

NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction

 , ,
, , 

Abstract
1. Introduction
1.1. Heart Failure with Preserved Ejection Fraction (HFpEF)
1.2. NADPH Oxidase (NOX) Enzymes

2. NADPH Oxidases in Cardiovascular Disease
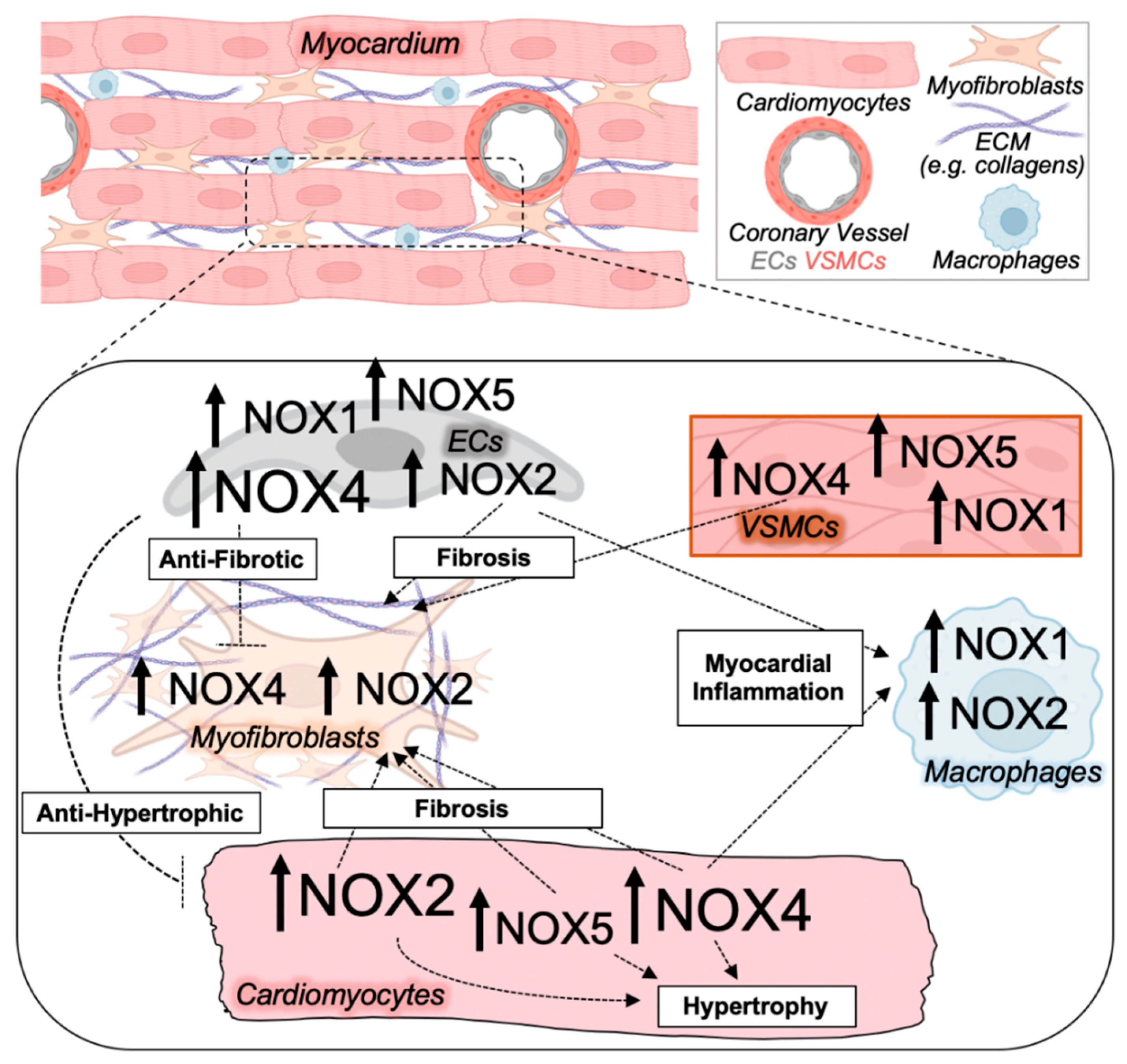
2.1. NOX Isoforms in Cardiac Hypertrophy, Fibrosis, and Diastolic Dysfunction
2.2. NADPH Oxidases in Non-Myocyte Cardiac Cell Types
2.2.1. NOX Enzymes in the Vasculature
2.2.2. NOX Enzymes in Fibroblasts
2.2.3. NOX Enzymes in Immune Cells
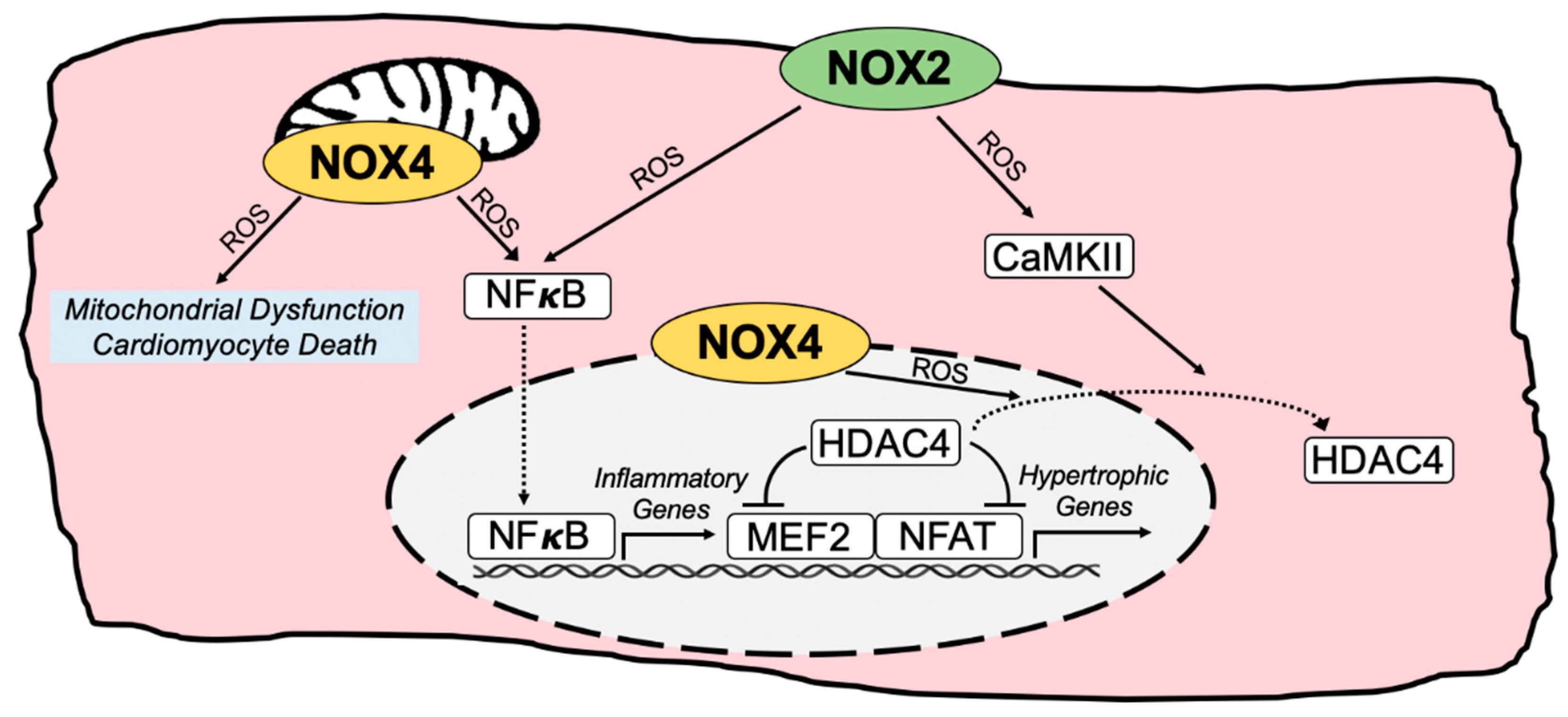
3. NOX-Regulated Pathogenic Redox Signaling in Cardiomyocytes

3.1. Redox Regulation of Hypertrophic and Maladaptive Gene Expression Programs
3.2. Sarcoplasmic Reticulum Calcium Handling and Myocardial Contractility
3.3. Mitochondrial ROS and Cardiomyocyte Death
4. NADPH Oxidases and Nitric Oxide (NO) Signaling
4.1. Nitric Oxide Synthase (NOS) Uncoupling and cGMP/Protein Kinase G (PKG) Signaling

4.2. Nitrosative Stress and Protein S-Nitrosylation
5. Clinical Implications and Future Perspectives
5.1. Renin-Angiotensin-Aldosterone System (RAAS) Antagonists
5.2. Nitric Oxide Donors and cGMP/Protein Kinase G Signaling
5.3. Sodium-Glucose Cotransporter 2 Inhibitors
5.4. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin converting enzyme |
| AngII | angiotensin II |
| ARB | angiotensin receptor blocker |
| AT1R | angiotensin receptor type 1 |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| cGMP | cyclic GMP |
| eNOS | endothelial nitric oxide synthase |
| GDF6 | growth differentiation factor 6 |
| HDAC2, 4 | histone deacetylase 2, 4 |
| HFD | high-fat diet |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| iNOS | inducible nitric oxide synthase |
| IRE1𝛼 | inositol-requiring enzyme 1𝛼 |
| L-NAME | Nω-nitro-L-arginine |
| MEF2 | myocyte enhancer factor 2 |
| MRA | mineralocorticoid receptor antagonist |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NFAT | nuclear factor of activated T cells |
| NFκB | nuclear factor kappa B |
| nNOS | neuronal nitric oxide synthase |
| NOS | nitric oxide synthase |
| NOX | NADPH oxidase |
| PDGF | platelet-derived growth factor |
| PKG | protein kinase G |
| RAAS | renin-angiotensin-aldosterone system |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| ROS | reactive oxygen species |
| SAUNA | salty drinking water/unilateral nephrectomy/aldosterone |
| sGC | soluble guanylate cyclase |
| SGLT2 | sodium-glucose cotransporter 2 |
| TGF-β | transforming growth factor-beta |
| Treg | regulatory T cell |
| VSMC | vascular smooth muscle cell |
| Xbp1(s) | (spliced) X-box-binding protein 1. |
References
