
Airway Smooth Muscle Regulated by Oxidative Stress in COPD


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
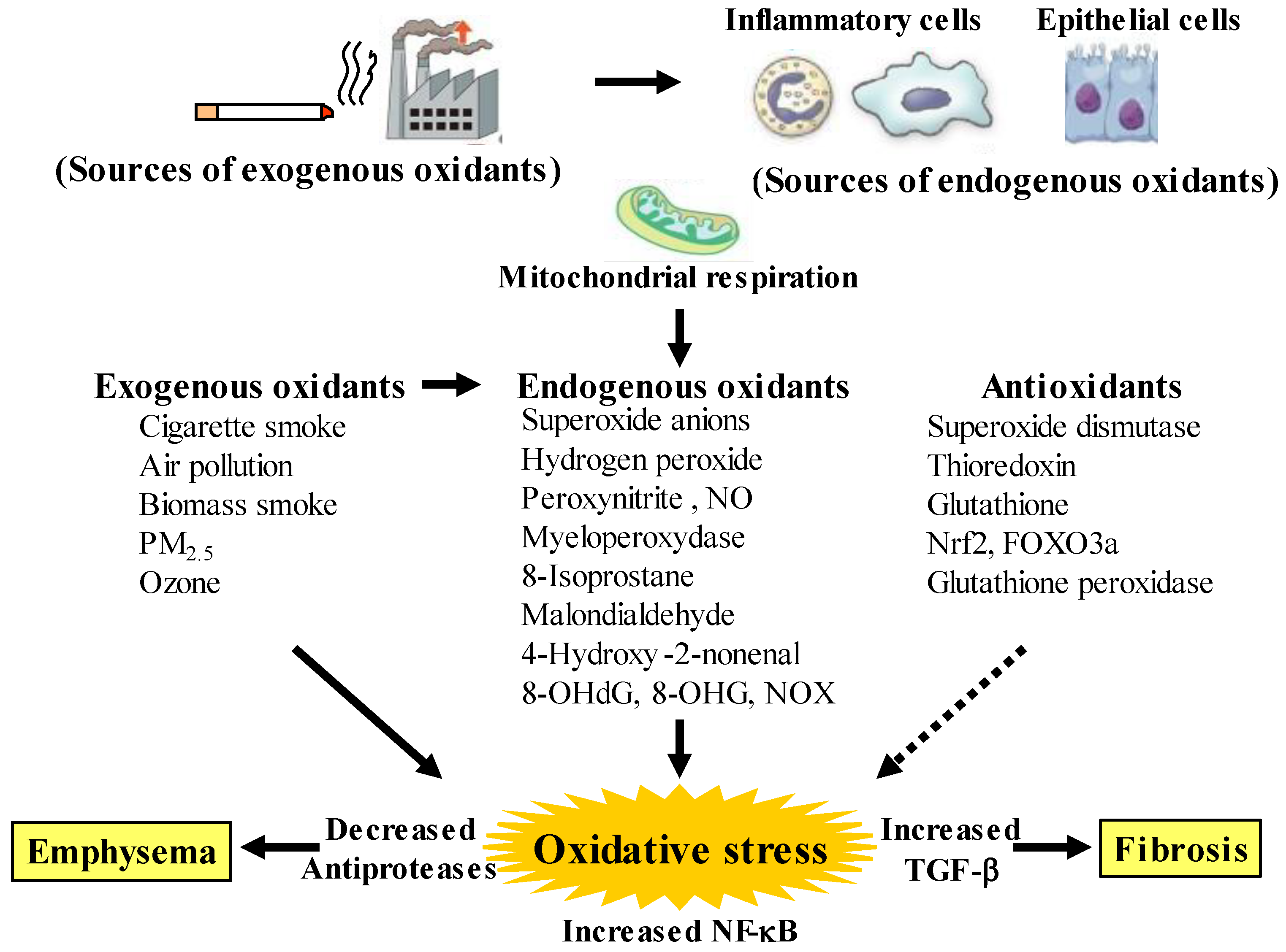
2. Oxidative Stress in COPD
2.1. Pathological Features
2.2. Oxidants Related to COPD
2.3. Antioxidants Related to COPD
2.4. Reduced Responsiveness to Corticosteroids Caused by Oxidative Stress
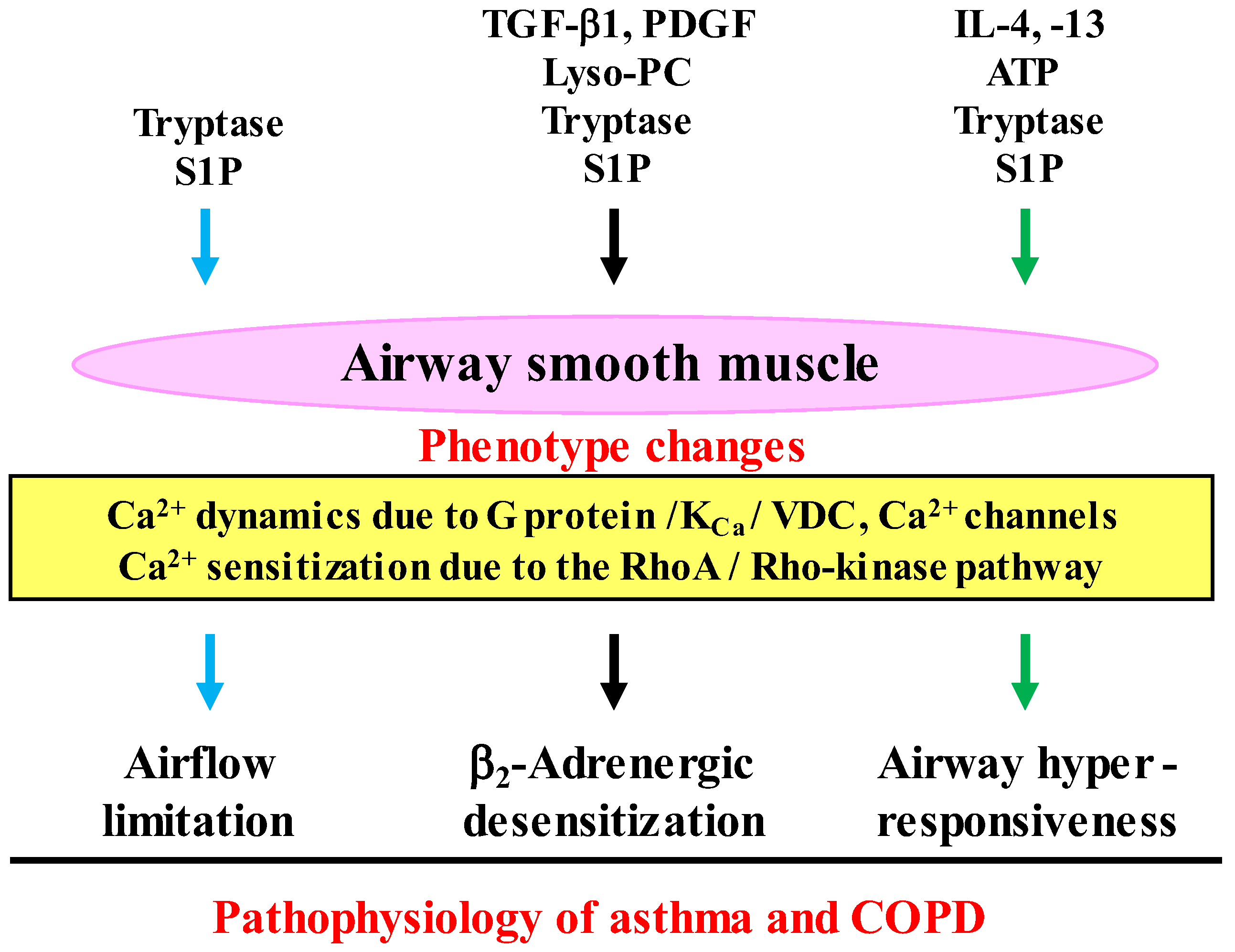
3. Dysfunction of Airway Smooth Muscle in COPD
3.1. Phenotype Changes
3.2. Airway Hyperresponsiveness
3.3. β2-Adrenerigic Desensitization
3.4. Airway Remodeling
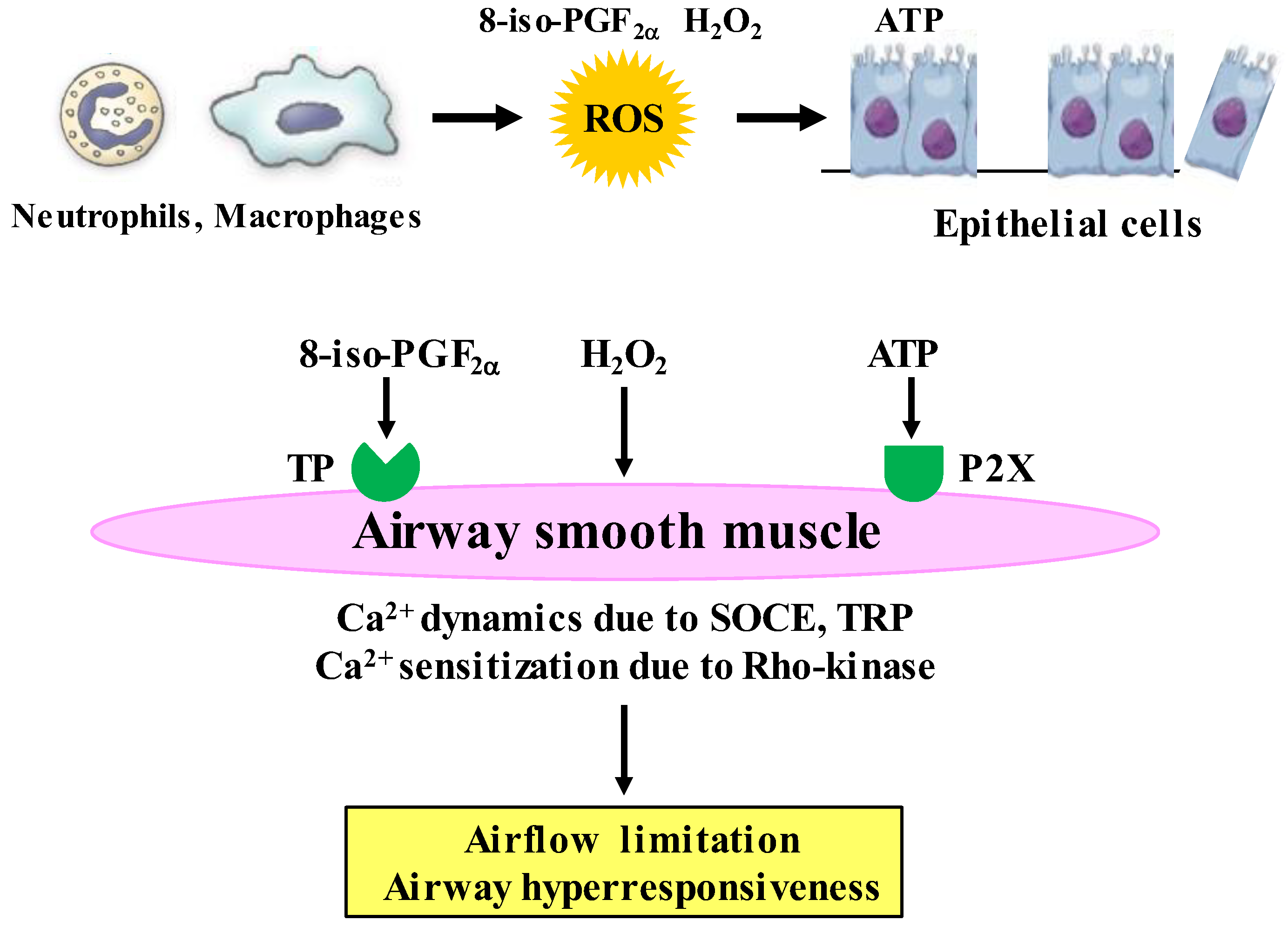
4. Airway Smooth Muscles Regulated by Oxidative Stress
4.1. Expression of Oxidants in Airway Smooth Muscle
4.2. Effects of Oxidative Stress on Contraction and Proliferation
4.3. Inhibitory Effects of Antioxidants on Oxidative Stress Induced Proliferation
4.4. Clinical Trials for Antioxidant Therapy
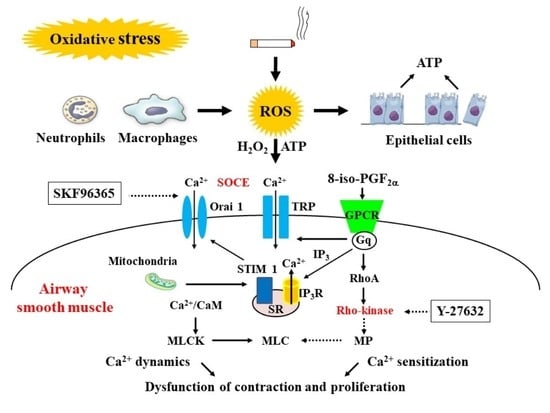
5. Calcium Signaling as Mechanisms of Oxidative Stress
5.1. Involvement of Ca2+ Dynamics in Oxidative Stress
5.2. Involvement of Ca2+ Sensitization in Oxidative Stress
6. Toward the Progress of Therapy for COPD
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Prevention, Diagnosis and Management of Chronic Obstructive Pulmonary Disease: 2023 Report. Available online: https://goldcopd.org/ (accessed on 1 December 2022).
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Burney, P.G.J.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef]
- Birnboim, H.C. DNA strand breaks in human leukocytes induced by superoxide anion, hydrogen peroxide and tumor promoters are repaired slowly compared to breaks induced by ionizing radiation. Carcinogenesis 1986, 7, 1511–1517. [Google Scholar] [CrossRef]
- Agustí, A.; Hogg, J.C. Update on the pathogenesis of chronic obstructive pulmonary disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular senescence as a mechanism and target in chronic lung diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Kumar, M.; Seeger, W.; Voswinckel, R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 323–333. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A.; Bel, E.; Thomas, M.; Vogelmeier, C.; Brusselle, G.; Holgate, S.; Humbert, M.; Jones, P.; Gibson, P.G.; Vestbo, J.; et al. Treatable traits: Toward precision medicine of chronic airway diseases. Eur. Respir. J. 2016, 47, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Domej, W.; Oettl, K.; Renner, W. Oxidative stress and free radicals in COPD–Implications and relevance for treatment. Int. J. Chron. Obs. Pulmon. Dis. 2014, 9, 1207–1224. [Google Scholar] [CrossRef]
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.F.; Donner, C.F.; Barnes, P.J.; et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Ravi, A.K.; Khurana, S.; Lemon, J.; Plumb, J.; Booth, G.; Healy, L.; Catley, M.; Vestbo, J.; Singh, D. Increased levels of soluble interleu-kin-6 receptor and CCL3 in COPD sputum. Respir. Res. 2014, 15, 103. [Google Scholar] [CrossRef]
- Shao, M.X.; Nakanaga, T.; Nadel, J.A. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L420–L427. [Google Scholar] [CrossRef]
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L962–L974. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D. End-stage chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 339–340. [Google Scholar] [CrossRef]
