
Hyperbaric Oxygen Therapy Alleviates Memory and Motor Impairments Following Traumatic Brain Injury via the Modulation of Mitochondrial-Dysfunction-Induced Neuronal Apoptosis in Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Study Design
2.3. Controlled Cortical Impact (CCI) Model
2.4. Hyperbaric Oxygen Therapy (HBOT)
2.5. Behavioral Tests
2.5.1. Morris Water Maze
2.5.2. Rotarod Test
2.6. Histology and Microscopy
2.6.1. Tissue Collection
2.6.2. Immunohistochemistry
2.7. Microscopy
2.8. Mitochondrial Function
2.8.1. Mitochondria Isolation from Rat Brain
2.8.2. Mitochondrial Respiration Measurements
2.9. Data Analysis
3. Results
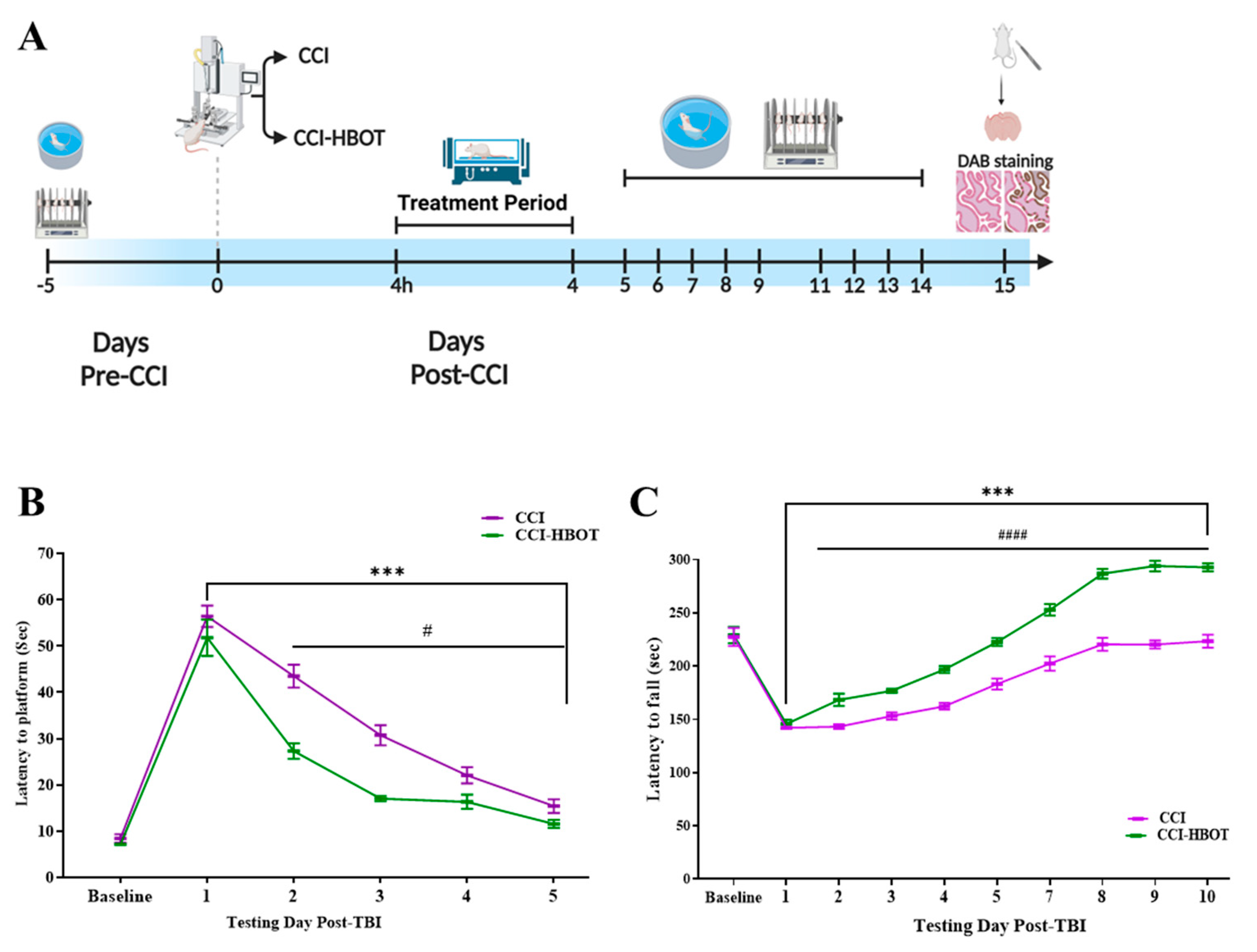
3.1. HBOT Controls the Development of Spatial Learning Impairment after CCI
3.2. HBOT Regulates the Progression of Motor Impairment Following CCI
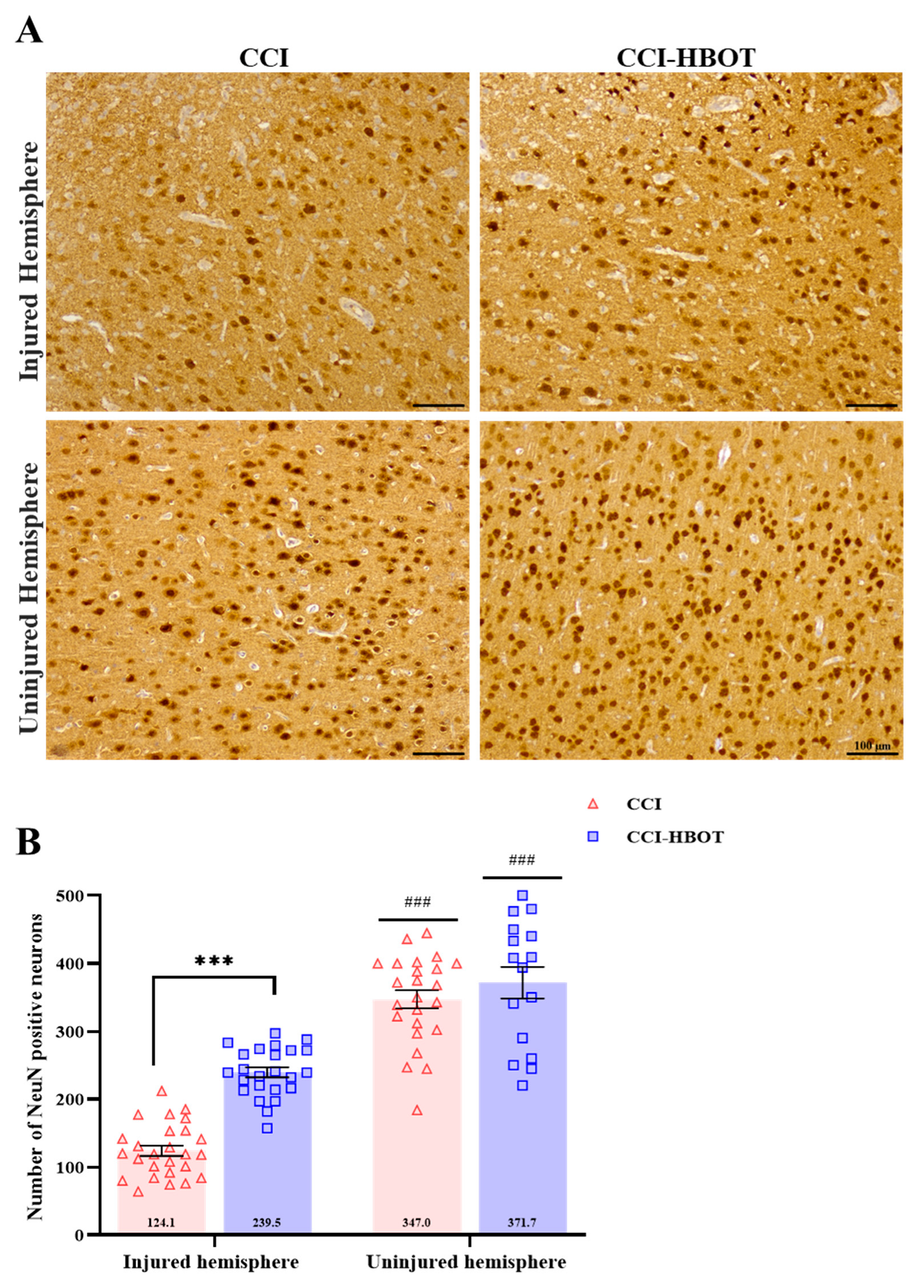
3.3. HBOT Prevents Neuronal Loss and Glial Cell Proliferation Following CCI
3.4. HBOT Restores Mitochondrial Respiration Following CCI
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TBI | Traumatic brain injury |
| CCI | Controlled cortical impact |
| HBOT | Hyperbaric oxygen therapy |
| ICP | Intracranial pressure |
| ROS | Reactive oxygen species |
| TSPO | Translocator protein |
| CI | Complex I |
| CII | Complex II |
| ET | Electron Transfer |
| mPTP | Mitochondrial permeability transition pore |
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the Global Incidence of Traumatic Brain Injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular Infiltration in Traumatic Brain Injury. J. Neuroinflammation 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 484040. [Google Scholar] [CrossRef]
- Freire, M.A.M.; Rocha, G.S.; Bittencourt, L.O.; Falcao, D.; Lima, R.R.; Cavalcanti, J.R.L.P. Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far? Biology 2023, 12, 1139. [Google Scholar] [CrossRef]
- Reilly, P.L.; Graham, D.I.; Adams, J.H.; Jennett, B. Patients with Head Injury Who Talk and Die. Lancet 1975, 2, 375–377. [Google Scholar] [CrossRef]
- Bayir, H.; Clark, R.S.B.; Kochanek, P.M. Traumatic Brain Injury in Infants and Children: Mechanisms of Secondary Damage and Treatment in the Intensive Care Unit. Crit. Care Clin. 2003, 19, 529–549. [Google Scholar] [CrossRef] [PubMed]
