
Optogenetics in Alzheimer’s Disease: Focus on Astrocytes


Abstract
:1. Introduction
2. Optogenetics as a Tool for Regulating the Activity of Nerve Cells
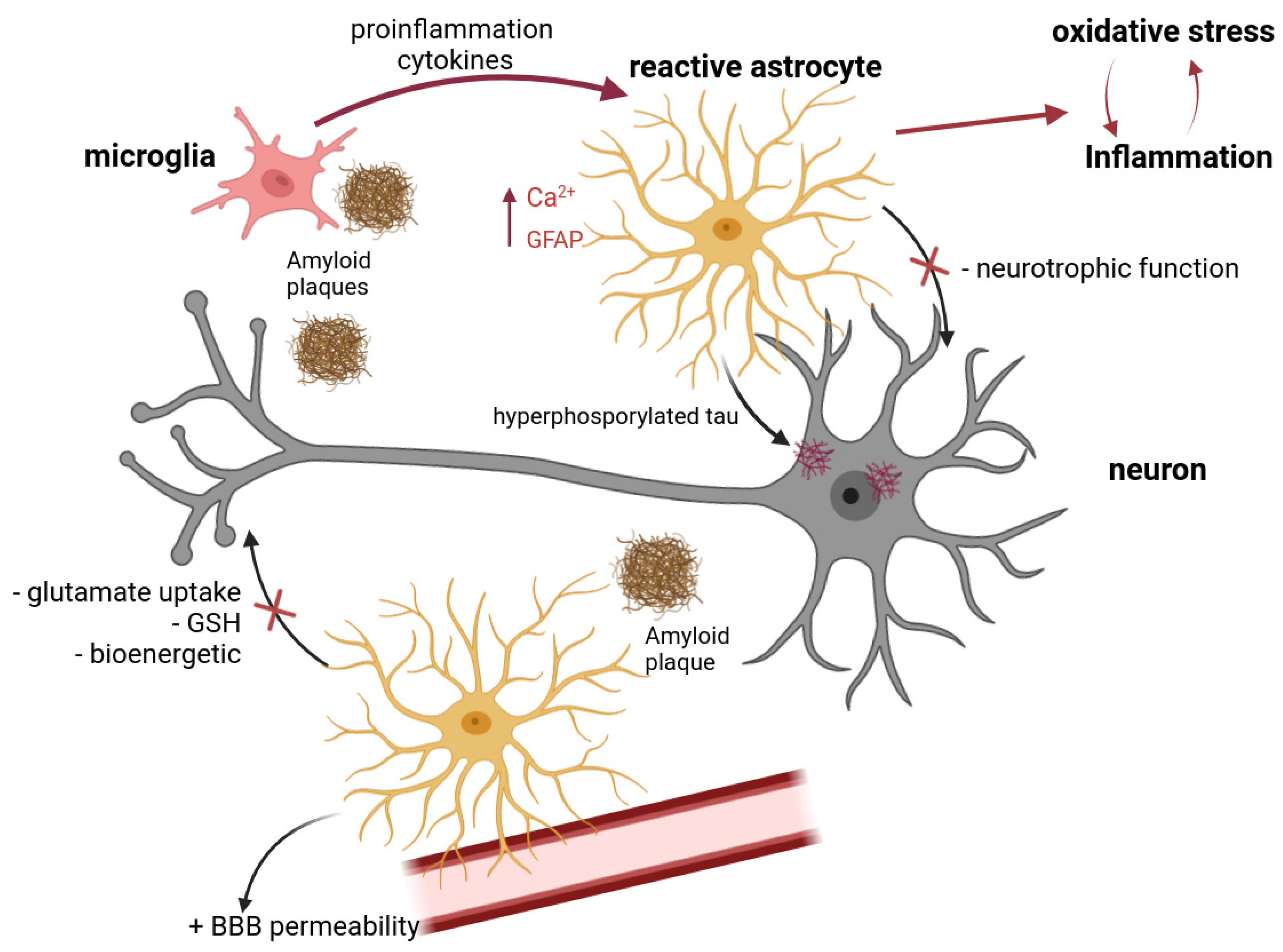
3. The Role of Astrocytes in the Pathogenesis of Alzheimer’s Disease
3.1. Production and Clearance of Amyloid Proteins
3.2. Neuroinflammation and Reactive Astrogliosis
3.3. Oxidative Stress
3.4. Interastrocytic Interactions. Calcium Dysregulation in AD
3.5. Interaction between Neurons and Astrocytes. Disorders of the Neuron–Astrocyte Interaction in AD. Excitotoxicity
4. Possibilities of Optogenetics for the Treatment of Alzheimer’s Disease
4.1. Optogenetic Tools for the Correction of Neurodegenerative Changes in AD
4.2. Application of Optogenetic Approaches to Stimulate Neurogenesis in the Adult Brain in AD
4.3. Optogenetics for Modeling AD
5. Problems and Prospects of Using Optogenetics for AD Correction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s disease |
| AMPA receptor, AMPAR | the α-amino-3-hydroxy-5-methyl-4-isoxazole-propionicacid receptor |
| APOE | Apolipoprotein E |
| APP | amyloid precursor protein |
| ATP | adenosine triphosphate |
| AQP4 | aquaporin 4 |
| BBB | blood–brain barrier |
| ChR2 | channelrhodopsin 2 |
| CNS | The central nervous system |
| CS | complement system |
| EAAT (GLAST-1 и GLT-1) | the excitatory amino acid transporter |
| ECB | endocannabinoids |
| ER | endoplasmic reticulum |
| EVs | extracellular vesicles |
| IL | interleukin |
| IP3 | inositol-3-phosphate |
| HR | halorhodopsin |
| GABA | gamma-aminobutyric acid |
| GFAP | glial fibrillar acidic protein |
| GPCR | G-protein-coupled receptors |
| LTP | long-term potentiation |
| MAC | membrane attack complex |
| MAPT | microtubule-associated protein tau |
| mGluR | metabotropic glutamate receptors |
| MMP | matrix metalloproteinases |
| NFT | нeйpoфибpилляpныe клyбки |
| NIR | near-infrared light |
| NSCs | neural stem cells |
| NMDA receptor, NDMAR | N-methyl-d-aspartate glutamate receptors |
| PK2 | prokineticin-2 |
| PLC | phospholipase C |
| ROS | reactive oxygen species |
| SGZ | subgranular zone of the dentate gyrus |
| TNF | tumor necrosis factor |
| UDP | uridine triphosphate |
References
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum (ER)–Mitochondria Interactions and Ca2+ Cross-Talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef]
- Diociaiuti, M.; Bonanni, R.; Cariati, I.; Frank, C.; D’Arcangelo, G. Amyloid Prefibrillar Oligomers: The Surprising Commonalities in Their Structure and Activity. Int. J. Mol. Sci. 2021, 22, 6435. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Sajad, M.; Kumar, R.; Thakur, S.C. History in Perspective: The Prime Pathological Players and Role of Phytochemicals in Alzheimer’s Disease. IBRO Neurosci. Rep. 2022, 12, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial Dysfunction, Oxidative Stress, Neuroinflammation, and Metabolic Alterations in the Progression of Alzheimer’s Disease: A Meta-Analysis of in Vivo Magnetic Resonance Spectroscopy Studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Kim, C.K.; Adhikari, A.; Deisseroth, K. Integration of Optogenetics with Complementary Methodologies in Systems Neuroscience. Nat. Rev. Neurosci. 2017, 18, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.K.; Li, D. Optogenetics: Basic Concepts and Their Development. In Optogenetics: Methods and Protocols; Kianianmomeni, A., Ed.; Humana Press: New York, NY, USA, 2016; pp. 1–17. [Google Scholar]
- Tan, P.; He, L.; Huang, Y.; Zhou, Y. Optophysiology: Illuminating Cell Physiology with Optogenetics. Physiol. Rev. 2022, 102, 1263–1325. [Google Scholar] [CrossRef]
- Packer, A.M.; Roska, B.; Häusser, M. Targeting Neurons and Photons for Optogenetics. Nat. Neurosci. 2013, 16, 805–815. [Google Scholar] [CrossRef]
- Bernstein, J.G.; Boyden, E.S. Optogenetic Tools for Analyzing the Neural Circuits of Behavior. Trends Cogn. Sci. 2011, 15, 592–600. [Google Scholar] [CrossRef]
- Heston, J.; Friedman, A.; Baqai, M.; Bavafa, N.; Aron, A.R.; Hnasko, T.S. Activation of Subthalamic Nucleus Stop Circuit Disrupts Cognitive Performance. eNeuro 2020, 7, 0159-20. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why Are Astrocytes Important? Neurochem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef] [PubMed]
- McConnell, H.L.; Mishra, A. Cells of the Blood–Brain Barrier: An Overview of the Neurovascular Unit in Health and Disease. In The Blood-Brain Barrier: Methods and Protocols; Stone, N., Ed.; Humana Press: New York, NY, USA, 2022; pp. 3–24. [Google Scholar]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte Function from Information Processing to Cognition and Cognitive Impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Murat, C.D.B.; García-Cáceres, C. Astrocyte Gliotransmission in the Regulation of Systemic Metabolism. Metabolites 2021, 11, 732. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Rodríguez-Giraldo, M.; González-Reyes, R.E.; Ramírez-Guerrero, S.; Bonilla-Trilleras, C.E.; Guardo-Maya, S.; Nava-Mesa, M.O. Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments. Int. J. Mol. Sci. 2022, 23, 13630. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.-M. The Role of Astrocytes in Amyloid Production and Alzheimer’s Disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte Dysfunction in Alzheimer Disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600. [Google Scholar] [CrossRef]
