
Differential Effects of Arsenic in Drinking Water on Mouse Hepatic and Intestinal Heme Oxygenase-1 Expression


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Animals and Treatments
2.3. Isolation of Intestinal Epithelial Cells and Preparation of Intestinal and Hepatic Microsomes
2.4. Total RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (RT-PCR) Analysis
2.5. Immunoblot Analysis
2.6. In Vitro Assay of HO Activity
2.7. LC-MS/MS Analysis
2.8. Quantification of Total Arsenic and Arsenic Species in Serum and Tissues
2.9. Statistical Analysis
3. Results
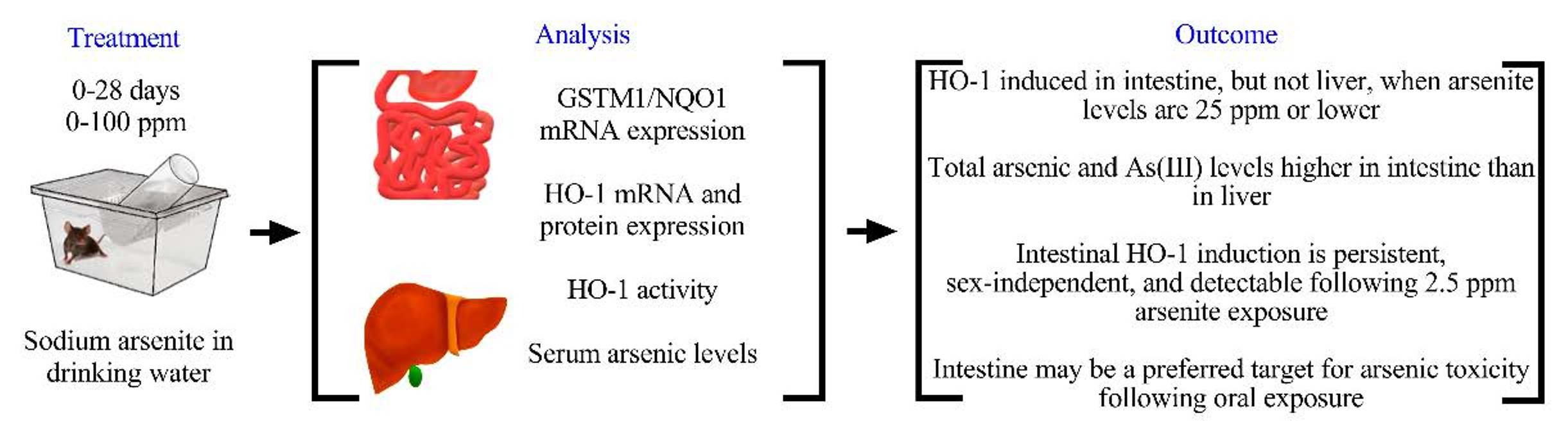
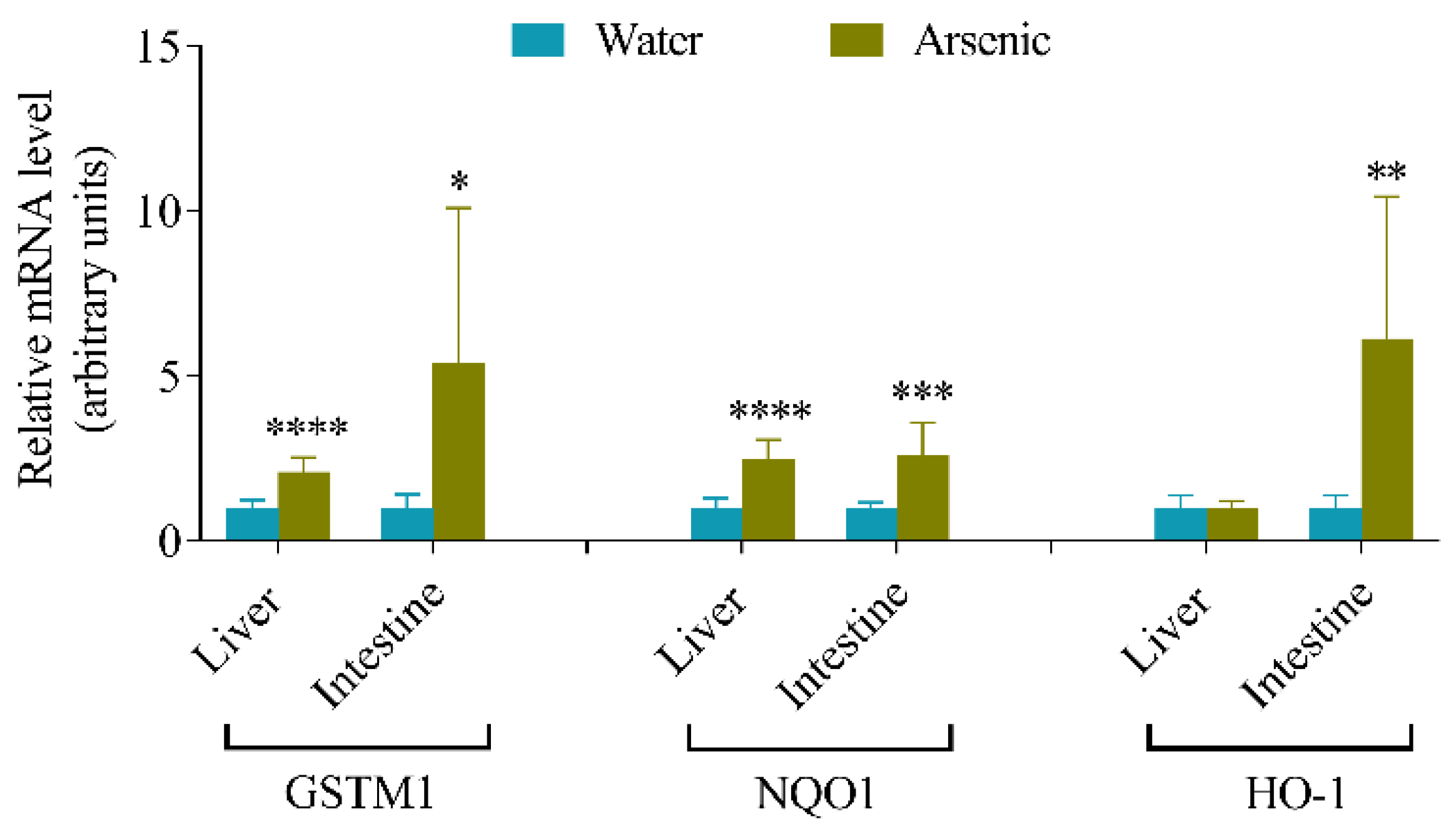
3.1. Effects of Arsenic Exposure via Drinking Water on the Expression of Oxidative Stress Responsive Genes in Mouse Liver and Intestine
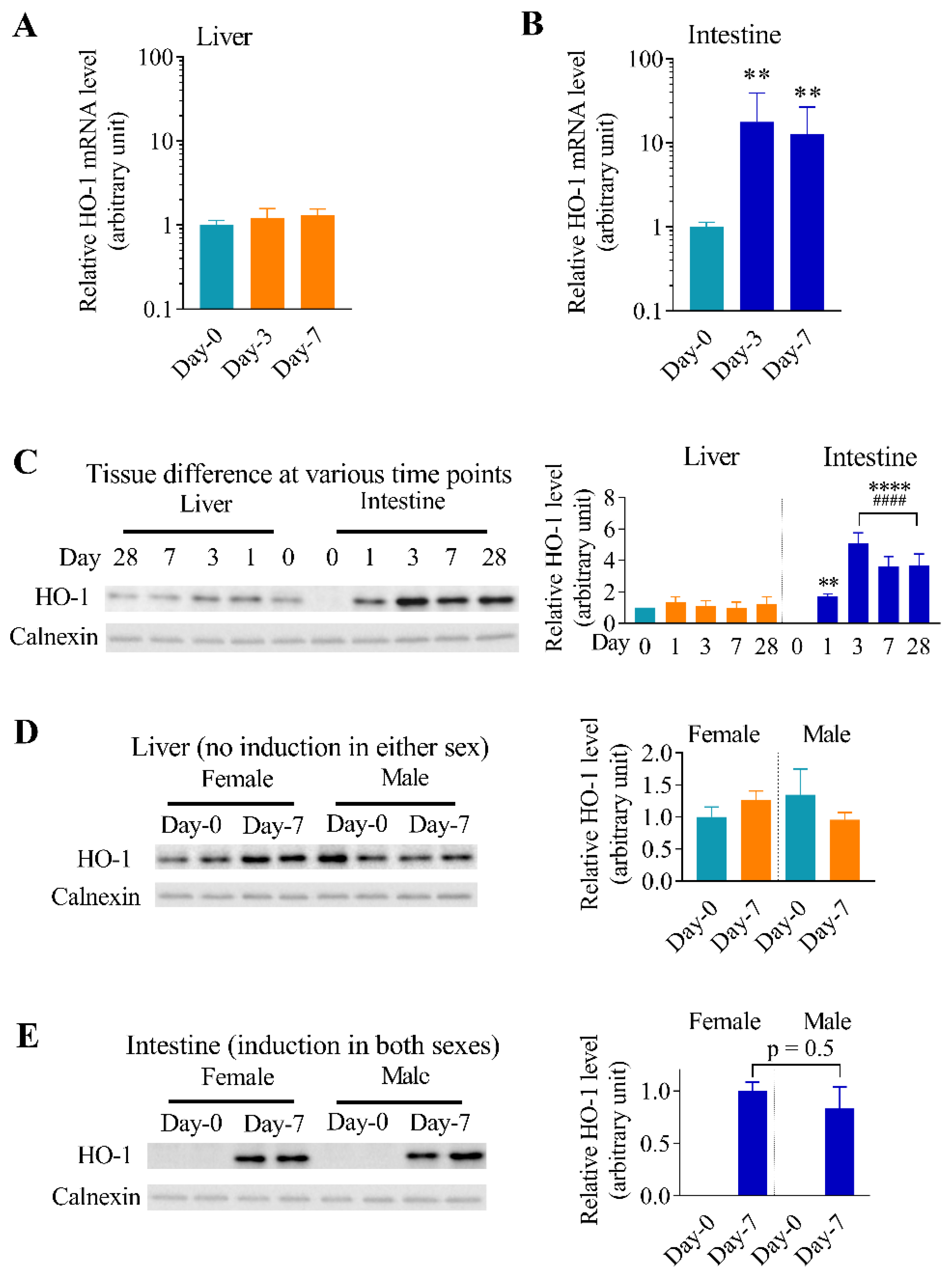