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Borlaug, B.A.; Kitzman, D.W.; McCulloch, A.D.; Blaxall, B.C.; Agarwal, R.; Chirinos, J.A.; Collins, S.; Deo, R.C.; Gladwin, M.T.; et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020, 141, 1001–1026. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.S.; Xu, H.; Matsouaka, R.A.; Bhatt, D.L.; Heidenreich, P.A.; Hernandez, A.F.; Devore, A.D.; Yancy, C.W.; Fonarow, G.C. Heart Failure With Preserved, Borderline, and Reduced Ejection Fraction: 5-Year Outcomes. J. Am. Coll. Cardiol. 2017, 70, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.A.A.; Velazquez, E.J. Heart Failure With Preserved Ejection Fraction: Time for a Reset. JAMA 2020, 324, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, S.E.; von Lueder, T.G.; Smiseth, O.A.; Wachtell, K.; Mistry, N.; Westheim, A.S.; Hopper, I.; Julius, S.; Pitt, B.; Reid, C.M.; et al. Medical Therapies for Heart Failure With Preserved Ejection Fraction. Hypertension 2020, 75, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Tribouilloy, C.; Rusinaru, D.; Mahjoub, H.; Souliere, V.; Levy, F.; Peltier, M.; Slama, M.; Massy, Z. Prognosis of heart failure with preserved ejection fraction: A 5 year prospective population-based study. Eur. Heart J. 2008, 29, 339–347. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure With Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef]
- LeWinter, M.M.; Meyer, M. Mechanisms of diastolic dysfunction in heart failure with a preserved ejection fraction: If it's not one thing it's another. Circ. Heart Fail 2013, 6, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Tharp, C.A.; Rubino, M.; McKinsey, T.A. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132, e148554. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Kane, G.C.; Karon, B.L.; Mahoney, D.W.; Redfield, M.M.; Roger, V.L.; Burnett, J.C., Jr.; Jacobsen, S.J.; Rodeheffer, R.J. Progression of left ventricular diastolic dysfunction and risk of heart failure. JAMA 2011, 306, 856–863. [Google Scholar] [CrossRef]
- Zile, M.R.; Gottdiener, J.S.; Hetzel, S.J.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Baicu, C.F.; Massie, B.M.; Carson, P.E.; Investigators, I.P. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation 2011, 124, 2491–2501. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef]
- Vitiello, D.; Harel, F.; Touyz, R.M.; Sirois, M.G.; Lavoie, J.; Myers, J.; Ducharme, A.; Racine, N.; O'Meara, E.; Gayda, M.; et al. Changes in cardiopulmonary reserve and peripheral arterial function concomitantly with subclinical inflammation and oxidative stress in patients with heart failure with preserved ejection fraction. Int. J. Vasc. Med. 2014, 2014, 917271. [Google Scholar] [CrossRef]