- Schaberg, T.; Klein, U.; Rau, M.; Eller, J.; Lode, H. Subpopulations of alveolar macrophages in smokers and nonsmokers: Relation to the expression of CD11/CD18 molecules and superoxide anion production. Am. J. Respir. Crit. Care Med. 1995, 151, 1551–1558. [Google Scholar] [CrossRef]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agustí, A.G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; van Schadewijk, A.A.; Crowther, A.J.; Hiemstra, P.S.; Stolk, J.; MacNee, W.; De Boer, W.I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydro-gen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. [Google Scholar] [CrossRef]
- Corradi, M.; Pignatti, P.; Manini, P.; Andreoli, R.; Goldoni, M.; Poppa, M.; Moscato, G.; Balbi, B.; Mutti, A. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur. Respir. J. 2004, 24, 1011–1017. [Google Scholar] [CrossRef]
- Bartoli, M.L.; Novelli, F.; Costa, F.; Malagrinò, L.; Melosini, L.; Bacci, E.; Cianchetti, S.; Dente, F.L.; Di Franco, A.; Vagaggini, B.; et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediat. Inflamm. 2011, 2011, 891752. [Google Scholar] [CrossRef]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003, 58, 294–298. [Google Scholar] [CrossRef]
- Zhu, A.; Ge, D.; Zhang, J.; Teng, Y.; Yuan, C.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Sputum myeloperoxidase in chronic obstructive pulmonary disease. Eur. J. Med. Res. 2014, 19, 12. [Google Scholar] [CrossRef]
- Antus, B. Oxidative Stress Markers in Sputum. Oxid. Med. Cell Longev. 2016, 2016, 2930434. [Google Scholar] [CrossRef]
- Deslee, G.; Adair-Kirk, T.L.; Betsuyaku, T.; Woods, J.C.; Moore, C.H.; Gierada, D.S.; Conradi, S.H.; Atkinson, J.J.; Toennies, H.M.; Battaile, J.T.; et al. Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2010, 43, 576–584. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Osoata, G.O.; Hanazawa, T.; Brindicci, C.; Ito, M.; Barnes, P.J.; Kharitonov, S.; Ito, K. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest 2009, 135, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.; Newbold, P.; White, P.; Thong, B.; Stone, H.; Stockley, R.A. 3-Chlorotyrosine in sputum of COPD patients: Relation-ship with airway inflammation. COPD. 2010, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Gorowiec, M.R.; Borthwick, L.A.; Parker, S.M.; Kirby, J.A.; Saretzki, G.C.; Fisher, A.J. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 1024–1032. [Google Scholar] [CrossRef]
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-β regulates Nox4, MnSOD and catalase expres-sion, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L295–L304. [Google Scholar] [CrossRef]
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300. [Google Scholar] [CrossRef]
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dis-mutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576. [Google Scholar] [CrossRef]
- Malhotra, D.; Thimmulappa, R.; Vij, N.; Navas-Acien, A.; Sussan, T.; Merali, S.; Zhang, L.; Kelsen, S.G.; Myers, A.; Wise, R.; et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: The role of Nrf2-regulated proteasomal activity. Am. J. Respir. Crit. Care Med. 2009, 180, 1196–1207. [Google Scholar] [CrossRef]
