

Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension



Abstract
:1. Introduction
2. Methods
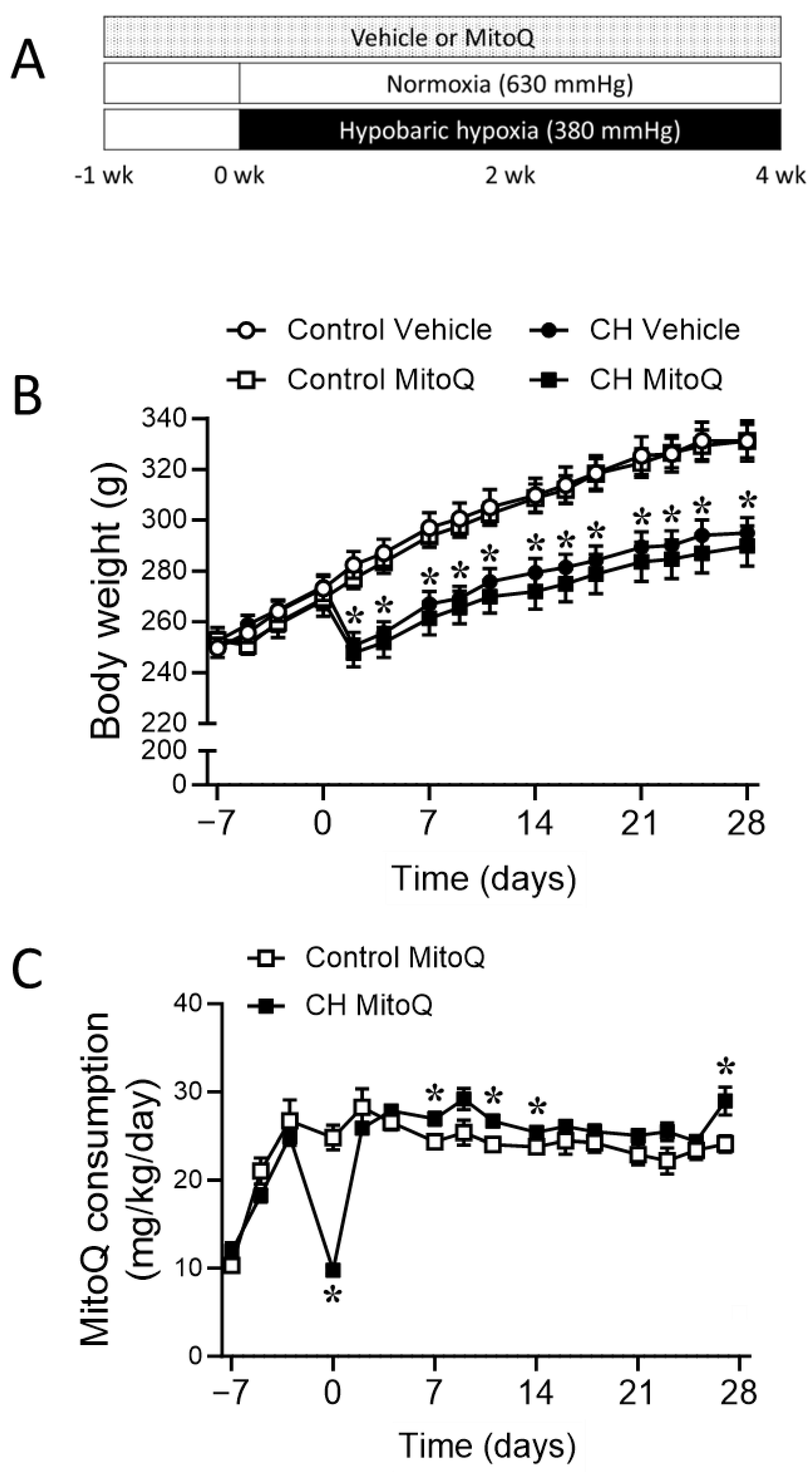
2.1. Animal Model
2.2. Contribution of mtROS to Chronic Hypoxia-Induced Pulmonary Hypertension
2.2.1. Experimental Groups
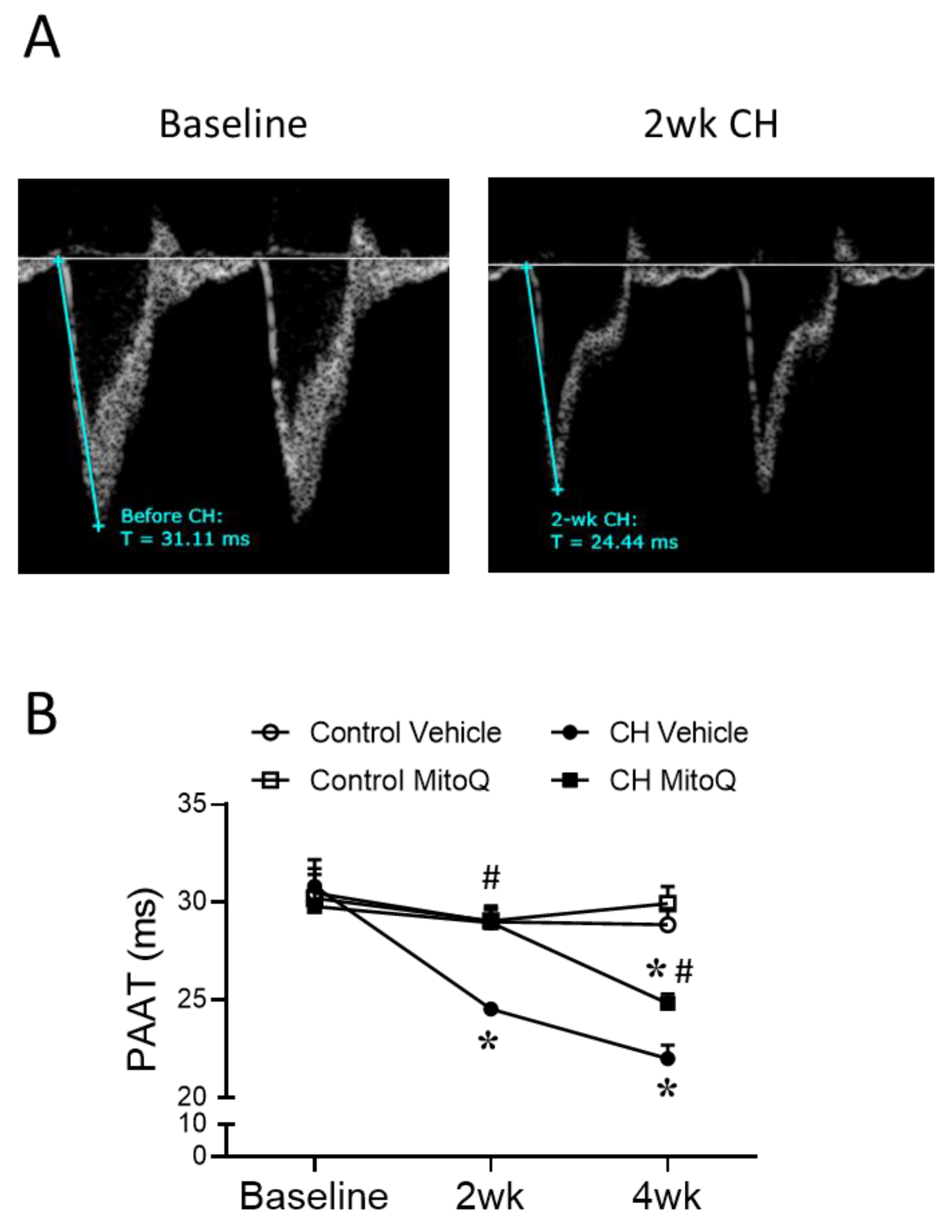
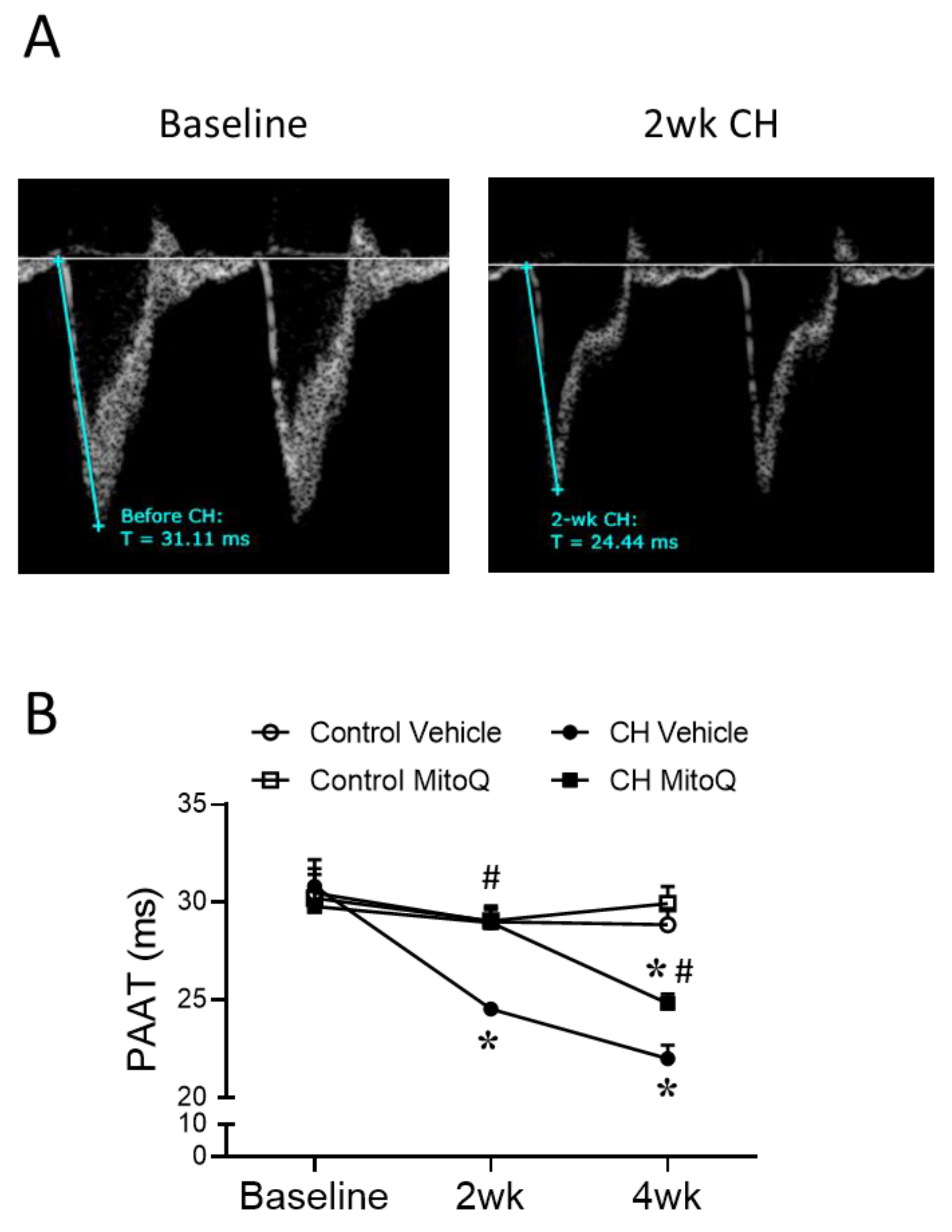
2.2.2. Echocardiography
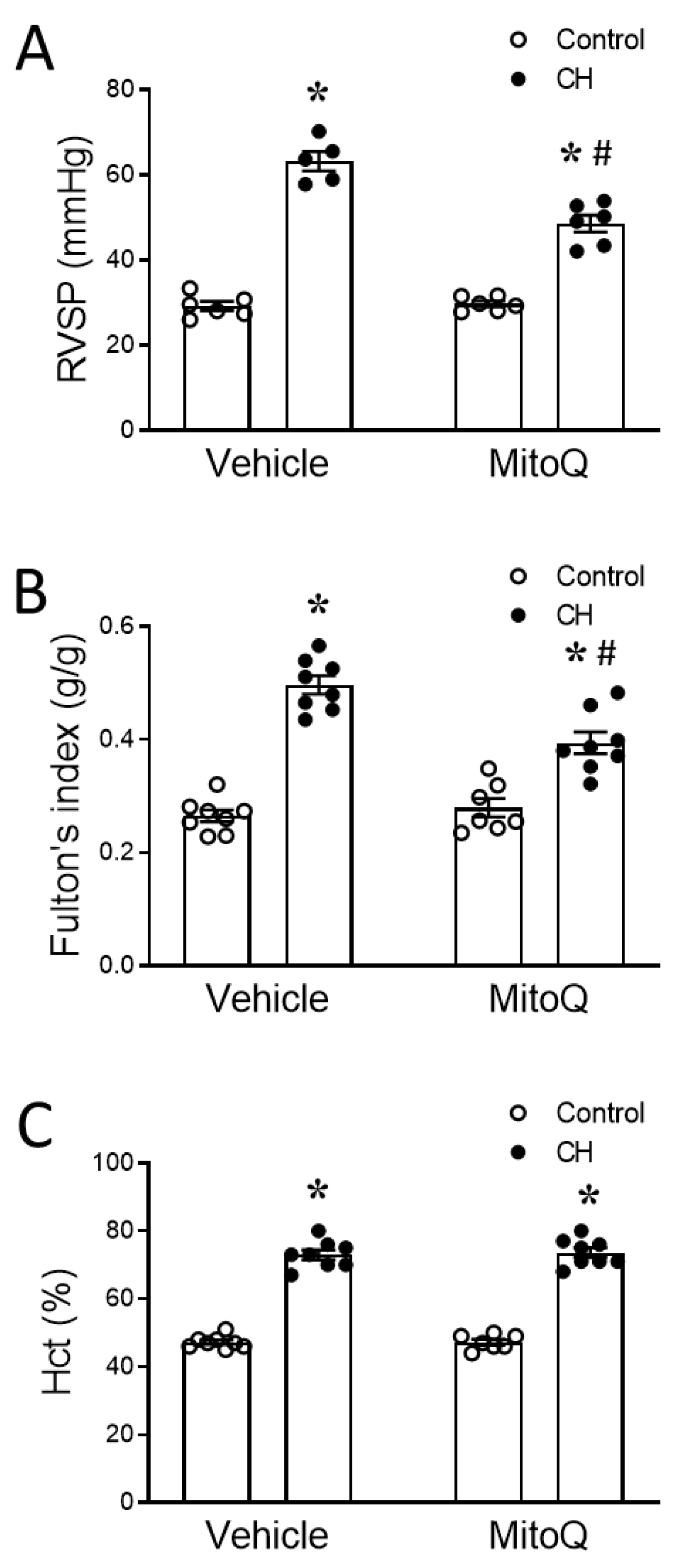
2.2.3. Right Ventricular and Systemic Arterial Blood Pressure Measurement
2.2.4. Hematocrit
2.2.5. Fulton’s Index
2.2.6. Lung Fixation and Assessment of Pulmonary Arterial Remodeling
2.3. Pressure Myography
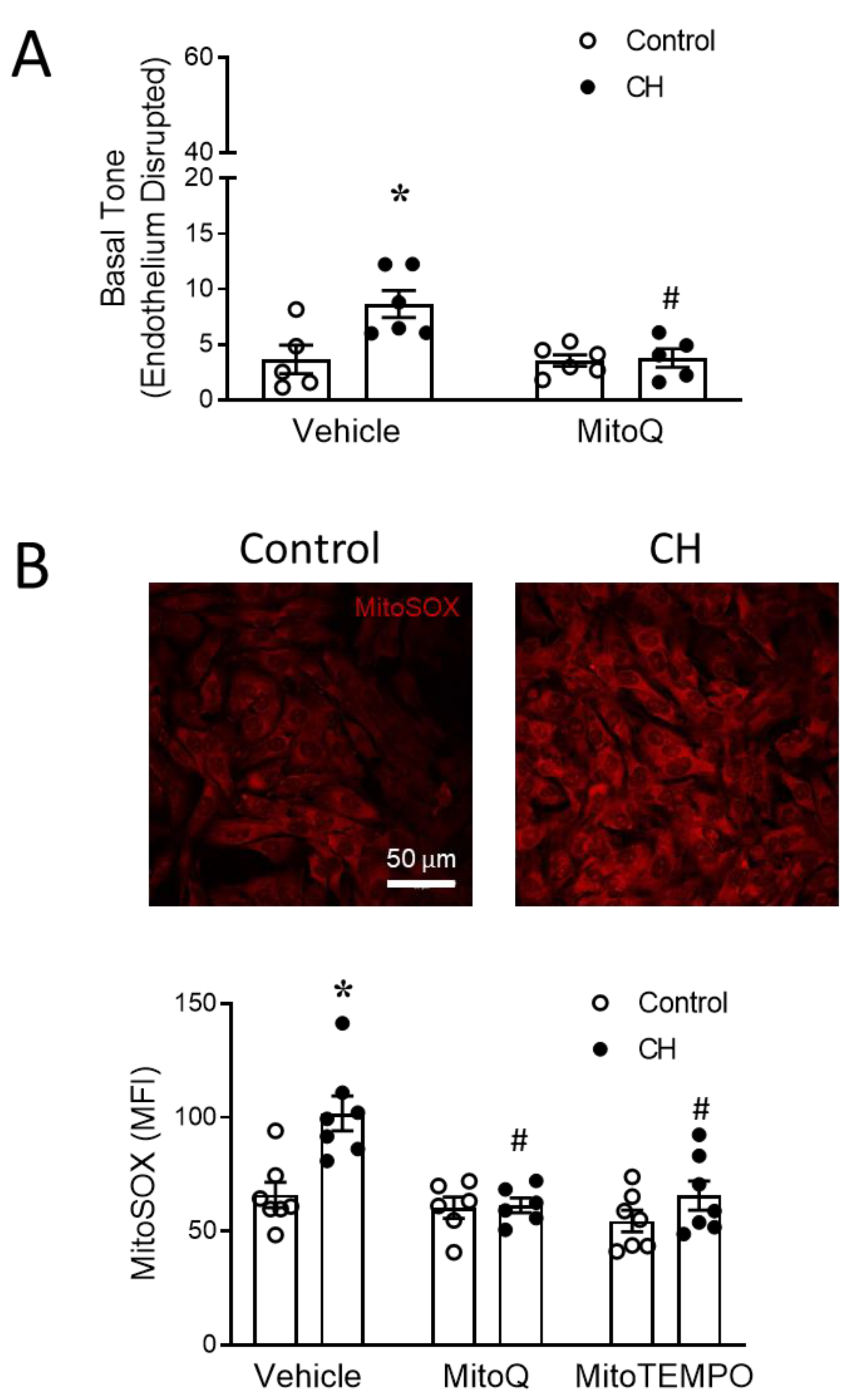
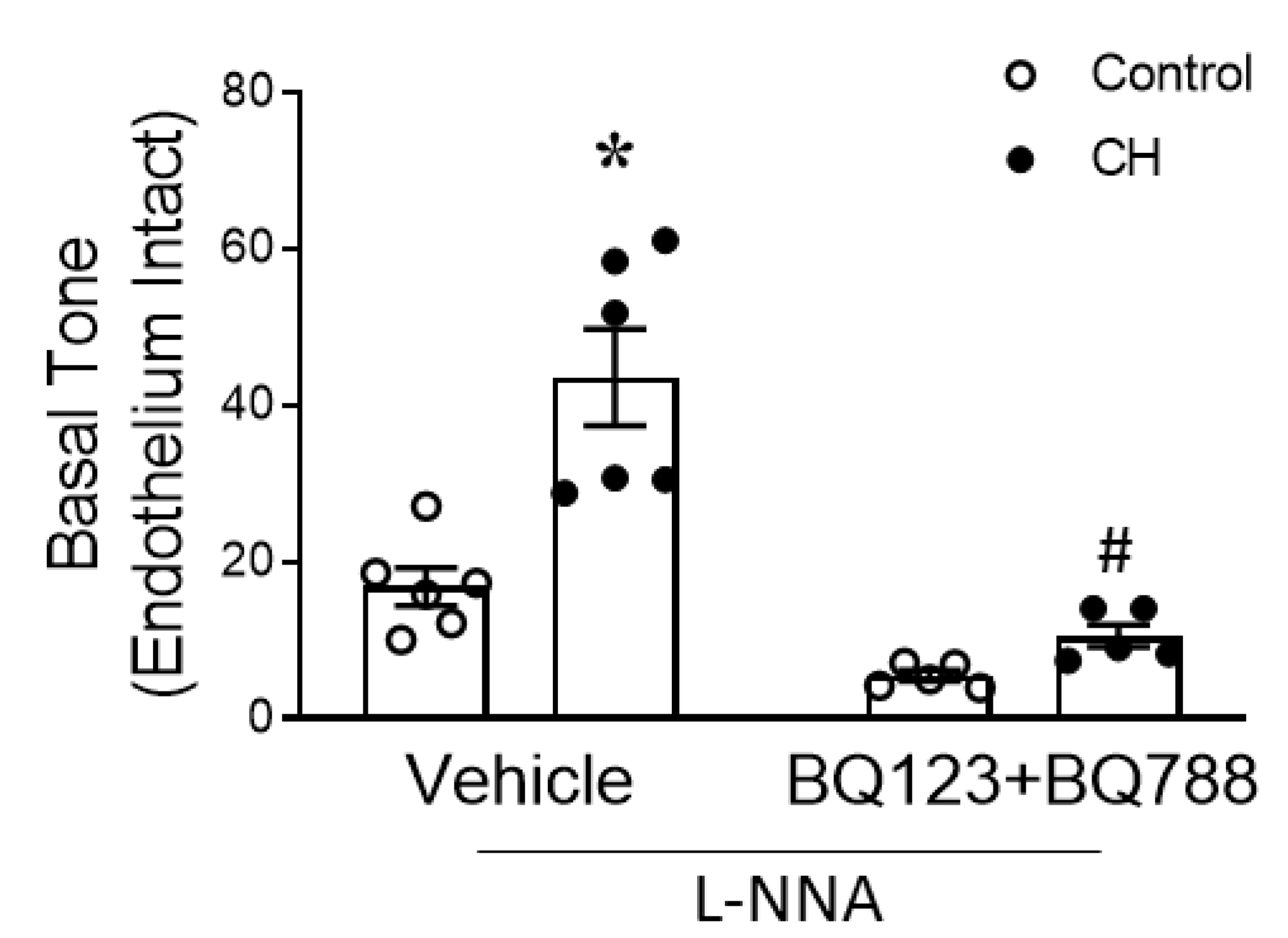
2.3.1. Basal Tone Measurement in Small Pulmonary Arteries
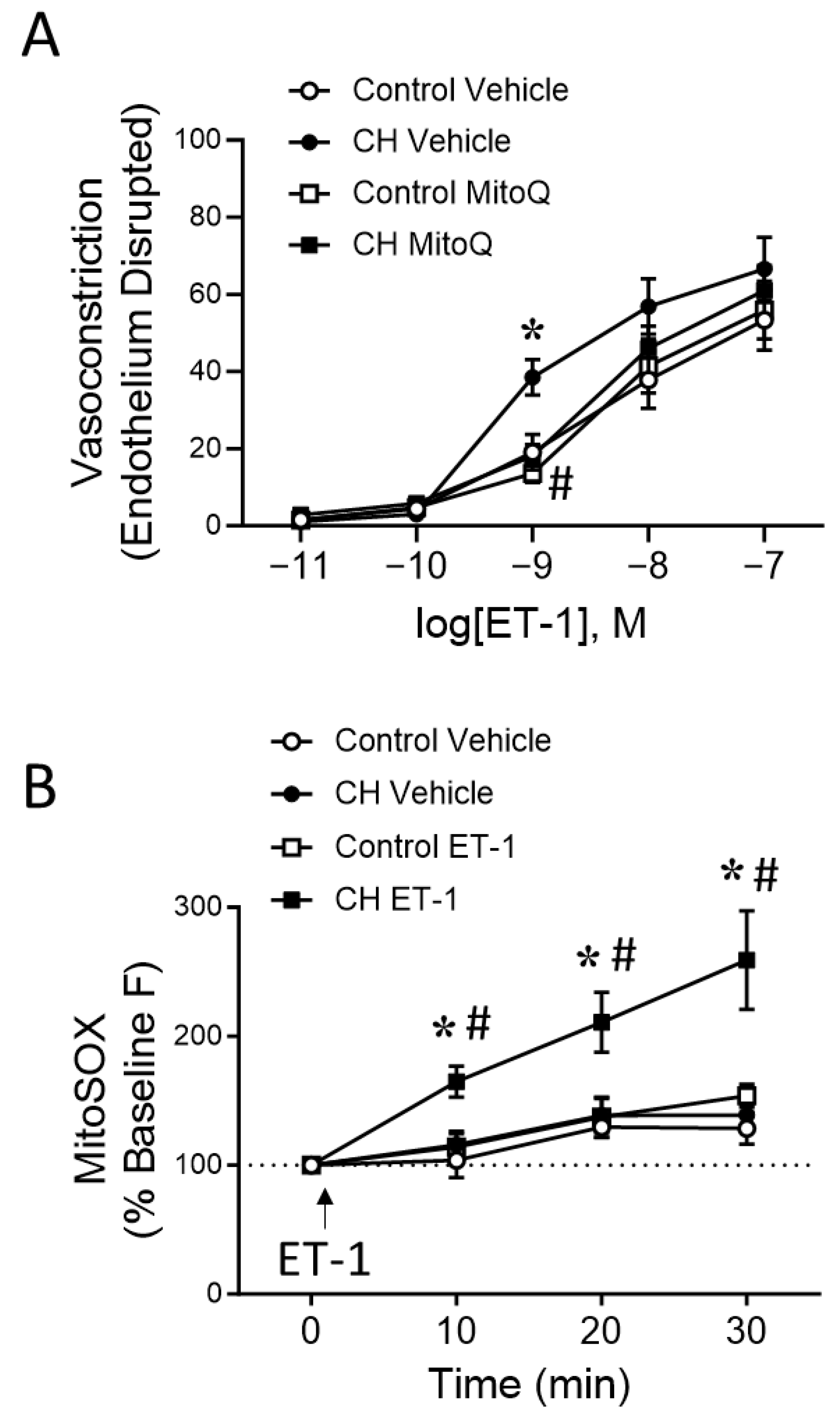
2.3.2. Vasoconstrictor Response to ET-1 in Small Endothelium-Disrupted PAs
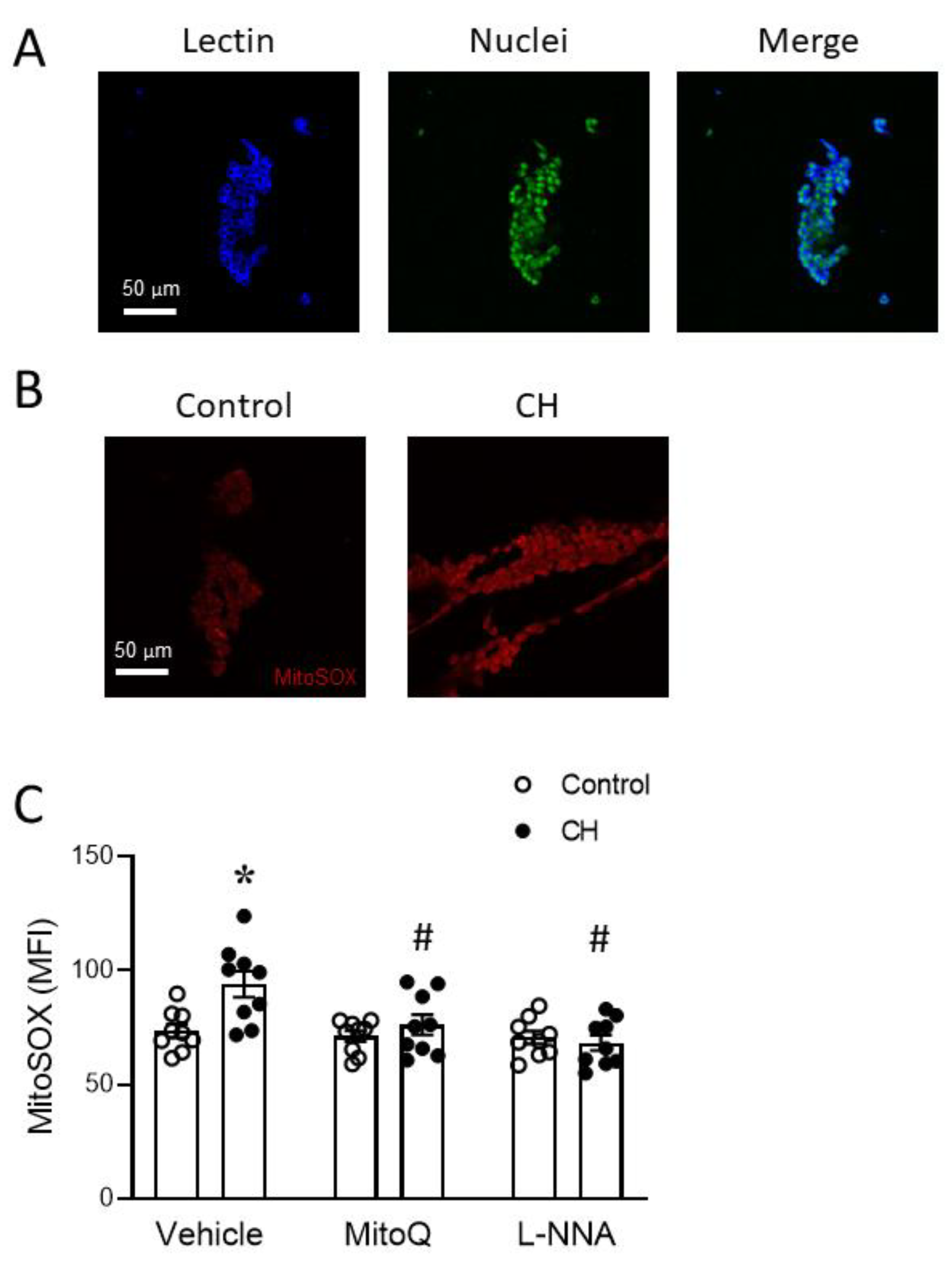
2.4. MtROS Detection in Pulmonary Arterial Smooth Muscle Cells (PASMCs)
2.4.1. Establishment of Primary PASMC Cultures
2.4.2. Basal mtROS Measurement in PASMCs
2.4.3. ET-1 Induced mtROS Production in PASMCs by Live Cell Imaging
2.4.4. Confocal Microscopy and Image Quantification
2.5. MtROS in Acutely Isolated Endothelial Sheets
2.6. Statistical Analysis
3. Results
3.1. Involvement of MtROS in CH-Induced Pulmonary Hypertension
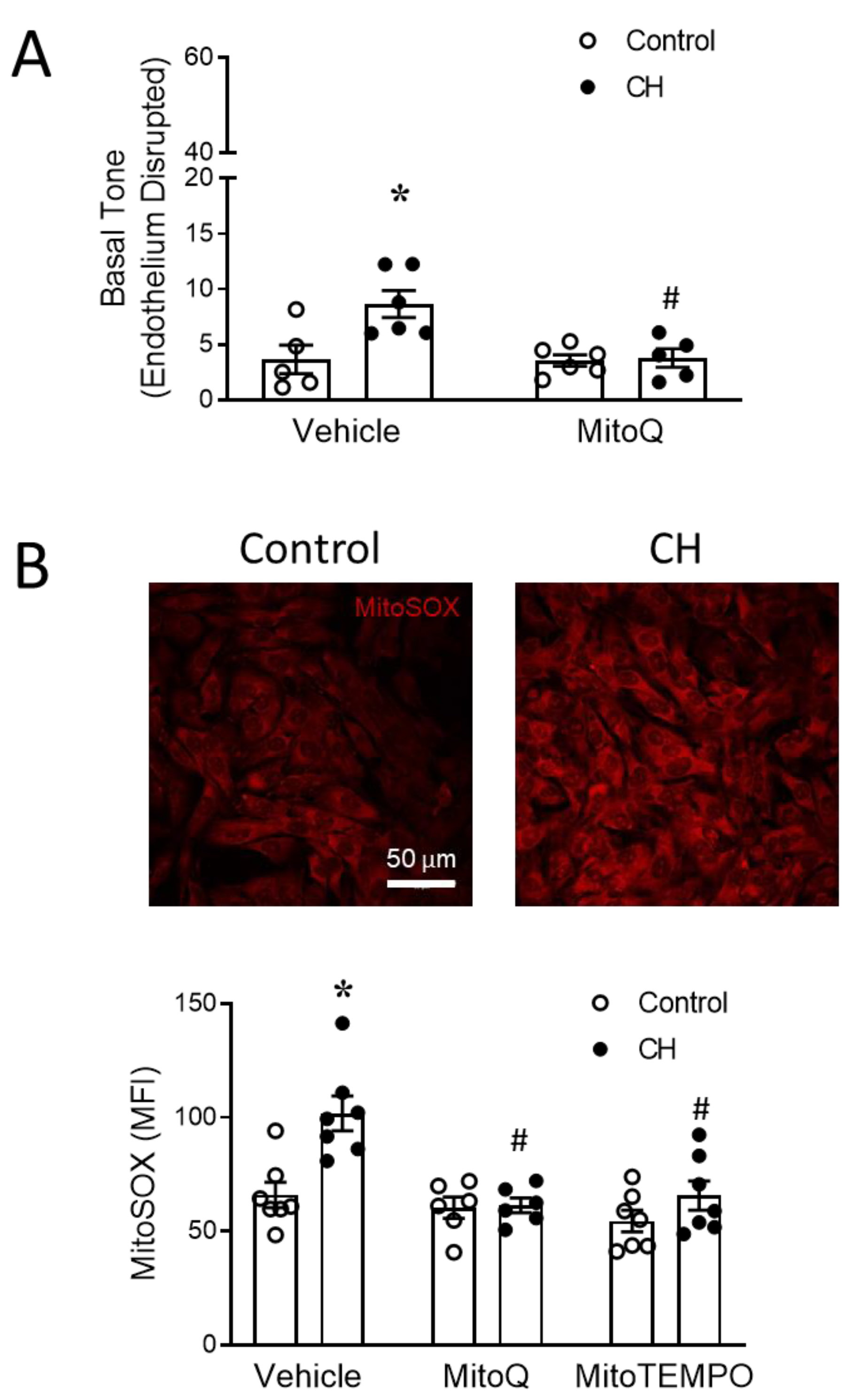
3.2. MtROS Mediate CH-Induced Increases in Pulmonary Arterial Tone
3.3. PASMC-Derived mtROS Contribute to Elevated PA Tone following CH
3.4. Endogenous ET-1 Contributes to Enhanced MtROS-Dependent Basal Tone following CH
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Naik, J.S.; Weise-Cross, L.; Detweiler, N.D.; Herbert, L.M.; Yellowhair, T.R.; Resta, T.C. Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLoS ONE 2017, 12, e0180455. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Sham, J.S.; Sylvester, J.T. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol. Res. 2000, 49, 549–560. [Google Scholar] [PubMed]
- Nagaoka, T.; Morio, Y.; Casanova, N.; Bauer, N.; Gebb, S.; McMurtry, I.; Oka, M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L665–L672. [Google Scholar] [CrossRef] [PubMed]