- Sharp, T.E., 3rd; Scarborough, A.L.; Li, Z.; Polhemus, D.J.; Hidalgo, H.A.; Schumacher, J.D.; Matsuura, T.R.; Jenkins, J.S.; Kelly, D.P.; Goodchild, T.T.; et al. Novel Gottingen Miniswine Model of Heart Failure With Preserved Ejection Fraction Integrating Multiple Comorbidities. JACC Basic Transl. Sci. 2021, 6, 154–170. [Google Scholar] [CrossRef]
- Zuo, L.; Chuang, C.C.; Hemmelgarn, B.T.; Best, T.M. Heart failure with preserved ejection fraction: Defining the function of ROS and NO. J. Appl. Physiol. 2015, 119, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Yan, L.; He, Q.; Xie, H.; Chen, M. Resveratrol Ameliorates Cardiac Remodeling in a Murine Model of Heart Failure With Preserved Ejection Fraction. Front. Pharmacol. 2021, 12, 646240. [Google Scholar] [CrossRef]
- Yang, H.J.; Kong, B.; Shuai, W.; Zhang, J.J.; Huang, H. MD1 deletion exaggerates cardiomyocyte autophagy induced by heart failure with preserved ejection fraction through ROS/MAPK signalling pathway. J. Cell Mol. Med. 2020, 24, 9300–9312. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lodi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovacs, A.; Fulop, G.A.; Falcao-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Galpha oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Sorop, O.; Heinonen, I.; van Kranenburg, M.; van de Wouw, J.; de Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; van Duin, R.W.B.; Stam, K.; van Geuns, R.J.; et al. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, T.; Yamamoto, E.; Sueta, D.; Fujisue, K.; Usuku, H.; Oike, F.; Takae, M.; Tabata, N.; Ito, M.; Yamanaga, K.; et al. Impact of Reactive Oxidative Metabolites Among New Categories of Nonischemic Heart Failure. J. Am. Heart Assoc. 2021, 10, e016765. [Google Scholar] [CrossRef]
- Hirata, Y.; Yamamoto, E.; Tokitsu, T.; Kusaka, H.; Fujisue, K.; Kurokawa, H.; Sugamura, K.; Maeda, H.; Tsujita, K.; Yamamuro, M.; et al. Reactive oxidative metabolites are associated with the severity of heart failure and predict future cardiovascular events in heart failure with preserved left ventricular ejection fraction. Int. J. Cardiol. 2015, 179, 305–308. [Google Scholar] [CrossRef]
- Xu, L.; Balzarolo, M.; Robinson, E.L.; Lorenz, V.; Verde, G.D.; Joray, L.; Mochizuki, M.; Kaufmann, B.A.; Valstar, G.; de Jager, S.C.A.; et al. NOX1 mediates metabolic heart disease in mice and is upregulated in monocytes of humans with diastolic dysfunction. Cardiovasc. Res. 2021, cvab349. [Google Scholar] [CrossRef]