3.2. Differential Effects of Oral Arsenic Exposure on Hepatic and Intestinal HO-1 Protein Expression
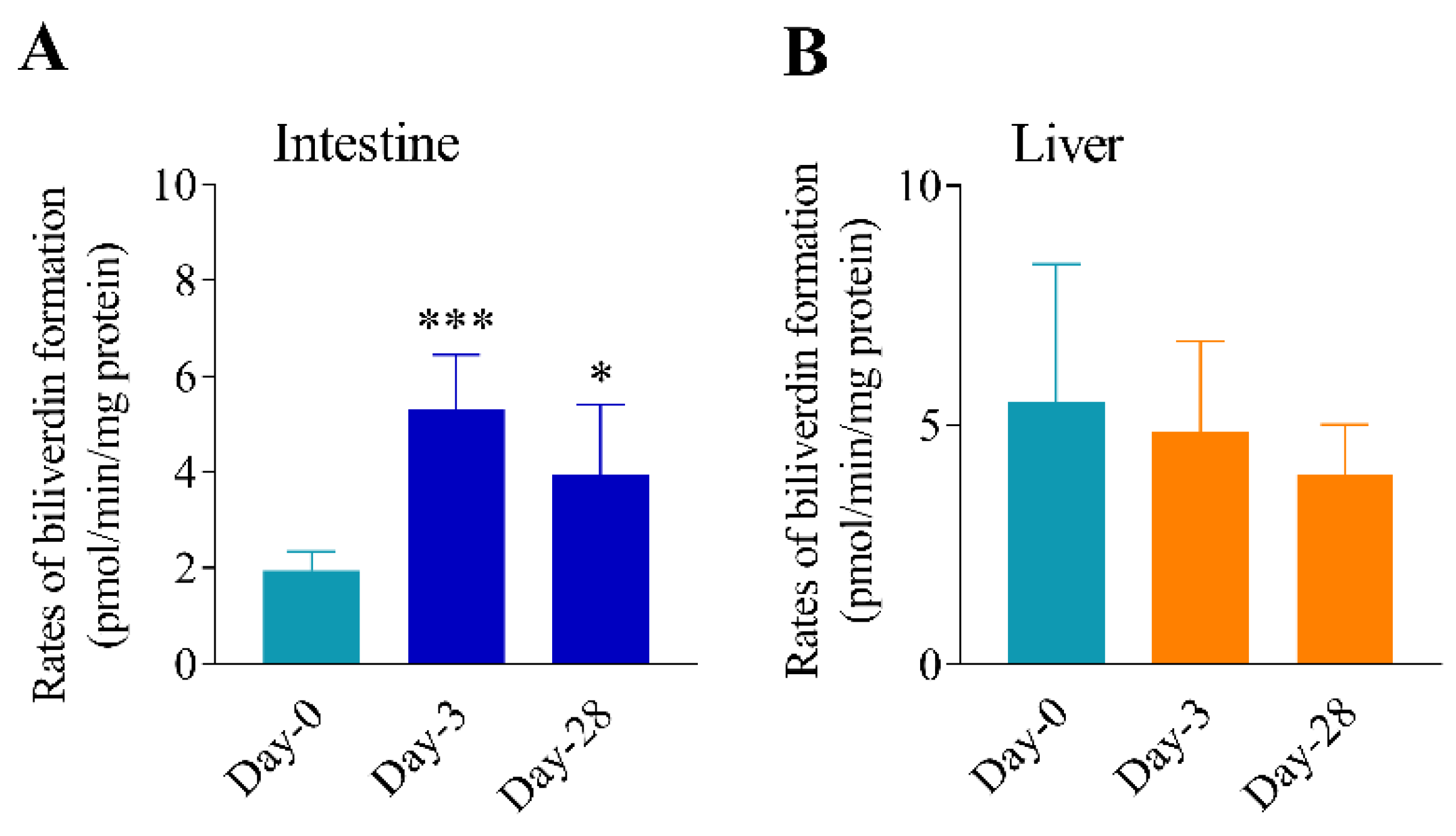
3.3. Differential Effects of Oral Arsenic Exposure on Hepatic and Intestinal HO Activity
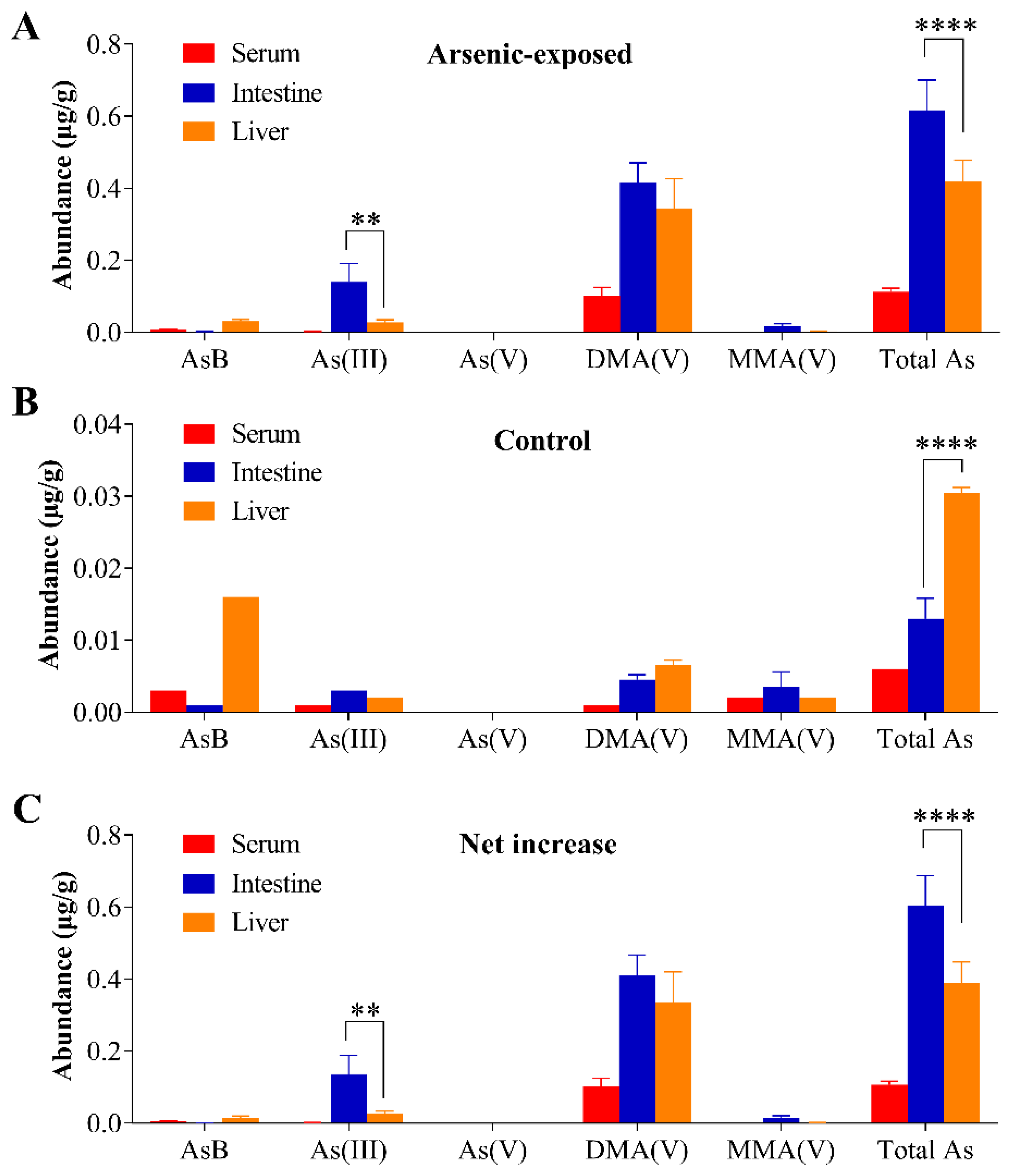
3.4. Tissue Arsenic Levels in Mice Exposed to Arsenic in Drinking Water
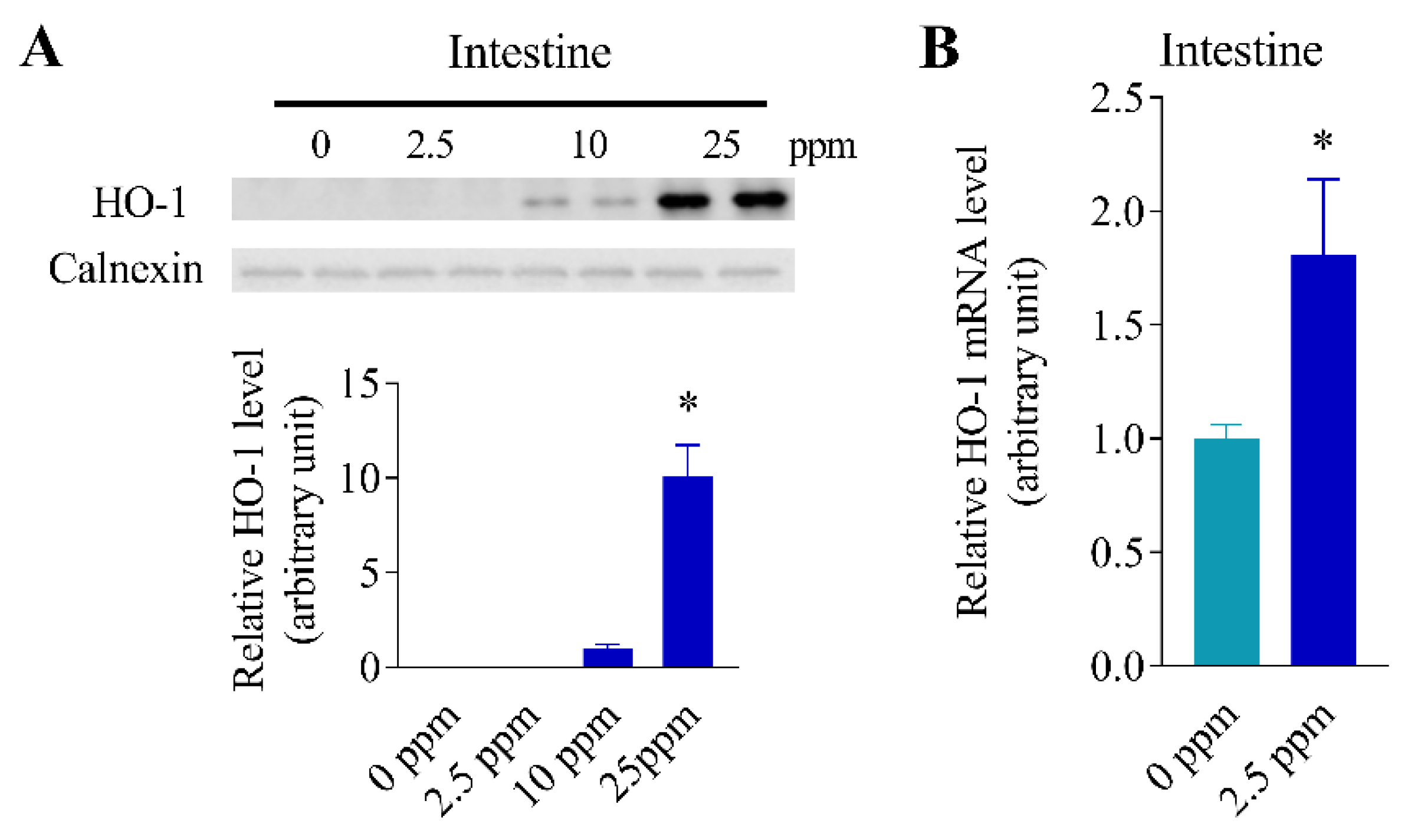