- Undem, C.; Luke, T.; Shimoda, L.A. Contribution of elevated intracellular calcium to pulmonary arterial myocyte alkalinization during chronic hypoxia. Pulm. Circ. 2016, 6, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L515–L529. [Google Scholar] [CrossRef]
- Li, H.; Chen, S.J.; Chen, Y.F.; Meng, Q.C.; Durand, J.; Oparil, S.; Elton, T.S. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J. Appl. Physiol. 1994, 77, 1451–1459. [Google Scholar] [CrossRef]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Yan, S.; Resta, T.C.; Jernigan, N.L. Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling. Antioxidants 2020, 9, 999. [Google Scholar] [CrossRef]
- Liu, J.Q.; Erbynn, E.M.; Folz, R.J. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: Role of superoxide and gp91phox. Chest 2005, 128, 594s–596s. [Google Scholar] [CrossRef]
- Knock, G.A.; Snetkov, V.A.; Shaifta, Y.; Connolly, M.; Drndarski, S.; Noah, A.; Pourmahram, G.E.; Becker, S.; Aaronson, P.I.; Ward, J.P. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca2+ sensitization. Free Radic. Biol. Med. 2009, 46, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Michael Hart, C.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Zhang, Y.; Codipilly, C.; Zaghloul, N.; Patel, D.; Wolin, M.; Miller, E.J. Extracellular superoxide dismutase overexpression can reverse the course of hypoxia-induced pulmonary hypertension. Mol. Med. 2012, 18, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Fike, C.D.; Dikalova, A.; Slaughter, J.C.; Kaplowitz, M.R.; Zhang, Y.; Aschner, J.L. Reactive oxygen species-reducing strategies improve pulmonary arterial responses to nitric oxide in piglets with chronic hypoxia-induced pulmonary hypertension. Antioxid. Redox Signal. 2013, 18, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Francis, B.N.; Hale, A.; Channon, K.M.; Wilkins, M.R.; Zhao, L. Effects of tetrahydrobiopterin oral treatment in hypoxia-induced pulmonary hypertension in rat. Pulm. Circ. 2014, 4, 462–470. [Google Scholar] [CrossRef]
