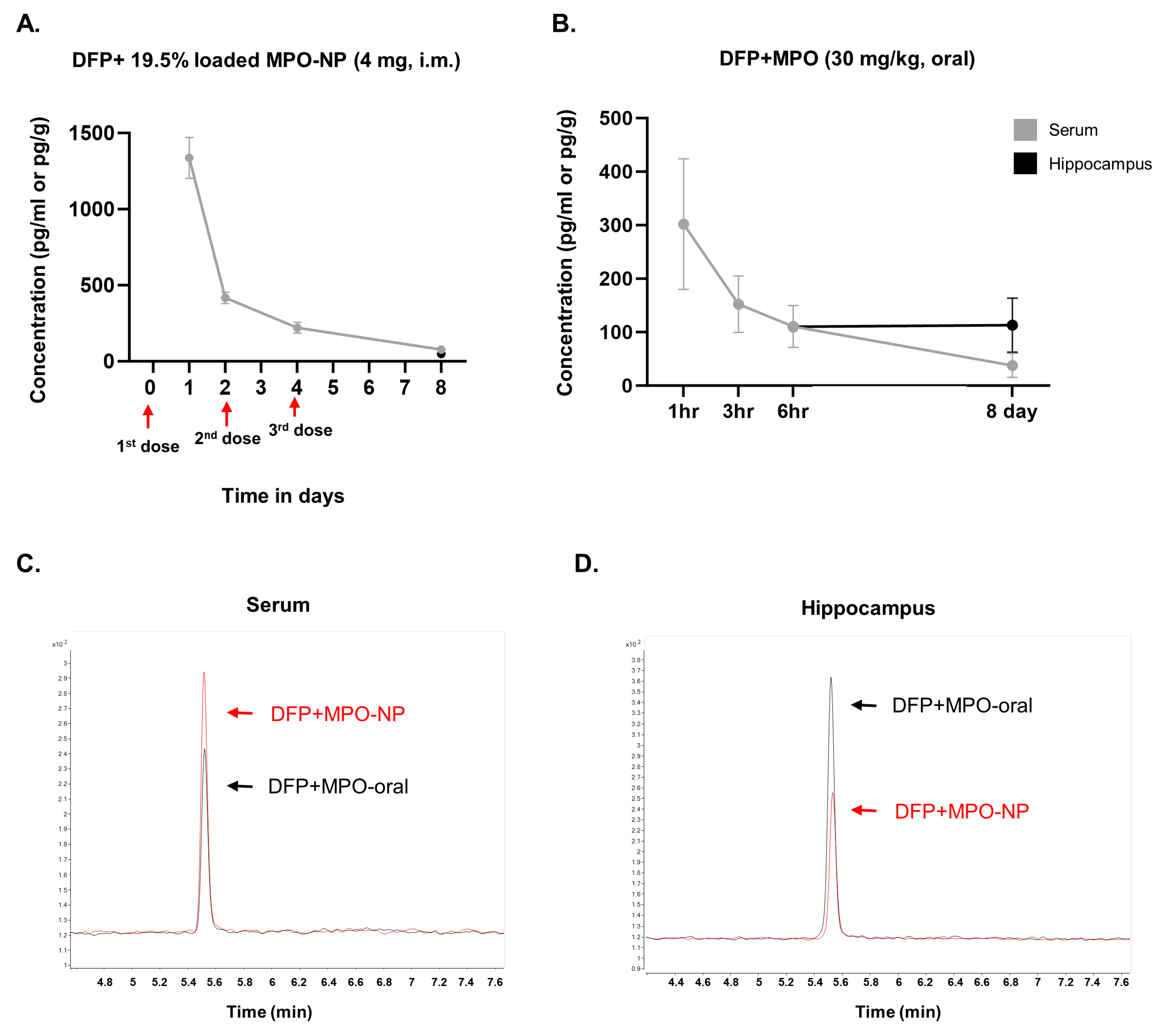
3.1. MPO-NP and MPO-Oral Concentrations in the Brain and Serum Post-DFP
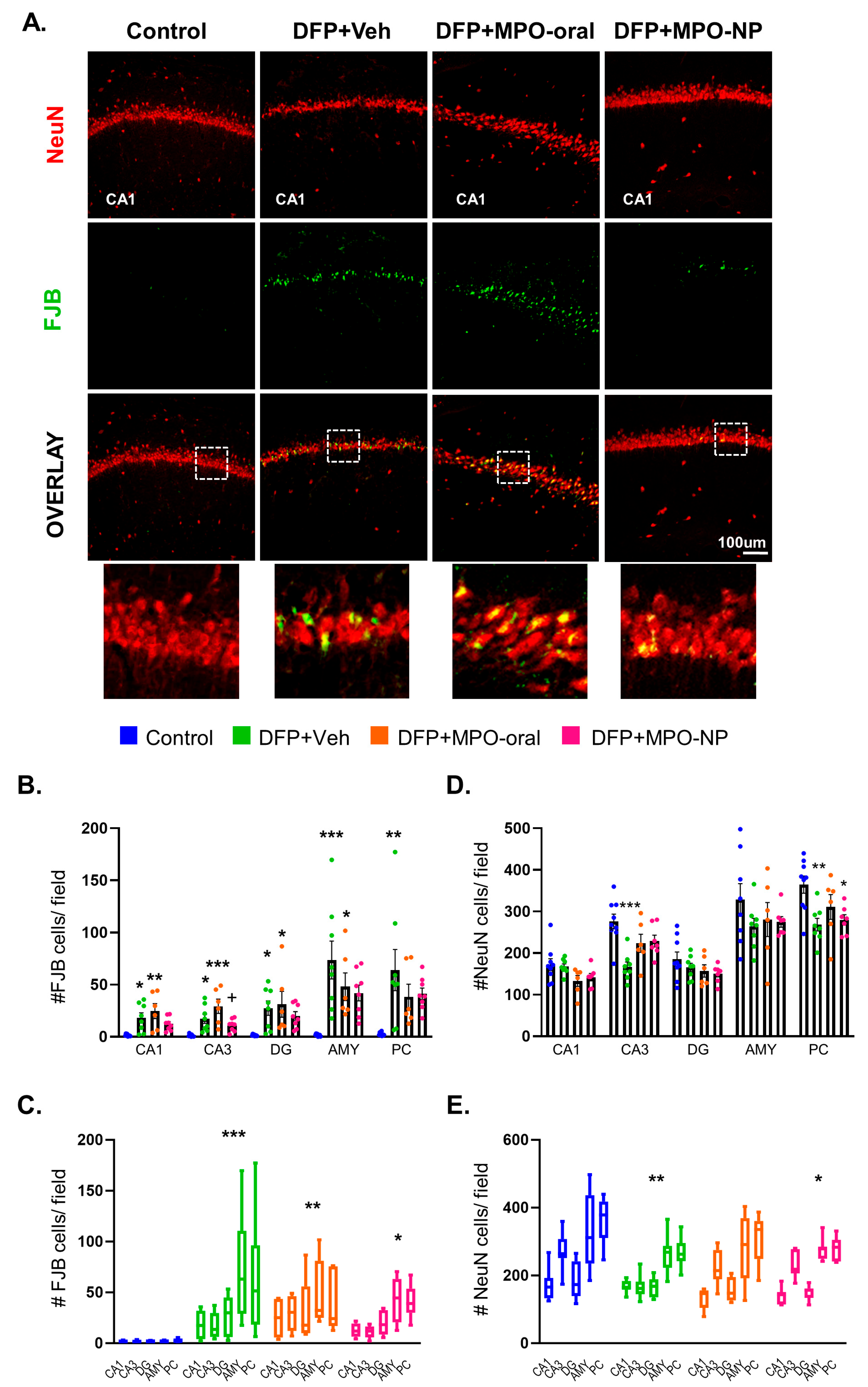
3.2. MPO-Oral Modifies Neuronal Loss Post-DFP
3.3. MPO-Oral Mitigates GABAergic Neuronal Loss in the CA1 Hippocampal Region Post-DFP
3.4. MPO-Oral and MPO-NP Treatment Did Not Affect NOX2 (GP91phox) Expression Post-DFP
3.5. DFP-Exposure Promoted Microglial Reactivity
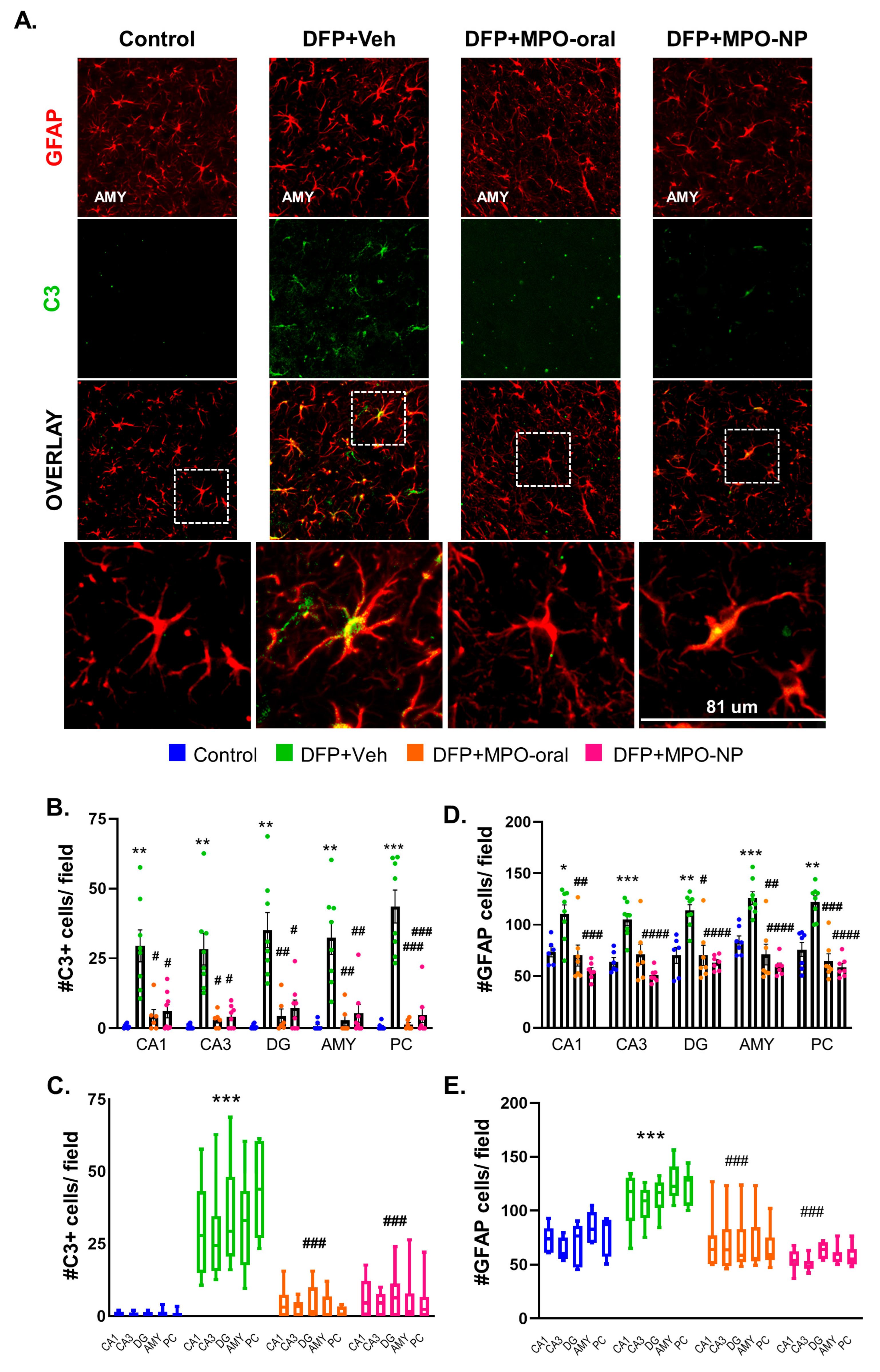
3.6. MPO-Oral and MPO-NP Attenuated Astrogliosis and Their Reactivity 8 Days Post-DFP
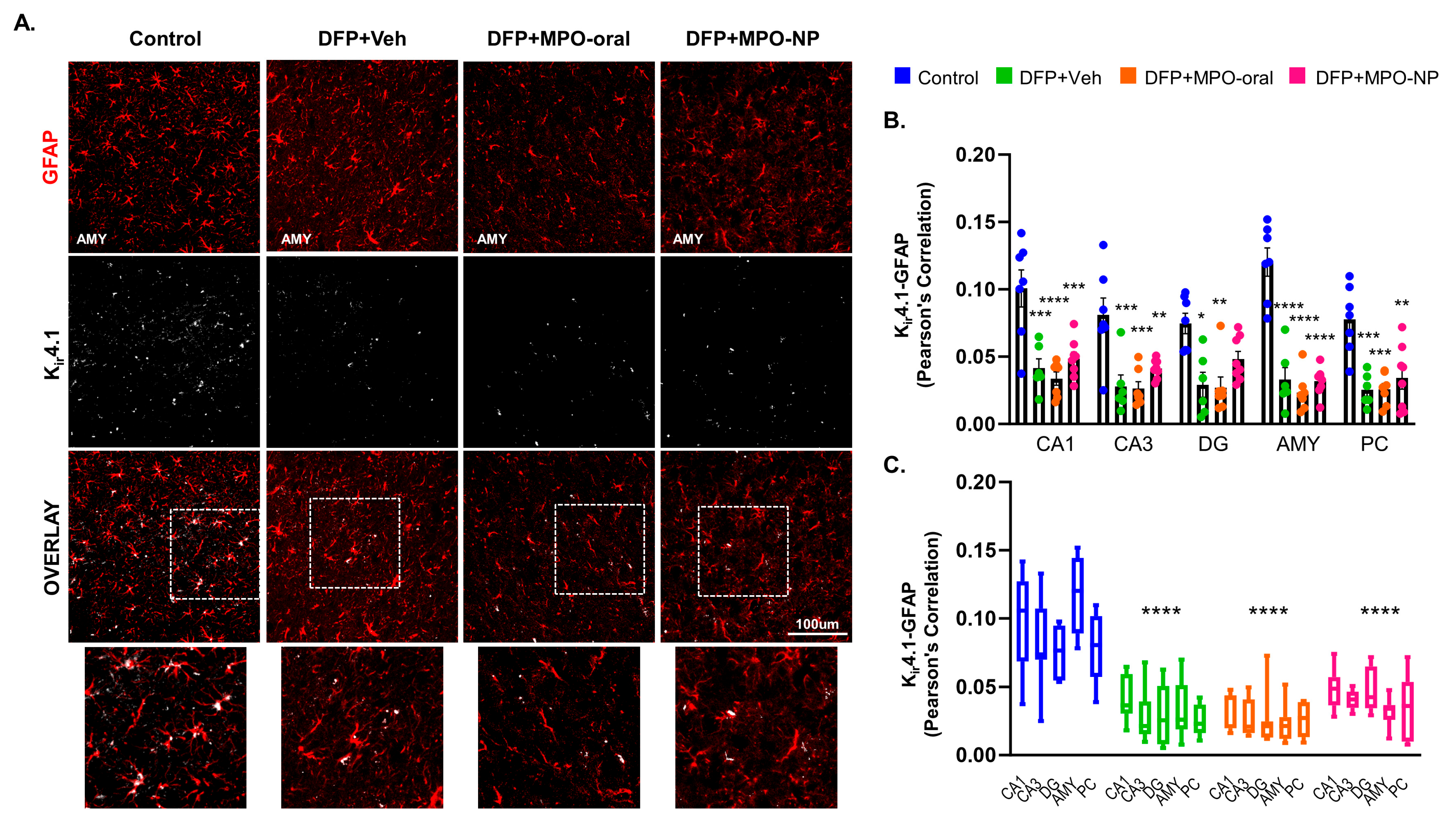
3.7. Kir4.1 Is Downregulated in Astrocytes after DFP Exposure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lothman, E.W.; Bertman, E.H., III. Epileptogenic effects of status epilepticus. Epilepsia 1993, 34, S59–S70. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Logroscino, G.; Cascino, G.; Annegers, J.F.; Hauser, W.A. Risk of unprovoked seizure after acute symptomatic seizure: Effect of status epilepticus. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1998, 44, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Dudek, F.E.; Staley, K.J. The time course and circuit mechanisms of acquired epileptogenesis. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Borowicz-Reutt, K.K.; Czuczwar, S.J. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol. Rep. 2020, 72, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Zilberter, Y.; Popova, I.; Zilberter, M. Unifying mechanism behind the onset of acquired epilepsy. Trends Pharmacol. Sci. 2022, 43, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Natrella, F.; Belmonte, G.; Miracco, C.; Cervellati, F.; Ciccoli, L.; Mariottini, A.; Rocchi, R.; Vatti, G.; Bua, A. NADPH oxidase activation and 4-hydroxy-2-nonenal/aquaporin-4 adducts as possible new players in oxidative neuronal damage presents in drug-resistant epilepsy. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 507–519. [Google Scholar] [CrossRef]
- Malkov, A.; Ivanov, A.I.; Latyshkova, A.; Bregestovski, P.; Zilberter, M.; Zilberter, Y. Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models. Ann. Neurol. 2019, 85, 907–920. [Google Scholar] [CrossRef]