3.5. Inducibility of Intestinal HO-1 Expression at Lower Arsenic Doses
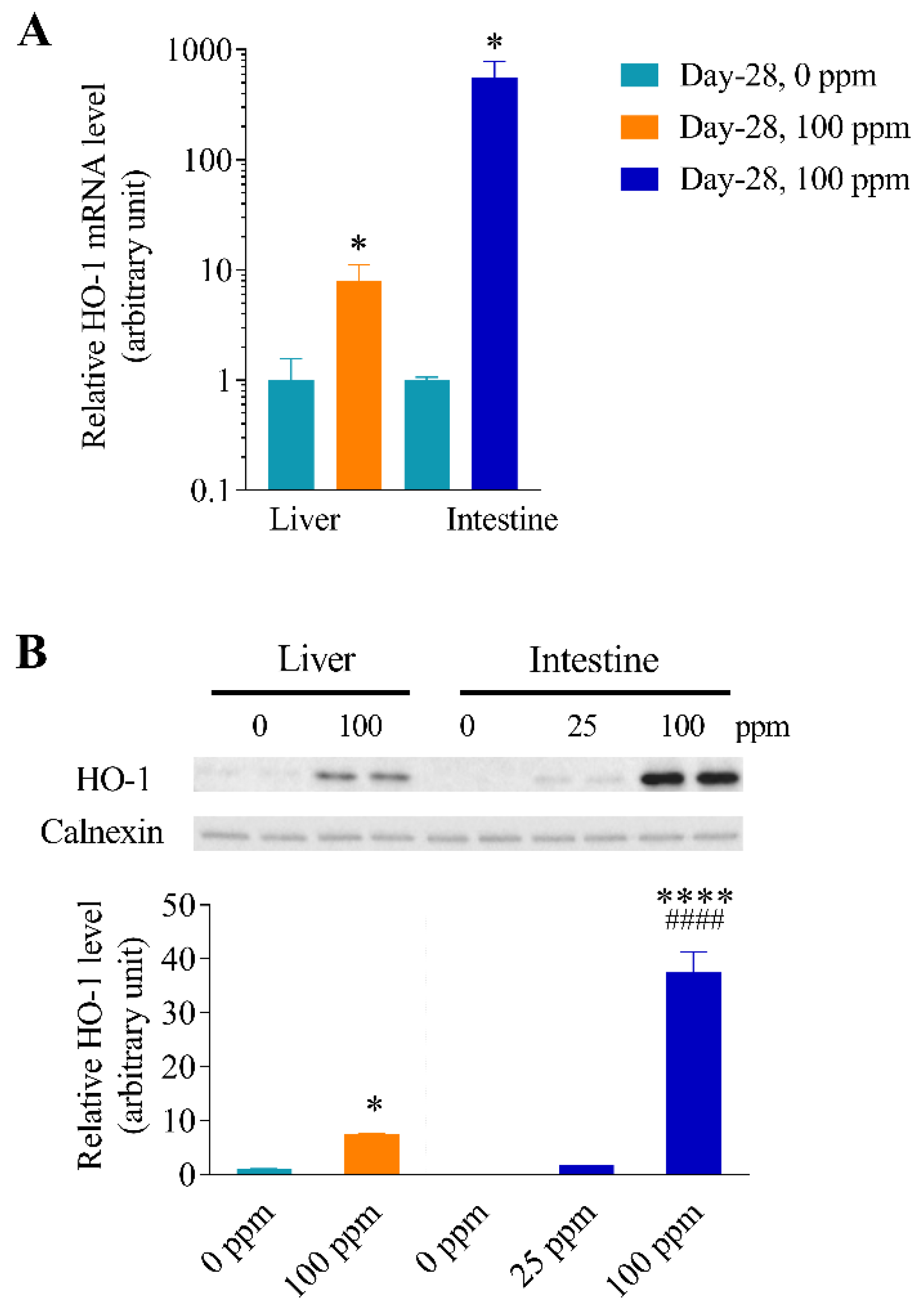
3.6. Differential Inducibility of Hepatic and Intestinal HO-1 Expression at a Higher Arsenic Dose
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mukhopadhyay, R.; Rosen, B.P.; Phung, L.T.; Silver, S. Microbial arsenic: From geocycles to genes and enzymes. FEMS Microbiol. Rev. 2002, 26, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef] [PubMed]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Scientific Opinion on arsenic in food. EFSA J. 2009, 7, 1351. [Google Scholar] [CrossRef]
- Kuo, C.C.; Moon, K.A.; Wang, S.L.; Silbergeld, E.; Navas-Acien, A. The Association of Arsenic Metabolism with Cancer, Cardiovascular Disease, and Diabetes: A Systematic Review of the Epidemiological Evidence. Environ. Health Perspect. 2017, 125, 087001. [Google Scholar] [CrossRef] [PubMed]
- USEPA. National primary drinking water regulations; arsenic and clarifications to compliance and new source contaminants monitoring. Fed. Reg. 2001, 66, 6975. [Google Scholar]
- Nielsen, M.G.; Lombard, P.J.; Schalk, L.F. Assessment of Arsenic Concentrations in Domestic Well Water, by Town, in Maine 2005-09; US Geological Survey: Washington, DC, USA, 2010.