- Saul, T.G.; Ducker, T.B. Effect of Intracranial Pressure Monitoring and Aggressive Treatment on Mortality in Severe Head Injury. J. Neurosurg. 1982, 56, 498–503. [Google Scholar] [CrossRef]
- Miller, J.D.; Becker, D.P.; Ward, J.D.; Sullivan, H.G.; Adams, W.E.; Rosner, M.J. Significance of Intracranial Hypertension in Severe Head Injury. J. Neurosurg. 1977, 47, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Bouma, G.J.; Muizelaar, J.P.; Bandoh, K.; Marmarou, A. Blood Pressure and Intracranial Pressure-Volume Dynamics in Severe Head Injury: Relationship with Cerebral Blood Flow. J. Neurosurg. 1992, 77, 15–19. [Google Scholar] [CrossRef]
- Jaggi, J.L.; Obrist, W.D.; Gennarelli, T.A.; Langfitt, T.W. Relationship of Early Cerebral Blood Flow and Metabolism to Outcome in Acute Head Injury. J. Neurosurg. 1990, 72, 176–182. [Google Scholar] [CrossRef]
- Obrist, W.D.; Langfitt, T.W.; Jaggi, J.L.; Cruz, J.; Gennarelli, T.A. Cerebral Blood Flow and Metabolism in Comatose Patients with Acute Head Injury. J. Neurosurg. 1984, 61, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.; Bergsneider, M.; Hattori, N.; Wu, H.-M.; Huang, S.-C.; Martin, N.A.; Glenn, T.C.; McArthur, D.L.; Hovda, D.A. Metabolic Crisis without Brain Ischemia Is Common after Traumatic Brain Injury: A Combined Microdialysis and Positron Emission Tomography Study. J. Cereb. Blood Flow. Metab. 2005, 25, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Chieregato, A.; Tanfani, A.; Compagnone, C.; Turrini, C.; Sarpieri, F.; Ravaldini, M.; Targa, L.; Fainardi, E. Global Cerebral Blood Flow and CPP after Severe Head Injury: A Xenon-CT Study. Intensive Care Med. 2007, 33, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Belli, A.; Sen, J.; Petzold, A.; Russo, S.; Kitchen, N.; Smith, M. Metabolic Failure Precedes Intracranial Pressure Rises in Traumatic Brain Injury: A Microdialysis Study. Acta Neurochir. 2008, 150, 461–469. [Google Scholar] [CrossRef]
- Khatri, N.; Sumadhura, B.; Kumar, S.; Kaundal, R.K.; Sharma, S.; Datusalia, A.K. The Complexity of Secondary Cascade Consequent to Traumatic Brain Injury: Pathobiology and Potential Treatments. Curr. Neuropharmacol. 2021, 19, 1984. [Google Scholar] [CrossRef]