- Fang, J.; Sheng, R.; Qin, Z.-H. NADPH oxidases in the central nervous system: Regional and cellular localization and the possible link to brain diseases. Antioxid. Redox Signal. 2021, 35, 951–973. [Google Scholar] [CrossRef]
- Gupta, R.C. Handbook of Toxicology of Chemical Warfare Agents; Academic Press: Cambridge, MA, USA, 2015; ISBN 0-12-800494-0. [Google Scholar]
- Chai, P.R.; Boyer, E.W.; Al-Nahhas, H.; Erickson, T.B. Toxic chemical weapons of assassination and warfare: Nerve agents VX and sarin. Toxicol. Commun. 2017, 1, 21–23. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.H., Jr.; Shih, T.-M. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci. Biobehav. Rev. 1997, 21, 559–579. [Google Scholar] [CrossRef]
- Suzuki, T.; Morita, H.; Ono, K.; Maekawa, K.; Nagai, R.; Yazaki, Y.; Nozaki, H.; Aikawa, N.; Shinozawa, Y.; Hori, S. Sarin poisoning in Tokyo subway. Lancet 1995, 345, 980–981. [Google Scholar] [CrossRef]
- Newmark, J. Therapy for acute nerve agent poisoning: An update. Neurol. Clin. Pract. 2019, 9, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, J.; Pouliot, W.; Bealer, S.L.; Roach, B.; Dudek, F.E. Antiseizure and neuroprotective effects of delayed treatment with midazolam in a rodent model of organophosphate exposure. Epilepsia 2019, 60, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Supasai, S.; González, E.A.; Rowland, D.J.; Hobson, B.; Bruun, D.A.; Guignet, M.A.; Soares, S.; Singh, V.; Wulff, H.; Saito, N. Acute administration of diazepam or midazolam minimally alters long-term neuropathological effects in the rat brain following acute intoxication with diisopropylfluorophosphate. Eur. J. Pharmacol. 2020, 886, 173538. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kuruba, R.; Reddy, D.S. Midazolam-resistant seizures and brain injury after acute intoxication of diisopropylfluorophosphate, an organophosphate pesticide and surrogate for nerve agents. J. Pharmacol. Exp. Ther. 2018, 367, 302–321. [Google Scholar] [CrossRef] [PubMed]
- Shrot, S.; Ramaty, E.; Biala, Y.; Bar-Klein, G.; Daninos, M.; Kamintsky, L.; Makarovsky, I.; Statlender, L.; Rosman, Y.; Krivoy, A. Prevention of organophosphate-induced chronic epilepsy by early benzodiazepine treatment. Toxicology 2014, 323, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Langley, M.R.; Harischandra, D.S.; Neal, M.L.; Jin, H.; Anantharam, V.; Joseph, J.; Brenza, T.; Narasimhan, B.; Kanthasamy, A. Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of Parkinson’s disease. J. Neuroimmune Pharmacol. 2016, 11, 259–278. [Google Scholar] [CrossRef]