- Boyden, E.S. A History of Optogenetics: The Development of Tools for Controlling Brain Circuits with Light. F1000 Biol. Rep. 2011, 3, 11. [Google Scholar] [CrossRef]
- White, M.; Mackay, M.; Whittaker, R.G. Taking Optogenetics into the Human Brain: Opportunities and Challenges in Clinical Trial Design. Open Access J. Clin. Trials 2020, 12, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Christenson Wick, Z.; Krook-Magnuson, E. Specificity, Versatility, and Continual Development: The Power of Optogenetics for Epilepsy Research. Front. Cell Neurosci. 2018, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K. Optogenetics: 10 Years of Microbial Opsins in Neuroscience. Nat. Neurosci. 2015, 18, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Chow, B.Y.; Zhou, H.; Klapoetke, N.C.; Chuong, A.; Rajimehr, R.; Yang, A.; Baratta, M.V.; Winkle, J.; Desimone, R.; et al. A High-Light Sensitivity Optical Neural Silencer: Development and Application to Optogenetic Control of Non-Human Primate Cortex. Front. Syst. Neurosci. 2011, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Mattis, J.; Tye, K.M.; Ferenczi, E.A.; Ramakrishnan, C.; O’Shea, D.J.; Prakash, R.; Gunaydin, L.A.; Hyun, M.; Fenno, L.E.; Gradinaru, V.; et al. Principles for Applying Optogenetic Tools Derived from Direct Comparative Analysis of Microbial Opsins. Nat. Methods 2012, 9, 159–172. [Google Scholar] [CrossRef]
- Spangler, S.M.; Bruchas, M.R. Optogenetic Approaches for Dissecting Neuromodulation and GPCR Signaling in Neural Circuits. Curr. Opin. Pharmacol. 2017, 32, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lohman, A.W.; Zhuravlova, Y.; Lu, X.; Wiens, M.D.; Hoi, H.; Yaganoglu, S.; Mohr, M.A.; Kitova, E.N.; Klassen, J.S.; et al. Optogenetic Control with a Photocleavable Protein, PhoCl. Nat. Methods 2017, 14, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Toettcher, J.E.; Weiner, O.D.; Lim, W.A. Using Optogenetics to Interrogate the Dynamic Control of Signal Transmission by the Ras/Erk Module. Cell 2013, 155, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Hsu, P.D.; Heidenreich, M.; Cong, L.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical Control of Mammalian Endogenous Transcription and Epigenetic States. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef]
- Motta-Mena, L.B.; Reade, A.; Mallory, M.J.; Glantz, S.; Weiner, O.D.; Lynch, K.W.; Gardner, K.H. An Optogenetic Gene Expression System with Rapid Activation and Deactivation Kinetics. Nat. Chem. Biol. 2014, 10, 196–202. [Google Scholar] [CrossRef]
- Cosentino, C.; Alberio, L.; Gazzarrini, S.; Aquila, M.; Romano, E.; Cermenati, S.; Zuccolini, P.; Petersen, J.; Beltrame, M.; Van Etten, J.L.; et al. Engineering of a Light-Gated Potassium Channel. Science 2015, 348, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.-G.; Ishizuka, T.; Yawo, H. Channelrhodopsins—Their Potential in Gene Therapy for Neurological Disorders. Neurosci. Res. 2013, 75, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Gourine, A.V.; Kasymov, V.; Marina, N.; Tang, F.; Figueiredo, M.F.; Lane, S.; Teschemacher, A.G.; Spyer, K.M.; Deisseroth, K.; Kasparov, S. Astrocytes Control Breathing Through PH-Dependent Release of ATP. Science 2010, 329, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Gerasimov, E.; Erofeev, A.; Borodinova, A.; Bolshakova, A.; Balaban, P.; Bezprozvanny, I.; Vlasova, O.L. Optogenetic Activation of Astrocytes—Effects on Neuronal Network Function. Int. J. Mol. Sci. 2021, 22, 9613. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.; Roshchin, M.; Bezprozvanny, I.; Smirnov, I.; Vlasova, O.; Balaban, P.; Borodinova, A. Bidirectional Regulation by “Star Forces”: Ionotropic Astrocyte’s Optical Stimulation Suppresses Synaptic Plasticity, Metabotropic One Strikes Back. Hippocampus 2023, 33, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal Glial Activation Coincides with Increased BACE1 Activation and Precedes Amyloid Plaque Deposition in APP[V717I] Transgenic Mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The Contribution of Activated Astrocytes to Aβ Production: Implications for Alzheimer’s Disease Pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Yoon, H.; Kim, J. Apolipoprotein E Metabolism and Functions in Brain and Its Role in Alzheimer’s Disease. Curr. Opin. Lipidol. 2017, 28, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Benech, P.; Greetham, L.; Stephan, D.; Jimenez, A.; Jullien, N.; García-González, L.; Tsvetkov, P.O.; Devred, F.; Sancho-Martinez, I.; et al. APOE4 Drives Inflammation in Human Astrocytes via TAGLN3 Repression and NF-ΚB Activation. Cell Rep. 2022, 40, 111200. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Drummond, E. APOE-Amyloid Interaction: Therapeutic Targets. Neurobiol. Dis. 2020, 138, 104784. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia Prevent Beta-Amyloid Plaque Formation in the Early Stage of an Alzheimer’s Disease Mouse Model with Suppression of Glymphatic Clearance. Alzheimers Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of Aquaporin-4 in APP/PS1 Mice Exacerbates Brain Aβ Accumulation and Memory Deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult Mouse Astrocytes Degrade Amyloid-β in Vitro and in Situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Liu, C.-C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Montoliu-Gaya, L.; Mulder, S.D.; Veerhuis, R.; Villegas, S. Effects of an Aβ-Antibody Fragment on Aβ Aggregation and Astrocytic Uptake Are Modulated by Apolipoprotein E and J Mimetic Peptides. PLoS ONE 2017, 12, e0188191. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.-F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-β Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, S.V.; Burmistrov, D.E.; Kondakova, E.V.; Sarimov, R.M.; Yarkov, R.S.; Franceschi, C.; Vedunova, M.V. An Emerging Role of Astrocytes in Aging/Neuroinflammation and Gut-Brain Axis with Consequences on Sleep and Sleep Disorders. Ageing Res. Rev. 2023, 83, 101775. [Google Scholar] [CrossRef]
- Guerriero, F.; Sgarlata, C.; Francis, M.; Maurizi, N.; Faragli, A.; Perna, S.; Rondanelli, M.; Rollone, M.; Ricevuti, G. Neuroinflammation, Immune System and Alzheimer Disease: Searching for the Missing Link. Aging Clin. Exp. Res. 2017, 29, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Nogueras-Ortiz, C.J.; Mahairaki, V.; Delgado-Peraza, F.; Das, D.; Avgerinos, K.; Eren, E.; Hentschel, M.; Goetzl, E.J.; Mattson, M.P.; Kapogiannis, D. Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells 2020, 9, 1618. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High Complement Levels in Astrocyte-Derived Exosomes of Alzheimer Disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Saviuk, M.; Vedunova, M.V. Necroptosis in CNS Diseases: Focus on Astrocytes. Front. Aging Neurosci. 2023, 14, 1016053. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo Proteins of Plasma Astrocyte-derived Exosomes in Alzheimer’s Disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, W.; Kooijman, L.; Orre, M.; Stassen, O.; Pekny, M.; Hol, E.M. GFAP and Vimentin Deficiency Alters Gene Expression in Astrocytes and Microglia in Wild-Type Mice and Changes the Transcriptional Response of Reactive Glia in Mouse Model for Alzheimer’s Disease. Glia 2015, 63, 1036–1056. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of Glia from Alzheimer’s Mice Reveals Inflammation and Dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe Reactive Astrocytes Precipitate Pathological Hallmarks of Alzheimer’s Disease via H2O2− Production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from Reactive Astrocytes Impairs Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic Inhibition in Dentate Gyrus Impairs Long-Term Potentiation and Memory in an Alzheimer’s Disease Model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef]
- Lee, K.-I.; Lee, H.-T.; Lin, H.-C.; Tsay, H.-J.; Tsai, F.-C.; Shyue, S.-K.; Lee, T.-S. Role of Transient Receptor Potential Ankyrin 1 Channels in Alzheimer’s Disease. J. Neuroinflamm. 2016, 13, 92. [Google Scholar] [CrossRef]
- Bai, J.-Z.; Lipski, J. Involvement of TRPV4 Channels in Aβ40-Induced Hippocampal Cell Death and Astrocytic Ca2+ Signalling. Neurotoxicology 2014, 41, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.; Luo, J.; Harischandra, D.S.; Gordon, R.; Sarkar, S.; Jin, H.; Anantharam, V.; Désaubry, L.; Kanthasamy, A.; Kanthasamy, A. Prokineticin-2 Promotes Chemotaxis and Alternative A2 Reactivity of Astrocytes. Glia 2018, 66, 2137–2157. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Astrocytes Provide Cysteine to Neurons by Releasing Glutathione. J. Neurochem. 2002, 74, 1434–1442. [Google Scholar] [CrossRef]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.-G.; Ransom, B.R.; Chen, X.-C.; Ye, Z.-C. Dual Pathways Mediate β-Amyloid Stimulated Glutathione Release from Astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Lee, C.J. Reactive Astrocytes in Alzheimer’s Disease: A Double-Edged Sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Speed, N.; Blair, I.A. Cyclooxygenase- and Lipoxygenase-Mediated DNA Damage. Cancer Metastasis Rev. 2011, 30, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH Oxidases: An Overview from Structure to Innate Immunity-Associated Pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- De Silva, T.M.; Miller, A.A. Cerebral Small Vessel Disease: Targeting Oxidative Stress as a Novel Therapeutic Strategy? Front. Pharmacol. 2016, 7, 61. [Google Scholar] [CrossRef]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH Oxidases: A Perspective on Reactive Oxygen Species Production in Tumor Biology. Antioxid. Redox Signal 2014, 20, 2873–2889. [Google Scholar] [CrossRef]