- Hartings, J.A. Spreading Depolarization Monitoring in Neurocritical Care of Acute Brain Injury. Curr. Opin. Crit. Care 2017, 23, 94–102. [Google Scholar] [CrossRef]
- Lauritzen, M.; Dreier, J.P.; Fabricius, M.; Hartings, J.A.; Graf, R.; Strong, A.J. Clinical Relevance of Cortical Spreading Depression in Neurological Disorders: Migraine, Malignant Stroke, Subarachnoid and Intracranial Hemorrhage, and Traumatic Brain Injury. J. Cereb. Blood Flow. Metab. 2011, 31, 17. [Google Scholar] [CrossRef]
- Pradhan, A.A.; Smith, M.L.; Zyuzin, J.; Charles, A. δ-Opioid Receptor Agonists Inhibit Migraine-Related Hyperalgesia, Aversive State and Cortical Spreading Depression in Mice. Br. J. Pharmacol. 2014, 171, 2375. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; William Shuttleworth, C.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, Analysis, and Interpretation of Spreading Depolarizations in Neurointensive Care: Review and Recommendations of the COSBID Research Group. J. Cereb. Blood Flow. Metab. 2017, 37, 1595. [Google Scholar] [CrossRef]
- Dreier, J.P. The Role of Spreading Depression, Spreading Depolarization and Spreading Ischemia in Neurological Disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef] [PubMed]
- Benaroya, H. Brain Energetics, Mitochondria, and Traumatic Brain Injury. Rev. Neurosci. 2020, 31, 363–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, C.H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants 2021, 10, 872. [Google Scholar] [CrossRef]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial Membrane Permeabilization in Neuronal Injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef]
- Palzur, E.; Edelman, D.; Sakas, R.; Soustiel, J.F. Etifoxine Restores Mitochondrial Oxidative Phosphorylation and Improves Cognitive Recovery Following Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 12881. [Google Scholar] [CrossRef] [PubMed]
- Camporesi, E.M.; Bosco, G. Mechanisms of Action of Hyperbaric Oxygen Therapy. Undersea Hyperb. Med. 2014, 41, 247–252. [Google Scholar]