- Villegas, L.R.; Kluck, D.; Field, C.; Oberley-Deegan, R.E.; Woods, C.; Yeager, M.E.; El Kasmi, K.C.; Savani, R.C.; Bowler, R.P.; Nozik-Grayck, E. Superoxide dismutase mimetic, MnTE-2-PyP, attenuates chronic hypoxia-induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid. Redox Signal. 2013, 18, 1753–1764. [Google Scholar] [CrossRef]
- Jefferson, J.A.; Simoni, J.; Escudero, E.; Hurtado, M.E.; Swenson, E.R.; Wesson, D.E.; Schreiner, G.F.; Schoene, R.B.; Johnson, R.J.; Hurtado, A. Increased oxidative stress following acute and chronic high altitude exposure. High Alt. Med. Biol. 2004, 5, 61–69. [Google Scholar] [CrossRef]
- Cracowski, J.L.; Cracowski, C.; Bessard, G.; Pepin, J.L.; Bessard, J.; Schwebel, C.; Stanke-Labesque, F.; Pison, C. Increased lipid peroxidation in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 1038–1042. [Google Scholar] [CrossRef]
- Sharp, J.; Farha, S.; Park, M.M.; Comhair, S.A.; Lundgrin, E.L.; Tang, W.H.; Bongard, R.D.; Merker, M.P.; Erzurum, S.C. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol. 2014, 2, 884–891. [Google Scholar] [CrossRef]
- Norton, C.E.; Sheak, J.R.; Yan, S.; Weise-Cross, L.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Augmented Pulmonary Vasoconstrictor Reactivity after Chronic Hypoxia Requires Src Kinase and Epidermal Growth Factor Receptor Signaling. Am. J. Respir. Cell Mol. Biol. 2020, 62, 61–73. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Dietrich, A.; Schermuly, R.T.; Ghofrani, H.A.; Gudermann, T.; Schulz, R.; Seeger, W.; Grimminger, F.; Weissmann, N. Regulation of hypoxic pulmonary vasoconstriction: Basic mechanisms. Eur. Respir. J. 2008, 32, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.N.; Al-Mehdi, A.B.; McMurtry, I.F. Mitochondria in hypoxic pulmonary vasoconstriction: Potential importance of compartmentalized reactive oxygen species signaling. Am. J. Respir. Crit. Care Med. 2013, 187, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef] [PubMed]