- Guo, Y.; Wen, J.; He, A.; Qu, C.; Peng, Y.; Luo, S.; Wang, X. iNOS contributes to heart failure with preserved ejection fraction through mitochondrial dysfunction and Akt S-nitrosylation. J. Advanced. Res. 2022. [Google Scholar] [CrossRef]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Matsushima, S.; Kuroda, J.; Zablocki, D.; Kitazono, T.; Sadoshima, J. The NADPH oxidase Nox4 and aging in the heart. Aging 2010, 2, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Zablocki, D.; Sadoshima, J. Angiotensin II and oxidative stress in the failing heart. Antioxid. Redox Signal. 2013, 19, 1095–1109. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Sumida, A.; Canugovi, C.; Lozhkin, A.; Hayami, T.; Madamanchi, N.R.; Runge, M.S. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol. 2019, 21, 101063. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Murdoch, C.; Shah, A.M. Involvement of NADPH oxidases in cardiac remodelling and heart failure. Am. J. Nephrol. 2007, 27, 649–660. [Google Scholar] [CrossRef]
- He, B.J.; Joiner, M.L.; Singh, M.V.; Luczak, E.D.; Swaminathan, P.D.; Koval, O.M.; Kutschke, W.; Allamargot, C.; Yang, J.; Guan, X.; et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 2011, 17, 1610–1618. [Google Scholar] [CrossRef]
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002, 105, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O'Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef]
- Maggiorani, D.; Manzella, N.; Edmondson, D.E.; Mattevi, A.; Parini, A.; Binda, C.; Mialet-Perez, J. Monoamine Oxidases, Oxidative Stress, and Altered Mitochondrial Dynamics in Cardiac Ageing. Oxid. Med. Cell Longev. 2017, 2017, 3017947. [Google Scholar] [CrossRef]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig. 2005, 115, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Otani, H.; Shimazu, T.; Yoshioka, K.; Fujita, M.; Katano, T.; Ito, S.; Iwasaka, T. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J. Mol. Cell Cardiol. 2011, 50, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef]
- Zhao, G.J.; Zhao, C.L.; Ouyang, S.; Deng, K.Q.; Zhu, L.; Montezano, A.C.; Zhang, C.; Hu, F.; Zhu, X.Y.; Tian, S.; et al. Ca(2+)-Dependent NOX5 (NADPH Oxidase 5) Exaggerates Cardiac Hypertrophy Through Reactive Oxygen Species Production. Hypertension 2020, 76, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G.; Heyworth, P.G.; Evans, T.; Curnutte, J.T.; Bokoch, G.M. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 1991, 254, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef]
- Hahn, N.E.; Meischl, C.; Kawahara, T.; Musters, R.J.; Verhoef, V.M.; van der Velden, J.; Vonk, A.B.; Paulus, W.J.; van Rossum, A.C.; Niessen, H.W.; et al. NOX5 expression is increased in intramyocardial blood vessels and cardiomyocytes after acute myocardial infarction in humans. Am. J. Pathol. 2012, 180, 2222–2229. [Google Scholar] [CrossRef]
- Harrison, C.B.; Trevelin, S.C.; Richards, D.A.; Santos, C.X.C.; Sawyer, G.; Markovinovic, A.; Zhang, X.; Zhang, M.; Brewer, A.C.; Yin, X.; et al. Fibroblast Nox2 (NADPH Oxidase-2) Regulates ANG II (Angiotensin II)-Induced Vascular Remodeling and Hypertension via Paracrine Signaling to Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 698–710. [Google Scholar] [CrossRef]