- Hwang, J.W.; Rajendrasozhan, S.; Yao, H.; Chung, S.; Sundar, I.K.; Huyck, H.L.; Pryhuber, G.S.; Kinnula, V.L.; Rahman, I. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 2011, 187, 987–998. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid. Med. Cell Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Glutathione peroxidase-1 as a novel therapeutic target for COPD. Redox Rep. 2013, 18, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Hardigan, A.A.; Wallace, A.M.; Mirochnitchenko, O.; Thankachen, J.; Arellanos, L.; Thompson, V.; D’Armiento, J.M.; Foronjy, R.F. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis. A key determinant of airway inflammation and alveolar destruction. Am. J. Respir. Cell Mol. Biol. 2013, 49, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-d with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef]
- Lewis, B.W.; Ford, M.L.; Rogers, L.K.; Britt, R.D., Jr. Oxidative stress promotes corticosteroid insensitivity in asthma and COPD. Antioxidants 2021, 10, 1335. [Google Scholar] [CrossRef]
- Mei, D.; Tan, W.S.D.; Wong, W.S.F. Pharmacological strategies to regain steroid sensitivity in severe asthma and COPD. Curr. Opin. Pharmacol. 2019, 46, 73–81. [Google Scholar] [CrossRef]
- Sakurai, H.; Morishima, Y.; Ishii, Y.; Yoshida, K.; Nakajima, M.; Tsunoda, Y.; Hayashi, S.Y.; Kiwamoto, T.; Matsuno, Y.; Kawaguchi, M.; et al. Sulforaphane ameliorates steroid insensitivity through an Nrf2-dependent pathway in cigarette smoke-exposed asthmatic mice. Free Radic. Biol. Med. 2018, 129, 473–485. [Google Scholar] [CrossRef]
- Kume, H. RhoA/Rho-kinase as a therapeutic target in asthma. Curr. Med. Chem. 2008, 15, 2876–2885. [Google Scholar] [CrossRef]
- Wright, D.B.; Trian, T.; Siddiqui, S.; Pascoe, C.D.; Johnson, J.R.; Dekkers, B.G.; Dakshinamurti, S.; Bagchi, R.; Burgess, J.K.; Kanabar, V.; et al. Phenotype modulation of airway smooth muscle in asthma. Pulm. Pharmacol. Ther. 2013, 26, 42–49. [Google Scholar] [CrossRef]
- Kume, H. Role of Airway Smooth Muscle in Inflammation Related to Asthma and COPD. Adv. Exp. Med. Biol. 2021, 1303, 139–172. [Google Scholar] [PubMed]
- Yan, F.; Gao, H.; Zhao, H.; Bhatia, M.; Zeng, Y. Roles of airway smooth muscle dysfunction in chronic obstructive pulmonary disease. J. Transl. Med. 2018, 16, 262. [Google Scholar] [CrossRef] [PubMed]
- Camoretti-Mercado, B.; Lockey, R.F. Airway smooth muscle pathophysiology in asthma. J. Allergy Clin. Immunol. 2021, 147, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Hojo, M.; Hashimoto, N. Eosinophil Inflammation and hyperresponsiveness in the airways as phenotypes of COPD, and usefulness of inhaled glucocorticosteroids. Front. Pharmacol. 2019, 10, 765. [Google Scholar] [CrossRef] [PubMed]
- Ramsdell, J.W.; Nachtwey, F.J.; Moser, K.M. Bronchial hyperreactivity in chronic obstructive bronchitis. Am. Rev. Respir. Dis. 1982, 126, 829–832. [Google Scholar] [PubMed]
- Opazo Saez, A.M.; Seow, C.Y.; Paré, P.D. Peripheral airway smooth muscle mechanics in obstructive airways disease. Am. J. Respir. Crit. Care Med. 2000, 161, 910–917. [Google Scholar] [CrossRef]
- Eum, S.Y.; Maghni, K.; Tolloczko, B.; Eidelman, D.H.; Martin, J.G. IL-13 may mediate allergen-induced hyperresponsiveness inde-pendently of IL-5 or eotaxin by effects on airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L576-84. [Google Scholar] [CrossRef]
- Manson, M.L.; Säfholm, J.; James, A.; Johnsson, A.K.; Bergman, P.; Al-Ameri, M.; Orre, A.C.; Kärrman-Mårdh, C.; Dahlén, S.E.; Adner, M. IL-13 and IL-4, but not IL-5 nor IL-17A, induce hyperresponsiveness in isolated human small airways. J. Allergy Clin. Immunol. 2020, 145, 808–817.e2. [Google Scholar] [CrossRef]