- Uddin, R.; Huda, N.H. Arsenic poisoning in bangladesh. Oman Med. J. 2011, 26, 207. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, K.T. Recent advances in arsenic carcinogenesis: Modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharmacol. 2001, 172, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.F. Arsenic toxicity and potential mechanisms of action. Toxicol. Lett. 2002, 133, 1–16. [Google Scholar] [CrossRef]
- Kitchin, K.T.; Ahmad, S.A. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol. Lett. 2003, 137, 3–13. [Google Scholar] [CrossRef]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, K.T.; Conolly, R. Arsenic-Induced Carcinogenesis Oxidative Stress as a Possible Mode of Action and Future Research Needs for More Biologically Based Risk Assessment. Chem. Res. Toxicol. 2010, 23, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J. Arsenic-induced oxidative stress and its reversibility. Free. Radic. Biol. Med. 2011, 51, 257–281. [Google Scholar] [CrossRef]
- Du, Y.; Villeneuve, N.F.; Wang, X.J.; Sun, Z.; Chen, W.; Li, J.; Lou, H.; Wong, P.K.; Zhang, D.D. Oridonin confers protection against arsenic-induced toxicity through activation of the Nrf2-mediated defensive response. Environ. Health Perspect. 2008, 116, 1154–1161. [Google Scholar] [CrossRef]
- Pi, J.; Diwan, B.A.; Sun, Y.; Liu, J.; Qu, W.; He, Y.; Styblo, M.; Waalkes, M.P. Arsenic-induced malignant transformation of human keratinocytes: Involvement of Nrf2. Free Radic. Biol. Med. 2008, 45, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Snyder, R. New developments in the regulation of heme metabolism and their implications. CRC Crit. Rev. Toxicol. 1984, 12, 241–314. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, K.T.; Del Razo, L.M.; Brown, J.L.; Anderson, W.L.; Kenyon, E.M. An integrated pharmacokinetic and pharmacodynamic study of arsenite action. 1. Heme oxygenase induction in rats. Teratog. Carcinog. Mutagenesis 1999, 19, 385–402. [Google Scholar] [CrossRef]
- Kenyon, E.M.; Del Razo, L.M.; Hughes, M.F.; Kitchin, K.T. An integrated pharmacokinetic and pharmacodynamic study of arsenite action 2. Heme oxygenase induction in mice. Toxicology 2005, 206, 389–401. [Google Scholar] [CrossRef]
- Liu, J.; Kadiiska, M.B.; Liu, Y.; Lu, T.; Qu, W.; Waalkes, M.P. Stress-related gene expression in mice treated with inorganic arsenicals. Toxicol. Sci. 2001, 61, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Sardana, M.K.; Drummond, G.S.; Sassa, S.; Kappas, A. The potent heme oxygenase inducing action of arsenic and parasiticidal arsenicals. Pharmacology 1981, 23, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Menzel, D.; Rasmussen, R.; Lee, E.; Meacher, D.; Said, B.; Hamadeh, H.; Vargas, M.; Greene, H.; Roth, R. Human lymphocyte heme oxygenase 1 as a response biomarker to inorganic arsenic. Biochem. Biophys. Res. Commun. 1998, 250, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, Y.; Duan, X.; Li, B. Heme oxygenase-1 (HO-1) assists inorganic arsenic-induced immune tolerance in murine dendritic cells. Chemosphere 2021, 264, 128452. [Google Scholar] [CrossRef]
- Liu, D.; Xu, G.; Bai, C.; Gu, Y.; Wang, D.; Li, B. Differential effects of arsenic species on Nrf2 and Bach1 nuclear localization in cultured hepatocytes. Toxicol. Appl. Pharmacol. 2021, 413, 115404. [Google Scholar] [CrossRef] [PubMed]
- Inesta-Vaquera, F.; Navasumrit, P.; Henderson, C.J.; Frangova, T.G.; Honda, T.; Dinkova-Kostova, A.T.; Ruchirawat, M.; Wolf, C.R. Application of the in vivo oxidative stress reporter Hmox1 as mechanistic biomarker of arsenic toxicity. Environ. Pollut. 2021, 270, 116053. [Google Scholar] [CrossRef] [PubMed]
- Vahter, M.; Norin, H. Metabolism of 74As-labeled trivalent and pentavalent inorganic arsenic in mice. Environ. Res. 1980, 21, 446–457. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Dunbar, D.; Kaminsky, L.S. Characterization of mouse small intestinal cytochrome P450 expression. Drug Metab. Dispos. 2003, 31, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Fasco, M.J.; Silkworth, J.B.; Dunbar, D.A.; Kaminsky, L.S. Rat small intestinal cytochromes P450 probed by warfarin metabolism. Mol. Pharmacol. 1993, 43, 226–233. [Google Scholar] [PubMed]
- Iwamori, S.; Sato, E.; Saigusa, D.; Yoshinari, K.; Ito, S.; Sato, H.; Takahashi, N. A novel and sensitive assay for heme oxygenase activity. Am. J. Physiol. Renal. Physiol. 2015, 309, F667–F671. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, G.; Lin, Z.; Luo, Y.; Fang, H.; Yang, L.; Xie, J.; Guo, L. Determination of arsenicals in mouse tissues after simulated exposure to arsenic from rice for sixteen weeks and the effects on histopathological features. Ecotoxicol. Environ. Saf. 2020, 200, 110742. [Google Scholar] [CrossRef]
- Kubachka, K.M.; Shockey, N.V.; Hanley, T.A.; Conklin, S.D.; Heitkemper, D.T. Elemental Analysis Manual for Food and Related Products, Section 4.11, Arsenic Speciation in Rice and Rice Products Using High Performance Liquid Chromatography−Inductively Coupled Plasma−Mass Spectrometric Determination; Version 1.1; United States Food and Drug Administration (FDA): Silver Spring, MD, USA, 2012.