- Lee, C.F.; Qiao, M.; Schroder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Park, J.Y.; Xie, L.H.; Tian, B.; Sadoshima, J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 2013, 112, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintron, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Zhai, P.; Liu, T.; Ikeda, S.; Nagarajan, N.; Oka, S.; Yokota, T.; Kinugawa, S.; Hsu, C.P.; et al. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Investig. 2016, 126, 3403–3416. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 2013, 113, 1253–1264. [Google Scholar] [CrossRef]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schroder, K.; Streckfuss-Bomeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP3 receptor-dependent Ca(2+) release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative Stress in Aging-Associated Cardiovascular Disease. Antioxid. Redox Signal. 2015, 23, 1389–1409. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.S.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W.; et al. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef]
- Hingtgen, S.D.; Tian, X.; Yang, J.; Dunlay, S.M.; Peek, A.S.; Wu, Y.; Sharma, R.V.; Engelhardt, J.F.; Davisson, R.L. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol. Genomics 2006, 26, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Nocella, C.; De Falco, E.; Palmerio, S.; Schirone, L.; Valenti, V.; Frati, G.; Carnevale, R.; Sciarretta, S. The Pathophysiological Role of NOX2 in Hypertension and Organ Damage. High Blood Press. Cardiovasc. Prev. 2016, 23, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.S.; Aasum, E.; Hafstad, A.D. The role of NADPH oxidases in diabetic cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.D.; Canugovi, C.; Vendrov, A.E.; Hayami, T.; Bowles, D.E.; Krause, K.H.; Madamanchi, N.R.; Runge, M.S. NADPH Oxidase 4 Regulates Inflammation in Ischemic Heart Failure: Role of Soluble Epoxide Hydrolase. Antioxid. Redox Signal. 2019, 31, 39–58. [Google Scholar] [CrossRef]
- van Berlo, J.H.; Maillet, M.; Molkentin, J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Investig. 2013, 123, 37–45. [Google Scholar] [CrossRef]
- Bishu, K.; Deswal, A.; Chen, H.H.; LeWinter, M.M.; Lewis, G.D.; Semigran, M.J.; Borlaug, B.A.; McNulty, S.; Hernandez, A.F.; Braunwald, E.; et al. Biomarkers in acutely decompensated heart failure with preserved or reduced ejection fraction. Am. Heart J. 2012, 164, 763–770.e3. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. Heart failure with preserved ejection fraction: Present status and future directions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Binder, C.; Poglitsch, M.; Duca, F.; Rettl, R.; Dachs, T.M.; Dalos, D.; Schrutka, L.; Seirer, B.; Ligios, L.C.; Capelle, C.; et al. Renin Feedback Is an Independent Predictor of Outcome in HFpEF. J. Pers. Med. 2021, 11, 370. [Google Scholar] [CrossRef]