- Rockswold, S.B.; Rockswold, G.L.; Zaun, D.A.; Zhang, X.; Cerra, C.E.; Bergman, T.A.; Liu, J. A Prospective, Randomized Clinical Trial to Compare the Effect of Hyperbaric to Normobaric Hyperoxia on Cerebral Metabolism, Intracranial Pressure, and Oxygen Toxicity in Severe Traumatic Brain Injury. J. Neurosurg. 2010, 112, 1080–1094. [Google Scholar] [CrossRef]
- Rockswold, S.B.; Rockswold, G.L.; Zaun, D.A.; Liu, J. A Prospective, Randomized Phase II Clinical Trial to Evaluate the Effect of Combined Hyperbaric and Normobaric Hyperoxia on Cerebral Metabolism, Intracranial Pressure, Oxygen Toxicity, and Clinical Outcome in Severe Traumatic Brain Injury. J. Neurosurg. 2013, 118, 1317–1328. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Sun, T.; Yu, H. lin Hyperbaric Oxygen Therapy for the Treatment of Traumatic Brain Injury: A Meta-Analysis. Neurol. Sci. 2016, 37, 693–701. [Google Scholar] [CrossRef]
- Palzur, E.; Zaaroor, M.; Vlodavsky, E.; Milman, F.; Soustiel, J.F. Neuroprotective Effect of Hyperbaric Oxygen Therapy in Brain Injury Is Mediated by Preservation of Mitochondrial Membrane Properties. Brain Res. 2008, 1221, 126–133. [Google Scholar] [CrossRef]
- Zhou, Z.; Daugherty, W.P.; Sun, D.; Levasseur, J.E.; Altememi, N.; Hamm, R.J.; Rockswold, G.L.; Bullock, M.R. Protection of Mitochondrial Function and Improvement in Cognitive Recovery in Rats Treated with Hyperbaric Oxygen Following Lateral Fluid-Percussion Injury. J. Neurosurg. 2007, 106, 687–694. [Google Scholar] [CrossRef]
- GoldenHyperbaric Oxygen Therapy for Reduction, Z.L.; Neubauer, R.; Golden, C.J.; Greene, L.; Marsh, J.; Mleko, A. Improvement in Cerebral Metabolism in Chronic Brain Injury after Hyperbaric Oxygen Therapy. Int. J. Neurosci. 2002, 112, 119–131. [Google Scholar]
- Neubauer, R.A.; James, P. Cerebral Oxygenation and the Recoverable Brain. Neurol. Res. 1998, 20 (Suppl. S1), S33–S36. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Cahill, J.; Zhang, J.H. Hyperbaric Oxygen and Cerebral Physiology. Neurol. Res. 2007, 29, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Lo, T.; Mychaskiw, G.; Colohan, A. Mechanisms of Hyperbaric Oxygen and Neuroprotection in Stroke. Pathophysiology 2005, 12, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Harch, P.G.; Andrews, S.R.; Fogarty, E.F.; Amen, D.; Pezzullo, J.C.; Lucarini, J.; Aubrey, C.; Taylor, D.V.; Staab, P.K.; Van