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H. Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in MitoPark transgenic mice. Antioxid. Redox Signal. 2017, 27, 1048–1066. [Google Scholar] [CrossRef]
- Dranka, B.P.; Gifford, A.; McAllister, D.; Zielonka, J.; Joseph, J.; O’Hara, C.L.; Stucky, C.L.; Kanthasamy, A.G.; Kalyanaraman, B. A novel mitochondrially-targeted apocynin derivative prevents hyposmia and loss of motor function in the leucine-rich repeat kinase 2 (LRRK2R1441G) transgenic mouse model of Parkinson’s disease. Neurosci. Lett. 2014, 583, 159–164. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1282–1294. [Google Scholar] [CrossRef]
- Amorim, R.; Benfeito, S.; Teixeira, J.; Cagide, F.; Oliveira, P.J.; Borges, F. Targeting mitochondria: The road to mitochondriotropic antioxidants and beyond. In Mitochondrial Biology and Experimental Therapeutics; Springer: Cham, Switzerland, 2018; pp. 333–358. [Google Scholar]
- Du, F.; Yu, Q.; Swerdlow, R.H.; Waites, C.L. Glucocorticoid-driven mitochondrial damage stimulates Tau pathology. Brain 2023, 146, 4378–4394. [Google Scholar] [CrossRef]
- Meyer, C.; Rao, N.S.; Vasanthi, S.S.; Pereira, B.; Gage, M.; Putra, M.; Holtkamp, C.; Huss, J.; Thippeswamy, T. Peripheral and central effects of NADPH oxidase inhibitor, mitoapocynin, in a rat model of diisopropylfluorophosphate (DFP) toxicity. Front. Cell. Neurosci. 2023, 17, 1195843. [Google Scholar] [CrossRef]
- Yang, T.; Zang, D.-W.; Shan, W.; Guo, A.-C.; Wu, J.-P.; Wang, Y.-J.; Wang, Q. Synthesis and evaluations of novel apocynin derivatives as anti-glioma agents. Front. Pharmacol. 2019, 10, 951. [Google Scholar] [CrossRef]
- Mahmoud, N.A.; Hassanein, E.H.; Bakhite, E.A.; Shaltout, E.S.; Sayed, A.M. Apocynin and its chitosan nanoparticles attenuated cisplatin-induced multiorgan failure: Synthesis, characterization, and biological evaluation. Life Sci. 2023, 314, 121313. [Google Scholar] [CrossRef]