- Dikalov, S.; Dikalova, A.; Bikineyeva, A.; Schmidt, H.; Harrison, D.; Griendling, K. Distinct Roles of Nox1 and Nox4 in Basal and Angiotensin II-Stimulated Superoxide and Hydrogen Peroxide Production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Togliatto, G.; Lombardo, G.; Brizzi, M.F. The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. Int. J. Mol. Sci. 2017, 18, 1988. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, A.; Palaiodimou, L.; Safouris, A.; Kargiotis, O.; Psychogios, K.; Kotsali-Peteinelli, V.; Foska, A.; Zouvelou, V.; Tzavellas, E.; Tzanetakos, D.; et al. Cerebral Amyloid Angiopathy—Related Inflammation: A Single-Center Experience and a Literature Review. J. Clin. Med. 2022, 11, 6731. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Gupta, S.; Parrino, T.E.; Knight, A.G.; Ebenezer, P.J.; Weidner, A.M.; LeVine, H.; Keller, J.N.; Markesbery, W.R. NOX Activity Is Increased in Mild Cognitive Impairment. Antioxid. Redox Signal 2010, 12, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Shimohama, S.; Tanino, H.; Kawakami, N.; Okamura, N.; Kodama, H.; Yamaguchi, T.; Hayakawa, T.; Nunomura, A.; Chiba, S.; Perry, G.; et al. Activation of NADPH Oxidase in Alzheimer’s Disease Brains. Biochem. Biophys. Res. Commun. 2000, 273, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Santhanam, A.V.R.; d’Uscio, L.V.; Katusic, Z.S. Regional Heterogeneity of Cerebral Microvessels and Brain Susceptibility to Oxidative Stress. PLoS ONE 2015, 10, e0144062. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef]
- Tarafdar, A.; Wolska, N.; Krisp, C.; Schlüter, H.; Pula, G. The Amyloid Peptide β Disrupts Intercellular Junctions and Increases Endothelial Permeability in a NADPH Oxidase 1-Dependent Manner. Redox Biol. 2022, 52, 102287. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.M.; van der Vies, S.M.; Rozemuller, A.J.M.; van Horssen, J.; de Vries, H.E. Amyloid Beta Induces Oxidative Stress-Mediated Blood–Brain Barrier Changes in Capillary Amyloid Angiopathy. Antioxid. Redox Signal 2011, 15, 1167–1178. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular Dysfunction and Neurodegeneration in Dementia and Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, S.; Furuno, A.; Ohshima, M.; Araki, T. Oxidative Stress–Dependent Phosphorylation Activates ZNRF1 to Induce Neuronal/Axonal Degeneration. J. Cell Biol. 2015, 211, 881–896. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, S.; Araki, T. NADPH Oxidases Promote Apoptosis by Activating ZNRF1 Ubiquitin Ligase in Neurons Treated with an Exogenously Applied Oxidant. Commun. Integr. Biol. 2016, 9, e1143575. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, S.; Takahashi, Y.; Shibata, M.; Araki, T. Selective Phosphorylation of Serine 345 on P47-Phox Serves as a Priming Signal of ROS-Mediated Axonal Degeneration. Exp. Neurol. 2022, 352, 114024. [Google Scholar] [CrossRef] [PubMed]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.-S. NOX4 Promotes Ferroptosis of Astrocytes by Oxidative Stress-Induced Lipid Peroxidation via the Impairment of Mitochondrial Metabolism in Alzheimer’s Diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef] [PubMed]
- Gage, M.C.; Thippeswamy, T. Inhibitors of Src Family Kinases, Inducible Nitric Oxide Synthase, and NADPH Oxidase as Potential CNS Drug Targets for Neurological Diseases. CNS Drugs 2021, 35, 1–20. [Google Scholar] [CrossRef]
- Gong, P.; Chen, Y.; Lin, A.; Zhang, H.; Zhang, Y.; Ye, R.D.; Yu, Y. P47phox Deficiency Improves Cognitive Impairment and Attenuates Tau Hyperphosphorylation in Mouse Models of AD. Alzheimers Res. Ther. 2020, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial Networks: A Step Further in Neuroglial and Gliovascular Interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [CrossRef]
- Newman, E.A.; Zahs, K.R. Calcium Waves in Retinal Glial Cells. Science 1997, 275, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Sanderson, M.J. Intercellular Ca2+ Waves: Mechanisms and Function. Physiol. Rev. 2012, 92, 1359–1392. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Decrock, E.; Wang, N.; Bol, M.; Vinken, M.; Bultynck, G.; Leybaert, L. The Dual Face of Connexin-Based Astroglial Ca2+ Communication: A Key Player in Brain Physiology and a Prime Target in Pathology. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 2211–2232. [Google Scholar] [CrossRef]
- Pannasch, U.; Rouach, N. Emerging Role for Astroglial Networks in Information Processing: From Synapse to Behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, C.G.; Green, C.R.; Nicholson, L.F.B. Upregulation in Astrocytic Connexin 43 Gap Junction Levels May Exacerbate Generalized Seizures in Mesial Temporal Lobe Epilepsy. Brain Res. 2002, 929, 105–116. [Google Scholar] [CrossRef]