- Oguma, T.; Ito, S.; Kondo, M.; Makino, Y.; Shimokata, K.; Honjo, H.; Kamiya, K.; Kume, H. Roles of P2X receptors and Ca2+ sensitization in extracellular adenosine triphosphate-induced hyperresponsiveness in airway smooth muscle. Clin. Exp. Allergy 2007, 37, 893–900. [Google Scholar] [CrossRef]
- Sekizawa, K.; Caughey, G.H.; Lazarus, S.C.; Gold, W.M.; Nadel, J.A. Mast cell tryptase causes airway smooth muscle hyperresponsiveness in dogs. J. Clin. Invest. 1989, 83, 175–179. [Google Scholar] [CrossRef]
- Kume, H.; Takeda, N.; Oguma, T.; Ito, S.; Kondo, M.; Ito, Y.; Shimokata, K. Sphingosine 1-phosphate causes airway hyper-reactivity by Rho-mediated myosin phosphatase inactivation. J. Pharmacol. Exp. Ther. 2007, 320, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Taki, F.; Kume, H.; Kobayashi, T.; Ohta, H.; Aratake, H.; Shimokata, K. Effects of Rho-kinase inactivation on eosinophilia and hyper-reactivity in murine airways by allergen challenges. Clin. Exp. Allergy 2007, 37, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Takagi, K. Inhibitory effects of Gs on desensitization of β-adrenergic receptors in tracheal smooth muscle. Am. J. Physiol. 1997, 273, L556–L564. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Takagi, K. Inhibition of β-adrenergic desensitization by KCa channels in human trachealis. Am. J Respir. Crit. Care Med. 1999, 159, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Ishikawa, T.; Oguma, T.; Ito, S. Shimokata K, Kotlikoff MI. Involvement of Ca2+ mobilization in tachyphylaxis to β-adrenergic receptors in trachealis. Am. J. Respir. Cell Mol. Biol. 2003, 29, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Fukunaga, K.; Oguma, T. Research and development of bronchodilators for asthma and COPD with a focus on G protein/KCa channel linkage and β2-adrenergic intrinsic efficacy. Pharmacol. Ther. 2015, 156, 75–89. [Google Scholar] [CrossRef]
- Kume, H.; Nishiyama, O.; Isoya, T.; Higashimoto, Y.; Tohda, Y.; Noda, Y. Involvement of allosteric effect and KCa channels in crosstalk between β2-adrenergic and muscarinic M2 receptors in airway smooth muscle. Int. J. Mol. Sci. 2018, 19, 1999. [Google Scholar] [CrossRef]
- Kume, H.; Takai, A.; Tokuno, H.; Tomita, T. Regulation of Ca2+-dependent K+-channel activity in tracheal myocytes by phos-phorylation. Nature 1989, 341, 152–154. [Google Scholar] [CrossRef]
- Kume, H.; Graziano, M.P.; Kotlikoff, M.I. Stimulatory and inhibitory regulation of calcium-activated potassium channels by guanine nucleotide-binding proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 11051–11055. [Google Scholar] [CrossRef]
- Kume, H.; Hall, I.P.; Washabau, R.J.; Takagi, K.; Kotlikoff, M.I. β-Adrenergic agonists regulate KCa channels in airway smooth muscle by cAMP-dependent and -independent mechanisms. J. Clin. Invest. 1994, 93, 371–379. [Google Scholar] [CrossRef]
- Guo, M.; Pascual, R.M.; Wang, S.; Fontana, M.F.; Valancius, C.A.; Panettieri, R.A., Jr.; Tilley, S.L.; Penn, R.B. Cytokines regulate beta-2-adrenergic receptor responsiveness in airway smooth muscle via multiple PKA- and EP2 receptor-dependent mechanisms. Biochemistry 2005, 44, 13771–13782. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kume, H.; Kondo, M.; Ito, Y.; Yamaki, K.; Shimokata, K. Inhibitory effects of interferon-γ on the heterologous desensitization of β-adrenoceptors by transforming growth factor-β1 in tracheal smooth muscle. Clin. Exp. Allergy 2003, 33, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, T.; Kume, H.; Oguma, T.; Makino, Y.; Shiraki, A.; Ito, Y.; Shimokata, K. Role of Ca2+ mobilization in desensitization of β-adrenoceptors by platelet-derived growth factor in airway smooth muscle. Eur. J. Pharmacol. 2008, 591, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Ito, S.; Ito, Y.; Yamaki, K. Role of lysophosphatidylcholine in the desensitization of β-adrenergic receptors by Ca2+ sensitization in tracheal smooth muscle. Am. J. Respir. Cell Mol. Biol. 2001, 25, 291–298. [Google Scholar] [CrossRef]
- Makino, Y.; Kume, H.; Oguma, T.; Sugishita, M.; Shiraki, A.; Hasegawa, Y.; Honjo, H.; Kamiya, K. Role of sphingosine-1-phosphate in β-adrenoceptor desensitization via Ca2+ sensitization in airway smooth muscle. Allergol. Int. 2012, 61, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kume, H.; Oguma, T.; Makino, Y.; Ito, Y.; Shimokata, K. Mast cell tryptase causes homologous desensitization of β-adrenoceptors by Ca2+ sensitization in tracheal smooth muscle. Clin. Exp. Allergy 2008, 38, 135–144. [Google Scholar] [CrossRef]
- Dekkers, B.G.; Schaafsma, D.; Nelemans, S.A.; Zaagsma, J.; Meurs, H. Extracellular matrix proteins differentially regulate airway smooth muscle phenotype and function. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1405–L1413. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.R.; Roth, M.; Tamm, M.; Hughes, M.; Ge, Q.; King, G.; Burgess, J.K.; Black, J.L. Airway smooth muscle cell proliferation is increased in asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Mahn, K.; Hirst, S.J.; Ying, S.; Holt, M.R.; Lavender, P.; Ojo, O.O.; Siew, L.; Simcock, D.E.; McVicker, C.G.; Kanabar, V.; et al. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodeling in bronchial asthma. Proc. Natl. Acad. Sci. USA 2009, 106, 10775–10780. [Google Scholar] [CrossRef]
- Sukkar, M.B.; Stanley, A.J.; Blake, A.E.; Hodgkin, P.D.; Johnson, P.R.; Armour, C.L.; Hughes, J.M. ‘Proliferative’ and ‘synthetic’ airway smooth muscle cells are overlapping populations. Immunol. Cell Biol. 2004, 82, 471–478. [Google Scholar] [CrossRef]
- Prakash, Y.S. Airway smooth muscle in airway reactivity and remodeling: What have we learned? Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L912–L933. [Google Scholar] [CrossRef] [PubMed]
- James, A.L.; Elliot, J.G.; Jones, R.L.; Carroll, M.L.; Mauad, T.; Bai, T.R.; Abramson, M.J.; McKay, K.O.; Green, F.H. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Noble, P.B.; Elliot, J.G.; James, A.L. Airway remodeling in COPD: It’s not asthma! Respirology 2016, 21, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Saunders, R.M.; Biddle, M.; Amrani, Y.; Brightling, C.E. Stressed out—The role of oxidative stress in airway smooth muscle dysfunction in asthma and COPD. Free Radic. Biol. Med. 2022, 185, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.E.; Luo, S.F.; Jou, M.J.; Lin, C.C.; Kou, Y.R.; Lee, I.T.; Hsieh, H.L.; Yang, C.M. Cigarette smoke extract induces cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs, AP-1, and NF-kB in human tracheal smooth muscle cells. Free Radic. Biol. Med. 2009, 46, 948–960. [Google Scholar] [CrossRef]
- Lin, C.C.; Lee, I.T.; Yang, Y.L.; Lee, C.W.; Kou, Y.R.; Yang, C.M. Induction of COX-2/PGE2/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: Role of TLR4-dependent NADPH oxidase activation. Free Radic. Biol. Med. 2010, 48, 240–254. [Google Scholar] [CrossRef]