- Achar, A.; Myers, R.; Ghosh, C. Drug delivery challenges in brain disorders across the blood–brain barrier: Novel methods and future considerations for improved therapy. Biomedicines 2021, 9, 1834. [Google Scholar] [CrossRef]
- Katti, D.S.; Lakshmi, S.; Langer, R.; Laurencin, C.T. Toxicity, biodegradation and elimination of polyanhydrides. Adv. Drug Deliv. Rev. 2002, 54, 933–961. [Google Scholar] [CrossRef]
- Torres, M.P.; Vogel, B.M.; Narasimhan, B.; Mallapragada, S.K. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2006, 76, 102–110. [Google Scholar] [CrossRef]
- Shieh, L.; Tamada, J.; Chen, I.; Pang, J.; Domb, A.; Langer, R. Erosion of a new family of biodegradable polyanhydrides. J. Biomed. Mater. Res. 1994, 28, 1465–1475. [Google Scholar] [CrossRef]
- Brenza, T.M.; Schlichtmann, B.W.; Bhargavan, B.; Vela Ramirez, J.E.; Nelson, R.D.; Panthani, M.G.; McMillan, J.M.; Kalyanaraman, B.; Gendelman, H.E.; Anantharam, V. Biodegradable polyanhydride-based nanomedicines for blood to brain drug delivery. J. Biomed. Mater. Res. Part A 2018, 106, 2881–2890. [Google Scholar] [CrossRef]
- Bernardino, P.N.; Luo, A.S.; Andrew, P.M.; Unkel, C.M.; Gonzalez, M.I.; Gelli, A.; Lein, P.J. Evidence implicating blood-brain barrier impairment in the pathogenesis of acquired epilepsy following acute organophosphate intoxication. UC Davis 2023. [Google Scholar] [CrossRef]
- Torres, M.P.; Determan, A.S.; Anderson, G.L.; Mallapragada, S.K.; Narasimhan, B. Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials 2007, 28, 108–116. [Google Scholar] [CrossRef]
- Ulery, B.D.; Phanse, Y.; Sinha, A.; Wannemuehler, M.J.; Narasimhan, B.; Bellaire, B.H. Polymer chemistry influences monocytic uptake of polyanhydride nanospheres. Pharm. Res. 2009, 26, 683–690. [Google Scholar] [CrossRef]
- Rao, N.S.; Meyer, C.; Vasanthi, S.S.; Massey, N.; Samidurai, M.; Gage, M.; Putra, M.; Almanza, A.N.; Wachter, L.; Thippeswamy, T. DFP-Induced Status Epilepticus Severity in Mixed-Sex Cohorts of Adult Rats Housed in the Same Room: Behavioral and EEG Comparisons. Front. Cell Dev. Biol. 2022, 10, 895092. [Google Scholar] [CrossRef]