- Collignon, F.; Wetjen, N.M.; Cohen-Gadol, A.A.; Cascino, G.D.; Parisi, J.; Meyer, F.B.; Marsh, W.R.; Roche, P.; Weigand, S.D. Altered Expression of Connexin Subtypes in Mesial Temporal Lobe Epilepsy in Humans. J. Neurosurg. 2006, 105, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Caltabiano, R.; Torrisi, A.; Condorelli, D.; Albanese, V.; Lanzafame, S. High Levels of Connexin 43 MRNA in High Grade Astrocytomas. Study of 32 Cases with in Situ Hybridization. Acta Histochem. 2010, 112, 529–535. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human Astrocytes: Structure and Functions in the Healthy Brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Roux, L.; Benchenane, K.; Rothstein, J.D.; Bonvento, G.; Giaume, C. Plasticity of Astroglial Networks in Olfactory Glomeruli. Proc. Natl. Acad. Sci. USA 2011, 108, 18442–18446. [Google Scholar] [CrossRef]
- Houades, V.; Koulakoff, A.; Ezan, P.; Seif, I.; Giaume, C. Gap Junction-Mediated Astrocytic Networks in the Mouse Barrel Cortex. J. Neurosci. 2008, 28, 5207–5217. [Google Scholar] [CrossRef]
- Claus, L.; Philippot, C.; Griemsmann, S.; Timmermann, A.; Jabs, R.; Henneberger, C.; Kettenmann, H.; Steinhäuser, C. Barreloid Borders and Neuronal Activity Shape Panglial Gap Junction-Coupled Networks in the Mouse Thalamus. Cereb. Cortex 2016, 28, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Houades, V.; Rouach, N.; Ezan, P.; Kirchhoff, F.; Koulakoff, A.; Giaume, C. Shapes of Astrocyte Networks in the Juvenile Brain. Neuron Glia Biol. 2006, 2, 3–14. [Google Scholar] [CrossRef]
- Tsai, H.-H.; Li, H.; Fuentealba, L.C.; Molofsky, A.V.; Taveira-Marques, R.; Zhuang, H.; Tenney, A.; Murnen, A.T.; Fancy, S.P.J.; Merkle, F.; et al. Regional Astrocyte Allocation Regulates CNS Synaptogenesis and Repair. Science 2012, 337, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Mattson, M.P. Alzheimer’s Amyloid β-Peptide Enhances ATP/Gap Junction-Mediated Calcium-Wave Propagation in Astrocytes. Neuromolecular Med. 2003, 3, 173–180. [Google Scholar] [CrossRef]
- Kelly, P.; Sanchez-Mico, M.V.; Hou, S.S.; Whiteman, S.; Russ, A.; Hudry, E.; Arbel-Ornath, M.; Greenberg, S.M.; Bacskai, B.J. Neuronally Derived Soluble Abeta Evokes Cell-Wide Astrocytic Calcium Dysregulation in Absence of Amyloid Plaques in Vivo. J. Neurosci. 2023, 43, 4926–4940. [Google Scholar] [CrossRef] [PubMed]
- Dragić, M.; Milićević, K.; Adžić, M.; Stevanović, I.; Ninković, M.; Grković, I.; Andjus, P.; Nedeljković, N. Trimethyltin Increases Intracellular Ca2+ Via L-Type Voltage-Gated Calcium Channels and Promotes Inflammatory Phenotype in Rat Astrocytes In Vitro. Mol. Neurobiol. 2021, 58, 1792–1805. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 Receptor Blockade Normalizes Network Dysfunction and Cognition in an Alzheimer’s Disease Model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Kosuri, P.; Arancio, O. Picomolar Amyloid-β Peptides Enhance Spontaneous Astrocyte Calcium Transients. J. Alzheimer’s Dis. 2013, 38, 49–62. [Google Scholar] [CrossRef]
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in Physiological Aging and Alzheimer’s Disease. Neuroscience 2016, 323, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid Beta Deregulates Astroglial MGluR5-Mediated Calcium Signaling via Calcineurin and Nf-KB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Casley, C.S.; Lakics, V.; Lee, H.; Broad, L.M.; Day, T.A.; Cluett, T.; Smith, M.A.; O’Neill, M.J.; Kingston, A.E. Up-Regulation of Astrocyte Metabotropic Glutamate Receptor 5 by Amyloid-β Peptide. Brain Res. 2009, 1260, 65–75. [Google Scholar] [CrossRef]
- Maulik, M.; Vasan, L.; Bose, A.; Dutta Chowdhury, S.; Sengupta, N.; Das Sarma, J. Amyloid-β Regulates Gap Junction Protein Connexin 43 Trafficking in Cultured Primary Astrocytes. J. Biol. Chem. 2020, 295, 15097–15111. [Google Scholar] [CrossRef]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Sáez, P.J.; Jiang, J.X.; Naus, C.C.; Sáez, J.C.; Giaume, C. Amyloid β-Induced Death in Neurons Involves Glial and Neuronal Hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial Connexin43 Contributes to Neuronal Suffering in a Mouse Model of Alzheimer’s Disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef]
- Larramona-Arcas, R.; González-Arias, C.; Perea, G.; Gutiérrez, A.; Vitorica, J.; García-Barrera, T.; Gómez-Ariza, J.L.; Pascua-Maestro, R.; Ganfornina, M.D.; Kara, E.; et al. Sex-Dependent Calcium Hyperactivity Due to Lysosomal-Related Dysfunction in Astrocytes from APOE4 versus APOE3 Gene Targeted Replacement Mice. Mol. Neurodegener. 2020, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H. Alzheimer’s Disease beyond Calcium Dysregulation: The Complex Interplay between Calmodulin, Calmodulin-Binding Proteins and Amyloid Beta from Disease Onset through Progression. Curr. Issues Mol. Biol. 2023, 45, 6246–6261. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Krivonosov, M.I.; Pakhomov, A.M.; Yarullina, L.E.; Gavrish, M.S.; Mishchenko, T.A.; Yarkov, R.S.; Vedunova, M.V. Unravelling the Collective Calcium Dynamics of Physiologically Aged Astrocytes under a Hypoxic State In Vitro. Int. J. Mol. Sci. 2023, 24, 12286. [Google Scholar] [CrossRef]