- Snow, J.B.; Norton, C.E.; Sands, M.A.; Weise-Cross, L.; Yan, S.; Herbert, L.M.; Sheak, J.R.; Gonzalez Bosc, L.V.; Walker, B.R.; Kanagy, N.L.; et al. Intermittent Hypoxia Augments Pulmonary Vasoconstrictor Reactivity through PKCβ/Mitochondrial Oxidant Signaling. Am. J. Respir. Cell Mol. Biol. 2020, 62, 732–746. [Google Scholar] [CrossRef]
- Supinski, G.S.; Murphy, M.P.; Callahan, L.A. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1095–R1102. [Google Scholar] [CrossRef]
- Maiti, A.K.; Spoorthi, B.C.; Saha, N.C.; Panigrahi, A.K. Mitigating peroxynitrite mediated mitochondrial dysfunction in aged rat brain by mitochondria-targeted antioxidant MitoQ. Biogerontology 2018, 19, 271–286. [Google Scholar] [CrossRef]
- Rehman, H.; Liu, Q.; Krishnasamy, Y.; Shi, Z.; Ramshesh, V.K.; Haque, K.; Schnellmann, R.G.; Murphy, M.P.; Lemasters, J.J.; Rockey, D.C.; et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 14–27. [Google Scholar] [PubMed]
- Nuzzo, A.M.; Camm, E.J.; Sferruzzi-Perri, A.N.; Ashmore, T.J.; Yung, H.W.; Cindrova-Davies, T.; Spiroski, A.M.; Sutherland, M.R.; Logan, A.; Austin-Williams, S.; et al. Placental Adaptation to Early-Onset Hypoxic Pregnancy and Mitochondria-Targeted Antioxidant Therapy in a Rodent Model. Am. J. Pathol. 2018, 188, 2704–2716. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Resta, T.C.; Walker, B.R. Contribution of oxygen radicals to altered NO-dependent pulmonary vasodilation in acute and chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L947–L955. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Yun, X.; Kliment, C.; Huetsch, J.; Damarla, M.; Shimoda, L.A. Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L893–L907. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Dranka, B.P.; Hardy, M.; Michalski, R.; Zielonka, J. HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes--the ultimate approach for intra- and extracellular superoxide detection. Biochim. Biophys. Acta 2014, 1840, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.Y.; Barnes, E.A.; Ying, L.; Chen, C.; Lee, L.; Alvira, C.M.; Cornfield, D.N. Pulmonary artery smooth muscle cell endothelin-1 expression modulates the pulmonary vascular response to chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L368–L377. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.S.; Albarwani, S.; Davies, A.R.; Kozlowski, R.Z. Endothelial cells freshly isolated from resistance-sized pulmonary arteries possess a unique K(+) current profile. Biochem. Biophys. Res. Commun. 1999, 263, 405–409. [Google Scholar] [CrossRef]