- Putra, M.; Gage, M.; Sharma, S.; Gardner, C.; Gasser, G.; Anantharam, V.; Thippeswamy, T. Diapocynin, an NADPH oxidase inhibitor, counteracts diisopropylfluorophosphate-induced long-term neurotoxicity in the rat model. Ann. N. Y. Acad. Sci. 2020, 1479, 75–93. [Google Scholar] [CrossRef]
- Gage, M.; Putra, M.; Gomez-Estrada, C.; Golden, M.; Wachter, L.; Gard, M.; Thippeswamy, T. Differential Impact of Severity and Duration of Status Epilepticus, Medical Countermeasures, and a Disease-Modifier, Saracatinib, on Brain Regions in the Rat Diisopropylfluorophosphate Model. Front. Cell. Neurosci. 2021, 426, 772868. [Google Scholar] [CrossRef]
- Gage, M.; Gard, M.; Thippeswamy, T. Characterization of Cortical Glial Scars in the Diisopropylfluorophosphate (DFP) Rat Model of Epilepsy. Front. Cell Dev. Biol. 2022, 10, 867949. [Google Scholar] [CrossRef]
- Young, K.; Morrison, H. Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J. Vis. Exp. 2018, 136, e57648. [Google Scholar]
- Rao, N.S.; Putra, M.; Meyer, C.; Almanza, A.N.; Thippeswamy, T. The effects of Src Tyrosine Kinase Inhibitor, Saracatinib, on the markers of epileptogenesis in a mixed-sex cohort of adult rats in the kainic acid model of epilepsy. Front. Mol. Neurosci. 2023, 16, 1294514. [Google Scholar] [CrossRef]
- Martinez, P.; Patel, H.; You, Y.; Jury, N.; Perkins, A.; Lee-Gosselin, A.; Taylor, X.; You, Y.; Viana Di Prisco, G.; Huang, X. Bassoon contributes to tau-seed propagation and neurotoxicity. Nat. Neurosci. 2022, 25, 1597–1607. [Google Scholar] [CrossRef]
- Cabungcal, J.-H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef]
- Schiavone, S.; Neri, M.; Trabace, L.; Turillazzi, E. The NADPH oxidase NOX2 mediates loss of parvalbumin interneurons in traumatic brain injury: Human autoptic immunohistochemical evidence. Sci. Rep. 2017, 7, 8752. [Google Scholar] [CrossRef]
- Liu, N.; Lin, M.-M.; Huang, S.-S.; Liu, Z.-Q.; Wu, J.-C.; Liang, Z.-Q.; Qin, Z.-H.; Wang, Y. NADPH and mito-apocynin treatment protects against ka-induced excitotoxic injury through autophagy pathway. Front. Cell Dev. Biol. 2021, 9, 612554. [Google Scholar] [CrossRef]