Meter, K.W. A Phase i Study of Low-Pressure Hyperbaric Oxygen Therapy for Blast-Induced Post-Concussion Syndrome and Post-Traumatic Stress Disorder. J. Neurotrauma 2012, 29, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Obenaus, A. Hyperbaric Oxygen Therapy for Traumatic Brain Injury. Med. Gas. Res. 2011, 1, 21. [Google Scholar] [CrossRef]
- Daugherty, W.P.; Levasseur, J.E.; Sun, D.; Rockswold, G.L.; Bullock, M.R. Effects of Hyperbaric Oxygen Therapy on Cerebral Oxygenation and Mitochondrial Function Following Moderate Lateral Fluid-Percussion Injury in Rats. J. Neurosurg. 2004, 101, 499–504. [Google Scholar] [CrossRef]
- Li, J.-S.; Zhang, W.; Kang, Z.-M.; Ding, S.-J.; Liu, W.-W.; Zhang, J.H.; Guan, Y.-T.; Sun, X.-J. Hyperbaric Oxygen Preconditioning Reduces Ischemia–Reperfusion Injury by Inhibition of Apoptosis via Mitochondrial Pathway in Rat Brain. Neuroscience 2009, 159, 1309–1315. [Google Scholar] [CrossRef]
- Council, N.R. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011; ISBN 978-0-309-15400-0. [Google Scholar]
- Dean, D.D.; Frank, J.A.; Turtzo, L.C. Controlled Cortical Impact in the Rat. Curr. Protoc. Neurosci. 2017, 81, 9.62.1–9.62.12. [Google Scholar] [CrossRef]
- Palzur, E.; Vlodavsky, E.; Mulla, H.; Arieli, R.; Feinsod, M.; Soustiel, J.F. Hyperbaric Oxygen Therapy for Reduction of Secondary Brain Damage in Head Injury: An Animal Model of Brain Contusion. J. Neurotrauma 2004, 21, 41–48. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris Water Maze: Procedures for Assessing Spatial and Related Forms of Learning and Memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A Rotarod Test for Evaluation of Motor Skill Learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef]
- Ben-Shabat, M.; Awad-Igbaria, Y.; Sela, S.; Gross, B.; Yagil, Y.; Yagil, C.; Palzur, E. Predisposition to Cortical Neurodegenerative Changes in Brains of Hypertension Prone Rats. J. Transl. Med. 2023, 21, 51. [Google Scholar] [CrossRef] [PubMed]
- SUIT-020 Fluo Mt D033—Bioblast. Available online: https://wiki.oroboros.at/index.php/SUIT-020_Fluo_mt_D033 (accessed on 17 February 2022).