- Ke, B.; Tan, X.; Ren, L.; Fan, Y.; Zhang, Y.; Li, F.; Sun, Q.; Liu, T.; Jia, L.; Wang, Y.; et al. Aldosterone dysregulation predicts the risk of mortality and rehospitalization in heart failure with a preserved ejection fraction. Sci. China Life Sci. 2022, 65, 631–642. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E. Treatment of Heart Failure With Preserved Ejection Fraction: Reflections on Its Treatment With an Aldosterone Antagonist. JAMA Cardiol. 2016, 1, 7–8. [Google Scholar] [CrossRef]
- Catena, C.; Verheyen, N.; Pilz, S.; Kraigher-Krainer, E.; Tomaschitz, A.; Sechi, L.A.; Pieske, B. Plasma aldosterone and left ventricular diastolic function in treatment-naive patients with hypertension: Tissue-Doppler imaging study. Hypertension 2015, 65, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Gimenes, R.; Gimenes, C.; Rosa, C.M.; Xavier, N.P.; Campos, D.H.S.; Fernandes, A.A.H.; Cezar, M.D.M.; Guirado, G.N.; Pagan, L.U.; Chaer, I.D.; et al. Influence of apocynin on cardiac remodeling in rats with streptozotocin-induced diabetes mellitus. Cardiovasc. Diabetol. 2018, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, H.; Shen, E.; Wan, L.; Arnold, J.M.; Peng, T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010, 59, 2033–2042. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Li, X.B.; Guo, S.J.; Chu, S.L.; Gao, P.J.; Zhu, D.L.; Niu, W.Q.; Jia, N. Apocynin attenuates oxidative stress and cardiac fibrosis in angiotensin II-induced cardiac diastolic dysfunction in mice. Acta Pharmacol. Sin. 2013, 34, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Park, M.Y.; Suh, Y.L.; Park, J.B. NAD(P)H oxidase inhibitor prevents blood pressure elevation and cardiovascular hypertrophy in aldosterone-infused rats. Biochem. Biophys. Res. Commun. 2004, 313, 812–817. [Google Scholar] [CrossRef]
- Bidwell, P.A.; Liu, G.S.; Nagarajan, N.; Lam, C.K.; Haghighi, K.; Gardner, G.; Cai, W.F.; Zhao, W.; Mugge, L.; Vafiadaki, E.; et al. HAX-1 regulates SERCA2a oxidation and degradation. J. Mol. Cell. Cardiol. 2018, 114, 220–233. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Hansen, S.S.; Lund, J.; Santos, C.X.C.; Boardman, N.T.; Shah, A.M.; Aasum, E. NADPH Oxidase 2 Mediates Myocardial Oxygen Wasting in Obesity. Antioxidants 2020, 9, 171. [Google Scholar] [CrossRef]
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010, 70, 9287–9297. [Google Scholar] [CrossRef]