- Massey, N.; Shrestha, D.; Bhat, S.M.; Kondru, N.; Charli, A.; Karriker, L.A.; Kanthasamy, A.G.; Charavaryamath, C. Organic dust-induced mitochondrial dysfunction could be targeted via cGAS-STING or cytoplasmic NOX-2 inhibition using microglial cells and brain slice culture models. Cell Tissue Res. 2021, 384, 465–486. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Chang, S.-J.; Yu, B.-C. Mitochondrial matters of the brain: Mitochondrial dysfunction and oxidative status in epilepsy. J. Bioenerg. Biomembr. 2010, 42, 457–459. [Google Scholar] [CrossRef]
- Marrero-Rosado, B.; de Araujo Furtado, M.; Schultz, C.R.; Stone, M.; Kundrick, E.; Walker, K.; O’Brien, S.; Du, F.; Lumley, L.A. Soman-induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia 2018, 59, 2206–2218. [Google Scholar] [CrossRef]
- Putra, M.; Sharma, S.; Gage, M.; Gasser, G.; Hinojo-Perez, A.; Olson, A.; Gregory-Flores, A.; Puttachary, S.; Wang, C.; Anantharam, V. Inducible nitric oxide synthase inhibitor, 1400W, mitigates DFP-induced long-term neurotoxicity in the rat model. Neurobiol. Dis. 2020, 133, 104443. [Google Scholar] [CrossRef]
- Schwaller, B.; Tetko, I.V.; Tandon, P.; Silveira, D.C.; Vreugdenhil, M.; Henzi, T.; Potier, M.-C.; Celio, M.R.; Villa, A.E.P. Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures. Mol. Cell. Neurosci. 2004, 25, 650–663. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Godoy, L.D.; Prizon, T.; Rossignoli, M.T.; Leite, J.P.; Liberato, J.L. Parvalbumin role in epilepsy and psychiatric comorbidities: From mechanism to intervention. Front. Integr. Neurosci. 2022, 16, 765324. [Google Scholar] [CrossRef]
- Liu, F.; Yang, L.; Sun, X.; Zhang, H.; Pan, W.; Wang, X.; Yang, J.; Ji, M.; Yuan, H. NOX2 mediated-parvalbumin interneuron loss might contribute to anxiety-like and enhanced fear learning behavior in a rat model of post-traumatic stress disorder. Mol. Neurobiol. 2016, 53, 6680–6689. [Google Scholar] [CrossRef]
- Schiavone, S.; Sorce, S.; Dubois-Dauphin, M.; Jaquet, V.; Colaianna, M.; Zotti, M.; Cuomo, V.; Trabace, L.; Krause, K.-H. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol. Psychiatry 2009, 66, 384–392. [Google Scholar] [CrossRef]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase-and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and beyond: Innate immune cells as regulators of brain development and behavioral function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Gage, M.; Putra, M.; Wachter, L.; Dishman, K.; Gard, M.; Gomez-Estrada, C.; Thippeswamy, T. Saracatinib, a Src tyrosine kinase inhibitor, as a disease modifier in the rat DFP model: Sex differences, neurobehavior, gliosis, neurodegeneration, and nitro-oxidative stress. Antioxidants 2022, 11, 61. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Green, T.R.; Murphy, S.M.; Moreno-Montano, M.P.; Audinat, E.; Rowe, R.K. Reactive morphology of dividing microglia following kainic acid administration. Front. Neurosci. 2022, 16, 972138. [Google Scholar] [CrossRef]
- Green, T.R.; Murphy, S.M.; Ortiz, J.B.; Rowe, R.K. Age-at-injury influences the glial response to traumatic brain injury in the cortex of male juvenile rats. Front. Neurol. 2022, 12, 804139. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, T.; Bosco, D.B.; Xie, M.; Zheng, J.; Dheer, A.; Ying, Y.; Wu, Q.; Lennon, V.A.; Wu, L.-J. The complement C3-C3aR pathway mediates microglia–astrocyte interaction following status epilepticus. Glia 2021, 69, 1155–1169. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arboledas, A.; Acharya, M.M.; Tenner, A.J. The role of complement in synaptic pruning and neurodegeneration. ImmunoTargets Ther. 2021, 10, 373–386. [Google Scholar] [CrossRef]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef]
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as guardians of neuronal excitability: Mechanisms underlying epileptogenesis. Front. Neurol. 2020, 11, 591690. [Google Scholar] [CrossRef]
- Kinboshi, M.; Ikeda, A.; Ohno, Y. Role of astrocytic inwardly rectifying potassium (Kir) 4.1 channels in epileptogenesis. Front. Neurol. 2020, 11, 626658. [Google Scholar] [CrossRef]
- Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97. [Google Scholar] [CrossRef]
- Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Nichols, C.G.; Maldonado, H.M.; Baksi, K.; Reichenbach, A.; Skatchkov, S.N.; Eaton, M.J. Downregulation of Kir4. 1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 2007, 55, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.-S.; McCarthy, K.D. Conditional knock-out of Kir4. 1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E.; Sterner, C.; Landoure, G. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 2009, 360, 1960–1970. [Google Scholar] [CrossRef]