- Vlodavsky, E.; Palzur, E.; Soustiel, J.F. Hyperbaric Oxygen Therapy Reduces Neuroinflammation and Expression of Matrix Metalloproteinase-9 in the Rat Model of Traumatic Brain Injury. Neuropathol. Appl. Neurobiol. 2006, 32, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Liu, K.; Li, L.; Li, X.; Zhao, P. The Effects of Hyperbaric Oxygen Therapy on Neuropathic Pain via Mitophagy in Microglia. Mol. Pain. 2017, 13, 1744806917710862. [Google Scholar] [CrossRef]
- Xia, A.; Huang, H.; You, W.; Liu, Y.; Wu, H.; Liu, S. The Neuroprotection of Hyperbaric Oxygen Therapy against Traumatic Brain Injury via NF-ΚB/MAPKs-CXCL1 Signaling Pathways. Exp. Brain Res. 2022, 240, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-H.; Zhang, X.-G.; Jiang, Z.-L.; Li, X.; Peng, L.-L.; Li, Y.-C.; Wang, Y. Neuroprotective Effects of Hyperbaric Oxygen Treatment on Traumatic Brain Injury in the Rat. J. Neurotrauma 2010, 27, 1733–1743. [Google Scholar] [CrossRef]
- Boussi-Gross, R.; Golan, H.; Fishlev, G.; Bechor, Y.; Volkov, O.; Bergan, J.; Friedman, M.; Hoofien, D.; Shlamkovitch, N.; Ben-Jacob, E.; et al. Hyperbaric Oxygen Therapy Can Improve Post Concussion Syndrome Years after Mild Traumatic Brain Injury—Randomized Prospective Trial. PLoS ONE 2013, 8, e79995. [Google Scholar] [CrossRef]
- Marcinkowska, A.B.; Mankowska, N.D.; Kot, J.; Winklewski, P.J. Impact of Hyperbaric Oxygen Therapy on Cognitive Functions: A Systematic Review. Neuropsychol. Rev. 2022, 32, 99. [Google Scholar] [CrossRef]
- Hadanny, A.; Golan, H.; Fishlev, G.; Bechor, Y.; Volkov, O.; Suzin, G.; Ben-Jacob, E.; Efrati, S. Hyperbaric Oxygen Can Induce Neuroplasticity and Improve Cognitive Functions of Patients Suffering from Anoxic Brain Damage. Restor. Neurol. Neurosci. 2015, 33, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.C.; Pickett, T.C. Motor Impairment after Severe Traumatic Brain Injury: A Longitudinal Multicenter Study. J. Rehabil. Res. Dev. 2007, 44, 975–982. [Google Scholar] [CrossRef]
- Fu, M.; Yu, X.; Lu, J.; Zuo, Y. Repetitive Motor Learning Induces Coordinated Formation of Clustered Dendritic Spines In Vivo. Nature 2012, 483, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Awad-Igbaria, Y.; Maaravi-Hesseg, R.; Admon, R.; Karni, A. Only Tomorrow: Delayed Effects of Teachers Attitude on Motor Skill Learning. Learn. Instr. 2022, 82, 101681. [Google Scholar] [CrossRef]
- Li, H.-Q.; Spitzer, N.C. Exercise Enhances Motor Skill Learning by Neurotransmitter Switching in the Adult Midbrain. Nat. Commun. 2020, 11, 2195. [Google Scholar] [CrossRef]
- Avraham, E.; Sacher, Y.; Maaravi-Hesseg, R.; Karni, A.; Doron, R. Skill-Learning by Observation-Training with Patients after Traumatic Brain Injury. Front. Hum. Neurosci. 2022, 16, 940075. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, Y.; Hanafy, K.A. Cell Death and Recovery in Traumatic Brain Injury. Neurotherapeutics 2020, 17, 446. [Google Scholar] [CrossRef]