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C.; Marber, M.; Monaghan, M.J.; Shah, A.M. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008, 51, 319–325. [Google Scholar] [CrossRef]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef]
- Grieve, D.J.; Byrne, J.A.; Siva, A.; Layland, J.; Johar, S.; Cave, A.C.; Shah, A.M. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J. Am. Coll. Cardiol. 2006, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, N.; Patel, V.B.; Wang, W.; Basu, R.; Oudit, G.Y. Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin. Sci. 2014, 127, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed]
- Ayuzawa, N.; Nagase, M.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Marumo, T.; Aiba, A.; Sakurai, T.; Shindo, T.; Fujita, T. Rac1-Mediated Activation of Mineralocorticoid Receptor in Pressure Overload-Induced Cardiac Injury. Hypertension 2016, 67, 99–106. [Google Scholar] [CrossRef]
- Essandoh, K.; Auchus, R.J.; Brody, M.J. Cardiac decompensation and promiscuous prenylation of small GTPases in cardiomyocytes in response to local mevalonate pathway disruption. J. Pathol. 2022, 256, 249–252. [Google Scholar] [CrossRef]
- Takemoto, M.; Node, K.; Nakagami, H.; Liao, Y.; Grimm, M.; Takemoto, Y.; Kitakaze, M.; Liao, J.K. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J. Clin. Investig. 2001, 108, 1429–1437. [Google Scholar] [CrossRef]
- Maack, C.; Kartes, T.; Kilter, H.; Schafers, H.J.; Nickenig, G.; Bohm, M.; Laufs, U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 2003, 108, 1567–1574. [Google Scholar] [CrossRef]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef]
- Zhang, M.; Mongue-Din, H.; Martin, D.; Catibog, N.; Smyrnias, I.; Zhang, X.; Yu, B.; Wang, M.; Brandes, R.P.; Schroder, K.; et al. Both cardiomyocyte and endothelial cell Nox4 mediate protection against hemodynamic overload-induced remodelling. Cardiovasc. Res. 2018, 114, 401–408. [Google Scholar] [CrossRef]
- Mongue-Din, H.; Patel, A.S.; Looi, Y.H.; Grieve, D.J.; Anilkumar, N.; Sirker, A.; Dong, X.; Brewer, A.C.; Zhang, M.; Smith, A.; et al. NADPH Oxidase-4 Driven Cardiac Macrophage Polarization Protects Against Myocardial Infarction-Induced Remodeling. JACC Basic. Transl. Sci. 2017, 2, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lee, C.F.; Wang, W.; Karamanlidis, G.; Kuroda, J.; Matsushima, S.; Sadoshima, J.; Tian, R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J. Am. Heart Assoc. 2014, 3, e000555. [Google Scholar] [CrossRef] [PubMed]
- Eaton, C.B.; Pettinger, M.; Rossouw, J.; Martin, L.W.; Foraker, R.; Quddus, A.; Liu, S.; Wampler, N.S.; Hank Wu, W.C.; Manson, J.E.; et al. Risk Factors for Incident Hospitalized Heart Failure With Preserved Versus Reduced Ejection Fraction in a Multiracial Cohort of Postmenopausal Women. Circ. Heart Fail. 2016, 9, e002883. [Google Scholar] [CrossRef] [PubMed]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R18–R36. [Google Scholar] [CrossRef]