- Heuser, K.; Nagelhus, E.A.; Taubøll, E.; Indahl, U.; Berg, P.R.; Lien, S.; Nakken, S.; Gjerstad, L.; Ottersen, O.P. Variants of the genes encoding AQP4 and Kir4. 1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 2010, 88, 55–64. [Google Scholar] [CrossRef]
- Putra, M.; Puttachary, S.; Liu, G.; Lee, G.; Thippeswamy, T. Fyn-tau ablation modifies PTZ-induced seizures and post-seizure hallmarks of early epileptogenesis. Front. Cell. Neurosci. 2020, 14, 592374. [Google Scholar] [CrossRef] [PubMed]
- Sicca, F.; Ambrosini, E.; Marchese, M.; Sforna, L.; Servettini, I.; Valvo, G.; Brignone, M.S.; Lanciotti, A.; Moro, F.; Grottesi, A. Gain-of-function defects of astrocytic Kir4. 1 channels in children with autism spectrum disorders and epilepsy. Sci. Rep. 2016, 6, 34325. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, C.; Grego, E.; S. Vasanthi, S.; Rao, N.S.; Massey, N.; Holtkamp, C.; Huss, J.; Showman, L.; Narasimhan, B.; Thippeswamy, T. The NADPH Oxidase Inhibitor, Mitoapocynin, Mitigates DFP-Induced Reactive Astrogliosis in a Rat Model of Organophosphate Neurotoxicity. Antioxidants 2023, 12, 2061. https://doi.org/10.3390/antiox12122061
Meyer C, Grego E, S. Vasanthi S, Rao NS, Massey N, Holtkamp C, Huss J, Showman L, Narasimhan B, Thippeswamy T. The NADPH Oxidase Inhibitor, Mitoapocynin, Mitigates DFP-Induced Reactive Astrogliosis in a Rat Model of Organophosphate Neurotoxicity. Antioxidants. 2023; 12(12):2061. https://doi.org/10.3390/antiox12122061
Chicago/Turabian StyleMeyer, Christina, Elizabeth Grego, Suraj S. Vasanthi, Nikhil S. Rao, Nyzil Massey, Claire Holtkamp, Joselyn Huss, Lucas Showman, Balaji Narasimhan, and Thimmasettappa Thippeswamy. 2023. "The NADPH Oxidase Inhibitor, Mitoapocynin, Mitigates DFP-Induced Reactive Astrogliosis in a Rat Model of Organophosphate Neurotoxicity" Antioxidants 12, no. 12: 2061. https://doi.org/10.3390/antiox12122061
APA StyleMeyer, C., Grego, E., S. Vasanthi, S., Rao, N. S., Massey, N., Holtkamp, C., Huss, J., Showman, L., Narasimhan, B., & Thippeswamy, T. (2023). The NADPH Oxidase Inhibitor, Mitoapocynin, Mitigates DFP-Induced Reactive Astrogliosis in a Rat Model of Organophosphate Neurotoxicity. Antioxidants, 12(12), 2061. https://doi.org/10.3390/antiox12122061