- Liu, X.; Hao, B.; Ma, A.; He, J.; Liu, X.; Chen, J. The expression of NOX4 in smooth muscles of small airway correlates with the disease severity of COPD. Biomed Res. Int. 2016, 2016, 2891810. [Google Scholar] [CrossRef]
- Hollins, F.; Sutcliffe, A.; Gomez, E.; Berair, R.; Russell, R.; Szyndralewiez, C.; Saunders, R.; Brightling, C. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir. Res. 2016, 17, 84. [Google Scholar] [CrossRef]
- Oostwoud, L.C.; Gunasinghe, P.; Seow, H.J.; Ye, J.M.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Apocynin and ebselen reduce influenza A virus-induced lung inflammation in cigarette smoke-exposed mice. Sci. Rep. 2016, 6, 20983. [Google Scholar] [CrossRef]
- Kojima, K.; Kume, H.; Ito, S.; Oguma, T.; Shiraki, A.; Kondo, M.; Ito, Y.; Shimokata, K. Direct effects of hydrogen peroxide on airway smooth muscle tone: Roles of Ca2+ influx and Rho-kinase. Eur. J. Pharmacol. 2007, 556, 151–156. [Google Scholar] [CrossRef]
- Shiraki, A.; Kume, H.; Oguma, T.; Makino, Y.; Ito, S.; Shimokata, K.; Honjo, H.; Kamiya, K. Role of Ca2+ mobilization and Ca2+ sensitization in 8-iso-PGF2α-induced contraction in airway smooth muscle. Clin. Exp. Allergy 2009, 39, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Sathish, V.; Freeman, M.R.; Long, E.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke and estrogen signaling in human airway smooth muscle. Cell Physiol. Biochem. 2015, 36, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Borkar, N.A.; Roos, B.; Prakash, Y.S.; Sathish, V.; Pabelick, C.M. Nicotinic α7 acetylcholine receptor (α7nAChR) in human airway smooth muscle. Arch. Biochem. Biophys. 2021, 706, 108897. [Google Scholar] [CrossRef] [PubMed]
- Wongtrakool, C.; Grooms, K.; Bijli, K.M.; Crothers, K.; Fitzpatrick, A.M.; Hart, C.M. Nicotine stimulates nerve growth factor in lung fibroblasts through an NFκB-dependent mechanism. PLoS ONE 2014, 9, e109602. [Google Scholar] [CrossRef] [PubMed]
- Thabut, G.; El-Benna, J.; Samb, A.; Corda, S.; Megret, J.; Leseche, G.; Vicaut, E.; Aubier, M.; Boczkowski, J. Tumor necrosis factor-alpha increases airway smooth muscle oxidants production through a NADPH oxidase-like system to enhance myosin light chain phosphorylation and contractility. J. Biol. Chem. 2002, 277, 22814–22821. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.K.; Lee, I.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Nox2/ROS-dependent human antigen R translocation contributes to TNF-α-induced SOCS-3 expression in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L521–L533. [Google Scholar] [CrossRef]
- Okonski, R.; Zheng, Y.M.; Di Mise, A.; Wang, Y.X. Reciprocal correlations of inflammatory and calcium signaling in asthma pathogenesis. Adv. Exp. Med. Biol. 2021, 1303, 319–331. [Google Scholar]
- Pera, T.; Gosens, R.; Lesterhuis, A.H.; Sami, R.; van der Toorn, M.; Zaagsma, J.; Meurs, H. Cigarette smoke and lipopolysaccharide induce a proliferative airway smooth muscle phenotype. Respir. Res. 2010, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yang, C.; Huang, L.; Chen, M.; Shi, J.; Ouyang, L.; Tang, T.; Zhang, W.; Li, Y.; Liang, R.; et al. Upregulation of TRPM7 augments cell proliferation and interleukin-8 release in airway smooth muscle cells of rats exposed to cigarette smoke. Mol. Med. Rep. 2016, 13, 4995–5004. [Google Scholar] [CrossRef]