- Koskenvuo, J.W.; Mirsky, R.; Zhang, Y.; Angeli, F.S.; Jahn, S.; Alastalo, T.P.; Schiller, N.B.; Boyle, A.J.; Chatterjee, K.; De Marco, T.; et al. A comparison of echocardiography to invasive measurement in the evaluation of pulmonary arterial hypertension in a rat model. Int. J. Cardiovasc. Imaging 2010, 26, 509–518. [Google Scholar] [CrossRef]
- Satwiko, M.G.; Ikeda, K.; Nakayama, K.; Yagi, K.; Hocher, B.; Hirata, K.; Emoto, N. Targeted activation of endothelin-1 exacerbates hypoxia-induced pulmonary hypertension. Biochem. Biophys. Res. Commun. 2015, 465, 356–362. [Google Scholar] [CrossRef]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Oparil, S. Endothelin and pulmonary hypertension. J. Cardiovasc. Pharmacol. 2000, 35, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Norton, C.E.; Broughton, B.R.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid. Redox Signal. 2013, 18, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16α-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef]
- Rudyk, O.; Rowan, A.; Prysyazhna, O.; Krasemann, S.; Hartmann, K.; Zhang, M.; Shah, A.M.; Ruppert, C.; Weiss, A.; Schermuly, R.T.; et al. Oxidation of PKGIα mediates an endogenous adaptation to pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13016–13025. [Google Scholar] [CrossRef]
- Waypa, G.B.; Marks, J.D.; Mack, M.M.; Boriboun, C.; Mungai, P.T.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 2002, 91, 719–726. [Google Scholar] [CrossRef]
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Zaldumbide, J.; Huetsch, J.C.; Kliment, C.; Acoba, M.G.; Kirsch, B.J.; Claypool, S.M.; et al. Regulation of mitochondrial fragmentation in microvascular endothelial cells isolated from the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L639–L652. [Google Scholar] [CrossRef]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelković, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 2020, 6, eaba0694. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, V.; Huang, L.; Kim, S.J.; Cheresh, P.; Shaaya, M.; Bandela, M.; Fu, P.; Feghali-Bostwick, C.; Di Paolo, G.; Kamp, D.W.; et al. Role of phospholipase D in bleomycin-induced mitochondrial reactive oxygen species generation, mitochondrial DNA damage, and pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L175–L187. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Nenan, S.; Leclerc, O.; Guenon, I.; Boichot, E.; Planquois, J.M.; Bertrand, C.P.; Lagente, V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir. Res. 2005, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. superoxide. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Adamcakova-Dodd, A.; Perry, S.S.; Tephly, L.A.; Keller, R.M.; Metwali, N.; Meyerholz, D.K.; Wang, Y.; Glogauer, M.; Thorne, P.S.; et al. Modulation of reactive oxygen species by Rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L846–L855. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.O.; Sjaastad, I.; Svindland, A.; Krobert, K.A.; Skjønsberg, O.H.; Christensen, G. Alveolar hypoxia induces left ventricular diastolic dysfunction and reduces phosphorylation of phospholamban in mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H507–H516. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Resta, T.C.; Chicoine, L.G.; Omdahl, J.L.; Walker, B.R. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am. J. Physiol. 1999, 276, H699–H708. [Google Scholar] [CrossRef] [PubMed]
- Resta, T.C.; O’Donaughy, T.L.; Earley, S.; Chicoine, L.G.; Walker, B.R. Unaltered vasoconstrictor responsiveness after iNOS inhibition in lungs from chronically hypoxic rats. Am. J. Physiol. 1999, 276, L122–L130. [Google Scholar] [CrossRef] [PubMed]
- Resta, T.C.; Walker, B.R. Chronic hypoxia selectively augments endothelium-dependent pulmonary arterial vasodilation. Am. J. Physiol. 1996, 270, H888–H896. [Google Scholar] [CrossRef] [PubMed]