- Gómez-Gonzalo, M.; Martin-Fernandez, M.; Martínez-Murillo, R.; Mederos, S.; Hernández-Vivanco, A.; Jamison, S.; Fernandez, A.P.; Serrano, J.; Calero, P.; Futch, H.S.; et al. Neuron-Astrocyte Signaling Is Preserved in the Aging Brain. Glia 2017, 65, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Pirttimaki, T.M.; Codadu, N.K.; Awni, A.; Pratik, P.; Nagel, D.A.; Hill, E.J.; Dineley, K.T.; Parri, H.R. A7 Nicotinic Receptor-Mediated Astrocytic Gliotransmitter Release: Aβ Effects in a Preclinical Alzheimer’s Mouse Model. PLoS ONE 2013, 8, e81828. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor Signalling Mediates Astrocytic Hyperactivity in Vivo in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed]
- Perez-Catalan, N.A.; Doe, C.Q.; Ackerman, S.D. The Role of Astrocyte-mediated Plasticity in Neural Circuit Development and Function. Neural Dev. 2021, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Rothhammer, V. Protective Functions of Reactive Astrocytes Following Central Nervous System Insult. Front. Immunol. 2020, 11, 573256. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite Synapses: Glia, the Unacknowledged Partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Dani, J.W.; Chernjavsky, A.; Smith, S.J. Neuronal Activity Triggers Calcium Waves in Hippocampal Astrocyte Networks. Neuron 1992, 8, 429–440. [Google Scholar] [CrossRef]
- Parri, H.R.; Gould, T.M.; Crunelli, V. Spontaneous Astrocytic Ca2+ Oscillations in Situ Drive NMDAR-Mediated Neuronal Excitation. Nat. Neurosci. 2001, 4, 803–812. [Google Scholar] [CrossRef]
- Nedergaard, M. Direct Signaling from Astrocytes to Neurons in Cultures of Mammalian Brain Cells. Science 1994, 263, 1768–1771. [Google Scholar] [CrossRef]
- Kang, J.; Jiang, L.; Goldman, S.A.; Nedergaard, M. Astrocyte-Mediated Potentiation of Inhibitory Synaptic Transmission. Nat. Neurosci. 1998, 1, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.-Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic Purinergic Signaling Coordinates Synaptic Networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Gundersen, V.; Galbete, J.L.; Seifert, G.; Steinhäuser, C.; Pilati, E.; Volterra, A. Astrocytes Contain a Vesicular Compartment That Is Competent for Regulated Exocytosis of Glutamate. Nat. Neurosci. 2004, 7, 613–620. [Google Scholar] [CrossRef]
- Coco, S.; Calegari, F.; Pravettoni, E.; Pozzi, D.; Taverna, E.; Rosa, P.; Matteoli, M.; Verderio, C. Storage and Release of ATP from Astrocytes in Culture. J. Biol. Chem. 2003, 278, 1354–1362. [Google Scholar] [CrossRef]
- Bazargani, N.; Attwell, D. Astrocyte Calcium Signaling: The Third Wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Glutamate-Dependent Astrocyte Modulation of Synaptic Transmission between Cultured Hippocampal Neurons. Eur. J. Neurosci. 1998, 10, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Pakhomov, A.M.; Krivonosov, M.I.; Yarkov, R.S.; Gavrish, M.S.; Shkirin, A.V.; Ivanchenko, M.V.; Vedunova, M.V. Novel Algorithm of Network Calcium Dynamics Analysis for Studying the Role of Astrocytes in Neuronal Activity in Alzheimer’s Disease Models. Int. J. Mol. Sci. 2022, 23, 15928. [Google Scholar] [CrossRef]
- Lines, J.; Baraibar, A.M.; Fang, C.; Martin, E.D.; Aguilar, J.; Lee, M.K.; Araque, A.; Kofuji, P. Astrocyte-neuronal Network Interplay Is Disrupted in Alzheimer’s Disease Mice. Glia 2022, 70, 368–378. [Google Scholar] [CrossRef]
- HYND, M. Glutamate-Mediated Excitotoxicity and Neurodegeneration in Alzheimer?S Disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of Glutamate Transporters Reveals a Major Role for Astroglial Transport in Excitotoxicity and Clearance of Glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient Astrocyte Metabolism Impairs Glutamine Synthesis and Neurotransmitter Homeostasis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma Frequency Entrainment Attenuates Amyloid Load and Modifies Microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhang, F.; Zhang, H.; Huang, X.; Wang, K.; Huang, T.; Yang, X.; Zou, L. Neuroprotective Effect of Optogenetics Varies with Distance from Channelrhodopsin-2 Expression in an Amyloid-β-Injected Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 583628. [Google Scholar] [CrossRef]
- Wang, K.-W.; Ye, X.-L.; Huang, T.; Yang, X.-F.; Zou, L.-Y. Optogenetics-Induced Activation of Glutamate Receptors Improves Memory Function in Mice with Alzheimer’s Disease. Neural Regen. Res. 2019, 14, 2147. [Google Scholar] [CrossRef]
- Yang, Q.; Song, D.; Xie, Z.; He, G.; Zhao, J.; Wang, Z.; Dong, Z.; Zhang, H.; Yang, L.; Jiang, M.; et al. Optogenetic Stimulation of CA3 Pyramidal Neurons Restores Synaptic Deficits to Improve Spatial Short-Term Memory in APP/PS1 Mice. Prog. Neurobiol. 2022, 209, 102209. [Google Scholar] [CrossRef]