- Schimmel, S.J.; Acosta, S.; Lozano, D. Neuroinflammation in Traumatic Brain Injury: A Chronic Response to an Acute Injury. Brain Circ. 2017, 3, 135. [Google Scholar] [CrossRef]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef]
- Lavrnja, I.; Parabucki, A.; Brkic, P.; Jovanovic, T.; Dacic, S.; Savic, D.; Pantic, I.; Stojiljkovic, M.; Pekovic, S. Repetitive Hyperbaric Oxygenation Attenuates Reactive Astrogliosis and Suppresses Expression of Inflammatory Mediators in the Rat Model of Brain Injury. Mediat. Inflamm. 2015, 2015, 498405. [Google Scholar] [CrossRef]
- McMillian, M.K.; Thai, L.; Hong, J.-S.; O’Callaghan, J.P.; Pennypacker, K.R. Brain Injury in a Dish: A Model for Reactive Gliosis. Trends Neurosci. 1994, 17, 138–142. [Google Scholar] [CrossRef]
- Zheng, R.Z.; Lee, K.Y.; Qi, Z.X.; Wang, Z.; Xu, Z.Y.; Wu, X.H.; Mao, Y. Neuroinflammation Following Traumatic Brain Injury: Take It Seriously or Not. Front. Immunol. 2022, 13, 855701. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Baylr, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, Y.; Huang, C.; Xia, A.; Wang, G.; Liu, S. Hyperbaric Oxygen Therapy Improves Neurological Function via the P38-MAPK/CCL2 Signaling Pathway Following Traumatic Brain Injury. Neuroreport 2021, 32, 1255. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Kang, N.; Li, P.; Liu, X.; Li, G.; Yang, J. Effect of Hyperbaric Oxygen Therapy on Polarization Phenotype of Rat Microglia After Traumatic Brain Injury. Front. Neurol. 2021, 12, 640816. [Google Scholar] [CrossRef]
- Geng, F.; Ma, Y.; Xing, T.; Zhuang, X.; Zhu, J.; Yao, L. Effects of Hyperbaric Oxygen Therapy on Inflammasome Signaling after Traumatic Brain Injury. Neuroimmunomodulation 2016, 23, 122–129. [Google Scholar] [CrossRef]
- Awad-Igbaria, Y.; Ferreira, N.; Keadan, A.; Sakas, R.; Edelman, D.; Shamir, A.; Francous-Soustiel, J.; Palzur, E. HBO Treatment Enhances Motor Function and Modulates Pain Development after Sciatic Nerve Injury via Protection the Mitochondrial Function. J. Transl. Med. 2023, 21, 545. [Google Scholar] [CrossRef]
- Fu, H.; Li, F.; Thomas, S.; Yang, Z. Hyperbaric Oxygenation Alleviates Chronic Constriction Injury (CCI)-Induced Neuropathic Pain and Inhibits GABAergic Neuron Apoptosis in the Spinal Cord. Scand. J. Pain. 2017, 17, 330–338. [Google Scholar] [CrossRef]
- Lim, T.K.Y.; Rone, M.B.; Lee, S.; Antel, J.P.; Zhang, J. Mitochondrial and Bioenergetic Dysfunction in Trauma-Induced Painful Peripheral Neuropathy. Mol. Pain. 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Barrientos, A. Mitochondrial Respiratory Chain Composition and Organization in Response to Changing Oxygen Levels. J. Life Sci. 2020, 2, 1–17. [Google Scholar] [CrossRef]