- Poderoso, J.J.; Carreras, M.C.; Lisdero, C.; Riobó, N.; Schöpfer, F.; Boveris, A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch. Biochem. Biophys. 1996, 328, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Callender, M.; Quintero, M.; Hollis, V.S.; Springett, R.J.; Moncada, S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 7630–7635. [Google Scholar] [CrossRef] [PubMed]
- Kourembanas, S.; McQuillan, L.P.; Leung, G.K.; Faller, D.V. Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J. Clin. Investig. 1993, 92, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.D.; Gonon, A.; Shimizu, M.; Sjöquist, P.O.; Pernow, J. Contribution of endothelin to the coronary vasoconstriction in the isolated rat heart induced by nitric oxide synthase inhibition. Acta Physiol. Scand. 1998, 163, 325–330. [Google Scholar] [CrossRef]
- Muramatsu, M.; Rodman, D.M.; Oka, M.; McMurtry, I.F. Endothelin-1 mediates nitro-L-arginine vasoconstriction of hypertensive rat lungs. Am. J. Physiol. 1997, 272, L807–L812. [Google Scholar] [CrossRef]
- Earley, S.; Resta, T.C. Estradiol attenuates hypoxia-induced pulmonary endothelin-1 gene expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L86–L93. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L801–L808. [Google Scholar] [CrossRef]
- Sun, X.; Kumar, S.; Sharma, S.; Aggarwal, S.; Lu, Q.; Gross, C.; Rafikova, O.; Lee, S.G.; Dasarathy, S.; Hou, Y.; et al. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Baki, L.; Baumgarten, C.M. Endothelin signalling regulates volume-sensitive Cl- current via NADPH oxidase and mitochondrial reactive oxygen species. Cardiovasc. Res. 2010, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028.e7. [Google Scholar] [CrossRef] [PubMed]
- Esterberg, R.; Linbo, T.; Pickett, S.B.; Wu, P.; Ou, H.C.; Rubel, E.W.; Raible, D.W. Mitochondrial calcium uptake underlies ROS generation during aminoglycoside-induced hair cell death. J. Clin. Investig. 2016, 126, 3556–3566. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Sham, J.S.; Shimoda, T.H.; Sylvester, J.T. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L884–L894. [Google Scholar] [CrossRef]