- Lee, Y.F.; Russ, A.N.; Zhao, Q.; Perle, S.J.; Maci, M.; Miller, M.R.; Hou, S.S.; Algamal, M.; Zhao, Z.; Li, H.; et al. Optogenetic Targeting of Astrocytes Restores Slow Brain Rhythm Function and Slows Alzheimer’s Disease Pathology. Sci. Rep. 2023, 13, 13075. [Google Scholar] [CrossRef]
- Suo, Q.; Deng, L.; Chen, T.; Wu, S.; Qi, L.; Liu, Z.; He, T.; Tian, H.-L.; Li, W.; Tang, Y.; et al. Optogenetic Activation of Astrocytes Reduces Blood-Brain Barrier Disruption via IL-10 In Stroke. Aging Dis. 2023, 14, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The Role of Adult Hippocampal Neurogenesis in Brain Health and Disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult Hippocampal Neurogenesis and Its Role in Alzheimer’s Disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Gadadhar, A.; Marr, R.; Lazarov, O. Presenilin-1 Regulates Neural Progenitor Cell Differentiation in the Adult Brain. J. Neurosci. 2011, 31, 2615–2623. [Google Scholar] [CrossRef]
- Gakhar-Koppole, N.; Hundeshagen, P.; Mandl, C.; Weyer, S.W.; Allinquant, B.; Müller, U.; Ciccolini, F. Activity Requires Soluble Amyloid Precursor Protein α to Promote Neurite Outgrowth in Neural Stem Cell-Derived Neurons via Activation of the MAPK Pathway. Eur. J. Neurosci. 2008, 28, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Teh, D.B.L.; Prasad, A.; Jiang, W.; Zhang, N.; Wu, Y.; Yang, H.; Han, S.; Yi, Z.; Yeo, Y.; Ishizuka, T.; et al. Driving Neurogenesis in Neural Stem Cells with High Sensitivity Optogenetics. Neuromolecular Med. 2020, 22, 139–149. [Google Scholar] [CrossRef]
- Giraldo, E.; Palmero-Canton, D.; Martinez-Rojas, B.; Sanchez-Martin, M.d.M.; Moreno-Manzano, V. Optogenetic Modulation of Neural Progenitor Cells Improves Neuroregenerative Potential. Int. J. Mol. Sci. 2020, 22, 365. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Vincent, P.F.Y.; Ziogas, N.K.; Xu, L.; Sadeghpour, S.; Curtin, J.; Alexandris, A.S.; Stewart, N.; Sima, R.; du Lac, S.; et al. Optogenetically Transduced Human ES Cell-Derived Neural Progenitors and Their Neuronal Progenies: Phenotypic Characterization and Responses to Optical Stimulation. PLoS ONE 2019, 14, e0224846. [Google Scholar] [CrossRef]
- Salmina, A.B.; Kapkaeva, M.R.; Vetchinova, A.S.; Illarioshkin, S.N. Novel Approaches Used to Examine and Control Neurogenesis in Parkinson′s Disease. Int. J. Mol. Sci. 2021, 22, 9608. [Google Scholar] [CrossRef]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-Mediated Activation of NeuroD1 and Retro-Elements during Adult Neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef]
- Cassé, F.; Richetin, K.; Toni, N. Astrocytes’ Contribution to Adult Neurogenesis in Physiology and Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 432. [Google Scholar] [CrossRef]
- Hedegaard, A.; Monzón-Sandoval, J.; Newey, S.E.; Whiteley, E.S.; Webber, C.; Akerman, C.J. Pro-Maturational Effects of Human IPSC-Derived Cortical Astrocytes upon IPSC-Derived Cortical Neurons. Stem Cell Rep. 2020, 15, 38–51. [Google Scholar] [CrossRef]
- Morgun, A.V.; Osipova, E.D.; Boitsova, E.B.; Shuvaev, A.N.; Malinovskaya, N.A.; Mosiagina, A.I.; Salmina, A.B. Neurogenic Potential of Implanted Neurospheres Is Regulated by Optogenetic Stimulation of Hippocampal Astrocytes Ex Vivo. Bull. Exp. Biol. Med. 2021, 170, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, U.; Faiz, M.; de Pablo, Y.; Sjöqvist, M.; Andersson, D.; Widestrand, Å.; Potokar, M.; Stenovec, M.; Smith, P.L.P.; Shinjyo, N.; et al. Astrocytes Negatively Regulate Neurogenesis Through the Jagged1-Mediated Notch Pathway. Stem Cells 2012, 30, 2320–2329. [Google Scholar] [CrossRef]
- Tiwari, P.; Tolwinski, N.S. Using Optogenetics to Model Cellular Effects of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4300. [Google Scholar] [CrossRef]
- Lim, C.H.; Kaur, P.; Teo, E.; Lam, V.Y.M.; Zhu, F.; Kibat, C.; Gruber, J.; Mathuru, A.S.; Tolwinski, N.S. Application of Optogenetic Amyloid-β Distinguishes between Metabolic and Physical Damages in Neurodegeneration. Elife 2020, 9, e52589. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Weitemier, A.Z.; Zeng, X.; He, L.; Wang, X.; Tao, Y.; Huang, A.J.Y.; Hashimotodani, Y.; Kano, M.; Iwasaki, H.; et al. Near-Infrared Deep Brain Stimulation via Upconversion Nanoparticle–Mediated Optogenetics. Science 2018, 359, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Purushothuman, S.; Johnstone, D.M.; Nandasena, C.; Mitrofanis, J.; Stone, J. Photobiomodulation with near Infrared Light Mitigates Alzheimer’s Disease-Related Pathology in Cerebral Cortex—Evidence from Two Transgenic Mouse Models. Alzheimers Res. Ther. 2014, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Photobiomodulation or Low-Level Laser Therapy. J. Biophotonics 2016, 9, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.; Hamblin, M.R. Photobiomodulation and the Brain: A New Paradigm. J. Opt. 2017, 19, 013003. [Google Scholar] [CrossRef]
- Salehpour, F.; Mahmoudi, J.; Kamari, F.; Sadigh-Eteghad, S.; Rasta, S.H.; Hamblin, M.R. Brain Photobiomodulation Therapy: A Narrative Review. Mol. Neurobiol. 2018, 55, 6601–6636. [Google Scholar] [CrossRef]