- Abiria, S.A.; Krapivinsky, G.; Sah, R.; Santa-Cruz, A.G.; Chaudhuri, D.; Zhang, J.; Adstamongkonkul, P.; DeCaen, P.G.; Clapham, D.E. TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, E6079–E6088. [Google Scholar] [CrossRef]
- Aravamudan, B.; Thompson, M.; Sieck, G.C.; Vassallo, R.; Pabelick, C.M.; Prakash, Y.S. Functional effects of cigarette smoke-induced changes in airway smooth muscle mitochondrial morphology. J. Cell Physiol. 2017, 232, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Huecksteadt, T.P.; Norman, K.; Sanders, K.; Murphy, T.M.; Chitano, P.; Wilson, K.; Hoidal, J.R.; Kennedy, T.P. Nox4 mediates TGF-β1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1543–L1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef] [PubMed]
- An, S.S.; Kim, J.; Ahn, K.; Trepat, X.; Drake, K.J.; Kumar, S.; Ling, G.; Purington, C.; Rangasamy, T.; Kensler, T.W.; et al. Cell stiffness, contractile stress and the role of extracellular matrix. Biochem. Biophys. Res. Commun. 2009, 382, 697–703. [Google Scholar] [CrossRef]
- Tsiligianni, I.G.; van der Molen, T. A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir. Res. 2010, 11, 171. [Google Scholar] [CrossRef]
- Biswas, S.; Hwang, J.W.; Kirkham, P.A.; Rahman, I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr. Med. Chem. 2013, 20, 1496–1530. [Google Scholar] [CrossRef]
- Culpitt, S.V.; Rogers, D.F.; Fenwick, P.S.; Shah, P.; De Matos, C.; Russell, R.E.; Barnes, P.J.; Donnelly, L.E. Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax 2003, 58, 942–946. [Google Scholar] [CrossRef]
- Fischer, A.; Johansson, I.; Blomberg, A.; Sundström, B. Adherence to a Mediterranean-like diet as a protective factor against COPD: A Nested Case-Control Study. COPD 2019, 16, 272–277. [Google Scholar] [CrossRef]
- Biswas, S.K.; Rahman, I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Mol. Asp. Med. 2009, 30, 60–76. [Google Scholar] [CrossRef]
- Grandjean, E.M.; Berthet, P.; Ruffmann, R.; Leuenberger, P. Efficacy of oral long-term N-acetylcysteine in chronic bronchopul-monary disease: A meta-analysis of published double-blind, placebo-controlled clinical trials. Clin. Ther. 2000, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.P.; Wen, F.Q.; Bai, C.X.; Wan, H.Y.; Kang, J.; Chen, P.; Yao, W.Z.; Ma, L.J.; Li, X.; Raiteri, L.; et al. PANTHEON study group. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2014, 2, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Decramer, M.; Rutten-van Mölken, M.; Dekhuijzen, P.N.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.; Olivieri, D.; Del Donno, M.; De Backer, W.; et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pul-monary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): A randomised placebo-controlled trial. Lancet 2005, 65, 1552–1560. [Google Scholar] [CrossRef]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Repine, J.E.; Bast, A.; Lankhorst, I. Oxidative stress in chronic obstructive pulmonary disease. Oxidative Stress Study Group. Am. J. Respir. Crit. Care Med. 1997, 156, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Basili, S.; Vieri, M.; Cordova, C.; Violi, F.; Fitzgerald, G.A. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2α-III, an index of oxidant stress. Am. J. Respir. Crit. Care Med. 1998, 158, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Thebault, S.; Roudbaraki, M.; Sydorenko, V.; Shuba, Y.; Lemonnier, L.; Slomianny, C.; Dewailly, E.; Bonnal, J.L.; Mauroy, B.; Skryma, R.; et al. α1-Adrenergic receptors activate Ca2+-permeable cationic channels in prostate cancer epithelial cells. J. Clin. Invest. 2003, 111, 1691–1701. [Google Scholar] [CrossRef]