- Zinkevich, N.S.; Fancher, I.S.; Gutterman, D.D.; Phillips, S.A. Roles of NADPH oxidase and mitochondria in flow-induced vasodilation of human adipose arterioles: ROS-induced ROS release in coronary artery disease. Microcirculation 2017, 24, e12380. [Google Scholar] [CrossRef]
- Joseph, L.C.; Kokkinaki, D.; Valenti, M.C.; Kim, G.J.; Barca, E.; Tomar, D.; Hoffman, N.E.; Subramanyam, P.; Colecraft, H.M.; Hirano, M.; et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight 2017, 2, e94248. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement | Time of Exposure | Control | CH | ||
|---|---|---|---|---|---|
| Vehicle | MitoQ | Vehicle | MitoQ | ||
| Cardiac output (mL/min/Kg) | Baseline | 277 ± 17 | 280 ± 27 | 275 ± 11 | 276 ± 24 |
| 2 weeks | 263 ± 11 | 266 ± 15 | 271 ± 15 | 266 ± 10 | |
| 4 weeks | 269 ± 16 | 270 ± 15 | 262 ± 20 | 260 ± 8 | |
| HR (bpm) | Baseline | 391 ± 4 | 387 ± 5 | 391 ± 12 | 389 ± 5 |
| 2 weeks | 364 ± 5 | 370 ± 8 | 379 ± 10 | 369 ± 6 | |
| 4 weeks | 374 ± 11 | 371 ± 11 | 380 ± 7 | 384 ± 7 | |
| Measurement | Control | CH | ||
|---|---|---|---|---|
| Vehicle | MitoQ | Vehicle | MitoQ | |
| MSAP (mmHg) | 113 ± 2 | 107 ± 2 | 117 ± 6 | 124 ± 2 * |
| HR (bpm) | 362 ± 6 | 348 ± 14 | 350 ± 9 | 360 ± 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Sheak, J.R.; Walker, B.R.; Jernigan, N.L.; Resta, T.C. Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension. Antioxidants 2023, 12, 2060. https://doi.org/10.3390/antiox12122060
Yan S, Sheak JR, Walker BR, Jernigan NL, Resta TC. Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension. Antioxidants. 2023; 12(12):2060. https://doi.org/10.3390/antiox12122060
Chicago/Turabian StyleYan, Simin, Joshua R. Sheak, Benjimen R. Walker, Nikki L. Jernigan, and Thomas C. Resta. 2023. "Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension" Antioxidants 12, no. 12: 2060. https://doi.org/10.3390/antiox12122060
APA StyleYan, S., Sheak, J. R., Walker, B. R., Jernigan, N. L., & Resta, T. C. (2023). Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension. Antioxidants, 12(12), 2060. https://doi.org/10.3390/antiox12122060