- Gong, X.; Mendoza-Halliday, D.; Ting, J.T.; Kaiser, T.; Sun, X.; Bastos, A.M.; Wimmer, R.D.; Guo, B.; Chen, Q.; Zhou, Y.; et al. An Ultra-Sensitive Step-Function Opsin for Minimally Invasive Optogenetic Stimulation in Mice and Macaques. Neuron 2020, 107, 38–51.e8. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of Region-Specific Astrocyte Subtypes at Single Cell Resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef]
- Borodinova, A.A.; Balaban, P.M.; Bezprozvanny, I.B.; Salmina, A.B.; Vlasova, O.L. Genetic Constructs for the Control of Astrocytes’ Activity. Cells 2021, 10, 1600. [Google Scholar] [CrossRef]
- von Jonquieres, G.; Mersmann, N.; Klugmann, C.B.; Harasta, A.E.; Lutz, B.; Teahan, O.; Housley, G.D.; Fröhlich, D.; Krämer-Albers, E.-M.; Klugmann, M. Glial Promoter Selectivity Following AAV-Delivery to the Immature Brain. PLoS ONE 2013, 8, e65646. [Google Scholar] [CrossRef]
- Taschenberger, G.; Tereshchenko, J.; Kügler, S. A MicroRNA124 Target Sequence Restores Astrocyte Specificity of GfaABC1D-Driven Transgene Expression in AAV-Mediated Gene Transfer. Mol. Ther. Nucleic Acids 2017, 8, 13–25. [Google Scholar] [CrossRef]
- Jollé, C.; Déglon, N.; Pythoud, C.; Bouzier-Sore, A.-K.; Pellerin, L. Development of Efficient AAV2/DJ-Based Viral Vectors to Selectively Downregulate the Expression of Neuronal or Astrocytic Target Proteins in the Rat Central Nervous System. Front. Mol. Neurosci. 2019, 12, 201. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| GABA | Glutamate | Purines | D-Serine |
|---|---|---|---|
| main inhibitory neurotransmitter in the CNS | main excitatory neurotransmitter in the CNS | ATP, adenosine—excitatory neurotransmitters | important in NMDAR modulation |
| ionotropic GABAA receptors and metabotropic GABAB receptors in neurons | N-methyl-D-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, kainate receptors, and metabotropic receptors in neurons | two families of receptors: P1 (subtypes A1, A2A, A2B and A3), which bind to adenosine, and P2 (ionotropic P2X (seven subtypes P2X1-7) and metabotropic P2Y receptors (8 subtypes)), which are activated by ATP/ADP-nucleotides | a physiological co-agonist of the N-methyl d-aspartate (NMDA) type of glutamate receptor |
| metabotropic GABAB receptors in astrocytes | glutamatergic transmission within glial cells occurs through metabotropic glutamate receptors (mGluR), which are divided into three groups: group I: mGluR1.5; group II: mGluR2, mGluR3, group III: mGluR4, mGluR6, mGluR 7, mGluR 8 | astrocytes express P2X1, P2X2, P2X3, P2X4, P2X5, and P2X7; P2Y1, P2Y2, P2Y4, P2Y6, and P2Y12 receptors; and functional adenosine receptors (A1, A2A, A2B) | |
| signal transformation from neurons or its amplification through astrocytes depends on the context Under normal conditions, hippocampal astrocytes contain very little GABA | Astroglia regulates extracellular glutamate homeostasis through Na+-dependent excitatory amino acid transporters 1 and 2 (excitatory amino acid transporter—EAAT): GLAST-1 and GLT-1, respectively. Astrocytes can release glutamate in a Ca2+-dependent and Ca2+-independent way. As a gliotransmitter, glutamate can have an inhibitory or excitatory effect on neurons. | ATP released by neurons can affect astrocytes’ purinergic receptors directly in the form of ATP or degradation products in the form of ADP, AMP, and adenosine, leading to an increase in astrocyte Ca2+ levels. ATP released from astrocytes is metabolized by extracellular ATPases with the formation of adenosine, which regulates synaptic transmission by affecting the A1 and A2A metabotropic receptors. | a putative gliotransmitter that is associated with learning and memory by affecting synaptic NMDARs |
| Astrocytes around amyloid plaques become reactive and produce and release GABA aberrantly and in large quantities | In AD, glutamate clearance is impaired. Increased release of glutamate is noted. High spontaneous and abnormal fluctuations in glutamate concentration are observed around Aβ plaques | Enhanced ATP release in hippocampal slices and astrocyte cultures is observed with the application of Aβ peptides | increased in experimental models of AD and in post-mortem samples |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitroshina, E.; Kalinina, E.; Vedunova, M. Optogenetics in Alzheimer’s Disease: Focus on Astrocytes. Antioxidants 2023, 12, 1856. https://doi.org/10.3390/antiox12101856
Mitroshina E, Kalinina E, Vedunova M. Optogenetics in Alzheimer’s Disease: Focus on Astrocytes. Antioxidants. 2023; 12(10):1856. https://doi.org/10.3390/antiox12101856
Chicago/Turabian StyleMitroshina, Elena, Elizaveta Kalinina, and Maria Vedunova. 2023. "Optogenetics in Alzheimer’s Disease: Focus on Astrocytes" Antioxidants 12, no. 10: 1856. https://doi.org/10.3390/antiox12101856
APA StyleMitroshina, E., Kalinina, E., & Vedunova, M. (2023). Optogenetics in Alzheimer’s Disease: Focus on Astrocytes. Antioxidants, 12(10), 1856. https://doi.org/10.3390/antiox12101856