- Schottlender, N.; Gottfried, I.; Ashery, U. Hyperbaric Oxygen Treatment: Effects on Mitochondrial Function and Oxidative Stress. Biomolecules 2021, 11, 1827. [Google Scholar] [CrossRef]
- Xing, P.; Ma, K.; Li, L.; Wang, D.; Hu, G.; Long, W. The Protection Effect and Mechanism of Hyperbaric Oxygen Therapy in Rat Brain with Traumatic Injury. Acta Cir. Bras. 2018, 33, 341–353. [Google Scholar] [CrossRef]
- Yang, L.; Hei, M.Y.; Dai, J.J.; Hu, N.; Xiang, X.Y. Effect of Hyperbaric Oxygenation on Mitochondrial Function of Neuronal in the Cortex of Neonatal Rats after Hypoxic-Ischemic Brain. Braz. J. Med. Biol. Res. 2016, 49, e5187. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Duan, R.; Sun, Y.; Li, Q. Hyperbaric Oxygen Therapy for Healthy Aging: From Mechanisms to Therapeutics. Redox Biol. 2022, 53, 102352. [Google Scholar] [CrossRef]
- Benedetti, S.; Lamorgese, A.; Piersantelli, M.; Pagliarani, S.; Benvenuti, F.; Canestrari, F. Oxidative Stress and Antioxidant Status in Patients Undergoing Prolonged Exposure to Hyperbaric Oxygen. Clin. Biochem. 2004, 37, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Karabiyikoglu, M.; Hua, Y.; Silbergleit, R.; He, Y.; Keep, R.F.; Xi, G. Hyperbaric Oxygen-Induced Attenuation of Hemorrhagic Transformation after Experimental Focal Transient Cerebral Ischemia. Stroke 2007, 38, 1362–1367. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakas, R.; Dan, K.; Edelman, D.; Abu-Ata, S.; Ben-Menashe, A.; Awad-Igbaria, Y.; Francois-Soustiel, J.; Palzur, E. Hyperbaric Oxygen Therapy Alleviates Memory and Motor Impairments Following Traumatic Brain Injury via the Modulation of Mitochondrial-Dysfunction-Induced Neuronal Apoptosis in Rats. Antioxidants 2023, 12, 2034. https://doi.org/10.3390/antiox12122034
Sakas R, Dan K, Edelman D, Abu-Ata S, Ben-Menashe A, Awad-Igbaria Y, Francois-Soustiel J, Palzur E. Hyperbaric Oxygen Therapy Alleviates Memory and Motor Impairments Following Traumatic Brain Injury via the Modulation of Mitochondrial-Dysfunction-Induced Neuronal Apoptosis in Rats. Antioxidants. 2023; 12(12):2034. https://doi.org/10.3390/antiox12122034
Chicago/Turabian StyleSakas, Reem, Katya Dan, Doron Edelman, Saher Abu-Ata, Aviv Ben-Menashe, Yaseen Awad-Igbaria, Jean Francois-Soustiel, and Eilam Palzur. 2023. "Hyperbaric Oxygen Therapy Alleviates Memory and Motor Impairments Following Traumatic Brain Injury via the Modulation of Mitochondrial-Dysfunction-Induced Neuronal Apoptosis in Rats" Antioxidants 12, no. 12: 2034. https://doi.org/10.3390/antiox12122034
APA StyleSakas, R., Dan, K., Edelman, D., Abu-Ata, S., Ben-Menashe, A., Awad-Igbaria, Y., Francois-Soustiel, J., & Palzur, E. (2023). Hyperbaric Oxygen Therapy Alleviates Memory and Motor Impairments Following Traumatic Brain Injury via the Modulation of Mitochondrial-Dysfunction-Induced Neuronal Apoptosis in Rats. Antioxidants, 12(12), 2034. https://doi.org/10.3390/antiox12122034