- Wang, J.; Shimoda, L.A.; Sylvester, J.T. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L848–L858. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Spinelli, A.M.; González-Cobos, J.C.; Zhang, X.; Motiani, R.K.; Rowan, S.; Zhang, W.; Garrett, J.; Vincent, P.A.; Matrougui, K.; Singer, H.A.; et al. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Arch. 2012, 464, 481–492. [Google Scholar] [CrossRef]
- Jiang, Q.; Fu, X.; Tian, L.; Chen, Y.; Yang, K.; Chen, X.; Zhang, J.; Lu, W.; Wang, J. NOX4 mediates BMP4-induced upregulation of TRPC1 and 6 protein expressions in distal pulmonary arterial smooth muscle cells. PLoS ONE 2014, 9, e107135. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Taggart, M.; Borthwick, L.; Fisher, A.; Brodlie, M.; Sassano, M.F.; Tarran, R.; Gray, M.A. Acute cigarette smoke or extract ex-posure rapidly activates TRPA1-mediated calcium influx in primary human airway smooth muscle cells. Sci. Rep. 2021, 11, 9643. [Google Scholar] [CrossRef] [PubMed]
- Wylam, M.E.; Sathish, V.; VanOosten, S.K.; Freeman, M.; Burkholder, D.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S. Mechanisms of cigarette smoke effects on human airway smooth muscle. PLoS ONE 2015, 10, e0128778. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial dysfunction in airway disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef]
- Ito, S.; Kume, H.; Honjo, H.; Katoh, H.; Kodama, I.; Yamaki, K.; Hayashi, H. Possible involvement of Rho kinase in Ca2+ sensitization and mobilization by MCh in tracheal smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1218–L1224. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Folkerts, G.; Nijkamp, F.P.; Henricks, P.A. ATP and the pathogenesis of COPD. Eur. J. Pharmacol. 2010, 638, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Uyeminami, D.L.; Kodavanti, U.P.; Crapo, J.D.; Chang, L.Y.; Pinkerton, K.E. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Rad. Biol. Med. 2002, 33, 1106–1114. [Google Scholar] [CrossRef]
- Churg, A.; Marshall, C.V.; Sin, D.D.; Bolton, S.; Zhou, S.; Thain, K.; Cadogan, E.B.; Maltby, J.; Soars, M.G.; Mallinder, P.R.; et al. Late intervention with a myeloperoxidase inhibitor stops progression of experimental COPD. Am. J. Respir. Crit. Care Med. 2012, 185, 34–43. [Google Scholar] [CrossRef]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mi-tochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kume, H.; Yamada, R.; Sato, Y.; Togawa, R. Airway Smooth Muscle Regulated by Oxidative Stress in COPD. Antioxidants 2023, 12, 142. https://doi.org/10.3390/antiox12010142
Kume H, Yamada R, Sato Y, Togawa R. Airway Smooth Muscle Regulated by Oxidative Stress in COPD. Antioxidants. 2023; 12(1):142. https://doi.org/10.3390/antiox12010142
Chicago/Turabian StyleKume, Hiroaki, Ryuki Yamada, Yuki Sato, and Ryuichi Togawa. 2023. "Airway Smooth Muscle Regulated by Oxidative Stress in COPD" Antioxidants 12, no. 1: 142. https://doi.org/10.3390/antiox12010142
APA StyleKume, H., Yamada, R., Sato, Y., & Togawa, R. (2023). Airway Smooth Muscle Regulated by Oxidative Stress in COPD. Antioxidants, 12(1), 142. https://doi.org/10.3390/antiox12010142