- Koller, B.H.; Snouwaert, J.N.; Douillet, C.; Jania, L.A.; El-Masri, H.; Thomas, D.J.; Styblo, M. Arsenic Metabolism in Mice Carrying a BORCS7/AS3MT Locus Humanized by Syntenic Replacement. Environ. Health Perspect. 2020, 128, 87003. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, D.N. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharmacol. 2005, 206, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.S.; Devesa, V.; Hernandez-Zavala, A.; Adair, B.M.; Walton, F.S.; Drobna, Z.; Thomas, D.J.; Styblo, M. Environmental arsenic as a disruptor of insulin signaling. In Metal Ions in Biology and Medicine: Proceedings of the … International Symposium on Metal Ions in Biology and Medicine Held … = Les Ions Metalliques en Biologie et en Medecine: … Symposium International Sur Les Ions Metalliques; NIH Public Access: Bethesda, MD, USA, 2008; Volume 10, pp. 1–7. [Google Scholar]
- Jo, W.J.; Loguinov, A.; Wintz, H.; Chang, M.; Smith, A.H.; Kalman, D.; Zhang, L.; Smith, M.T.; Vulpe, C.D. Comparative functional genomic analysis identifies distinct and overlapping sets of genes required for resistance to monomethylarsonous acid (MMAIII) and arsenite (AsIII) in yeast. Toxicol. Sci. 2009, 111, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Vahter, M.; Concha, G. Role of metabolism in arsenic toxicity. Pharmacol. Toxicol. 2001, 89, 1–5. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Fillebeen, C.; Gkouvatsos, K.; Fragoso, G.; Calvé, A.; Garcia-Santos, D.; Buffler, M.; Becker, C.; Schümann, K.; Ponka, P.; Santos, M.M. Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 2015, 100, e334. [Google Scholar] [CrossRef]
- Trakshel, G.M.; Kutty, R.K.; Maines, M.D. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J. Biol. Chem. 1986, 261, 11131–11137. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From metabolism to molecular therapy. Am. J. Respir. Cell Mol. Biol. 2009, 41, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Trakshel, G.M.; Kutty, R.K.; Maines, M.D. Cadmium-mediated inhibition of testicular heme oxygenase activity: The role of NADPH-cytochrome c (P-450) reductase. Arch. Biochem. Biophys. 1986, 251, 175–187. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef]
- Suliman, H.B.; Keenan, J.E.; Piantadosi, C.A. Mitochondrial quality-control dysregulation in conditional HO-1(-/-) mice. JCI Insight 2017, 2, e89676. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Fan, X.; Wu, X.; Han, W.; Amistadi, M.K.; Liu, P.; Zhang, D.; Chorover, J.; Ding, X.; Zhang, Q.-Y. Differential Effects of Arsenic in Drinking Water on Mouse Hepatic and Intestinal Heme Oxygenase-1 Expression. Antioxidants 2022, 11, 1835. https://doi.org/10.3390/antiox11091835
Li H, Fan X, Wu X, Han W, Amistadi MK, Liu P, Zhang D, Chorover J, Ding X, Zhang Q-Y. Differential Effects of Arsenic in Drinking Water on Mouse Hepatic and Intestinal Heme Oxygenase-1 Expression. Antioxidants. 2022; 11(9):1835. https://doi.org/10.3390/antiox11091835
Chicago/Turabian StyleLi, Hui, Xiaoyu Fan, Xiangmeng Wu, Weiguo Han, Mary Kay Amistadi, Pengfei Liu, Donna Zhang, Jon Chorover, Xinxin Ding, and Qing-Yu Zhang. 2022. "Differential Effects of Arsenic in Drinking Water on Mouse Hepatic and Intestinal Heme Oxygenase-1 Expression" Antioxidants 11, no. 9: 1835. https://doi.org/10.3390/antiox11091835
APA StyleLi, H., Fan, X., Wu, X., Han, W., Amistadi, M. K., Liu, P., Zhang, D., Chorover, J., Ding, X., & Zhang, Q.-Y. (2022). Differential Effects of Arsenic in Drinking Water on Mouse Hepatic and Intestinal Heme Oxygenase-1 Expression. Antioxidants, 11(9), 1835. https://doi.org/10.3390/antiox11091835