- Mighiu, A.S.; Recalde, A.; Ziberna, K.; Carnicer, R.; Tomek, J.; Bub, G.; Brewer, A.C.; Verheule, S.; Shah, A.M.; Simon, J.N.; et al. Inducibility, but not stability, of atrial fibrillation is increased by NOX2 overexpression in mice. Cardiovasc. Res. 2021, 117, 2354–2364. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef]
- Zhang, J.; Youn, J.Y.; Kim, A.Y.; Ramirez, R.J.; Gao, L.; Ngo, D.; Chen, P.; Scovotti, J.; Mahajan, A.; Cai, H. NOX4-Dependent Hydrogen Peroxide Overproduction in Human Atrial Fibrillation and HL-1 Atrial Cells: Relationship to Hypertension. Front. Physiol. 2012, 3, 140. [Google Scholar] [CrossRef]
- Reilly, S.N.; Jayaram, R.; Nahar, K.; Antoniades, C.; Verheule, S.; Channon, K.M.; Alp, N.J.; Schotten, U.; Casadei, B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation 2011, 124, 1107–1117. [Google Scholar] [CrossRef]
- Chen, W.J.; Chang, S.H.; Chan, Y.H.; Lee, J.L.; Lai, Y.J.; Chang, G.J.; Tsai, F.C.; Yeh, Y.H. Tachycardia-induced CD44/NOX4 signaling is involved in the development of atrial remodeling. J. Mol. Cell Cardiol. 2019, 135, 67–78. [Google Scholar] [CrossRef]
- Sovari, A.A.; Dudley, S.C., Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Frost, G.; Custodis, F.; Sussman, M.A.; Schafers, H.J.; Bohm, M.; Laufs, U. Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 2007, 50, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Batul, S.A.; Gopinathannair, R. Atrial Fibrillation in Heart Failure: A Therapeutic Challenge of Our Times. Korean Circ. J. 2017, 47, 644–662. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, R.; Chamberlain, A.M.; Roger, V.L.; Redfield, M.M. Temporal relationship and prognostic significance of atrial fibrillation in heart failure patients with preserved ejection fraction: A community-based study. Circulation 2013, 128, 1085–1093. [Google Scholar] [CrossRef]
- Reddy, Y.N.V.; Obokata, M.; Verbrugge, F.H.; Lin, G.; Borlaug, B.A. Atrial Dysfunction in Patients With Heart Failure With Preserved Ejection Fraction and Atrial Fibrillation. J. Am. Coll. Cardiol. 2020, 76, 1051–1064. [Google Scholar] [CrossRef]
- Packer, M.; Lam, C.S.P.; Lund, L.H.; Redfield, M.M. Interdependence of Atrial Fibrillation and Heart Failure With a Preserved Ejection Fraction Reflects a Common Underlying Atrial and Ventricular Myopathy. Circulation 2020, 141, 4–6. [Google Scholar] [CrossRef]
- Ariyaratnam, J.P.; Elliott, A.D.; Mishima, R.S.; Gallagher, C.; Lau, D.H.; Sanders, P. Heart failure with preserved ejection fraction: An alternative paradigm to explain the clinical implications of atrial fibrillation. Heart Rhythm. O2 2021, 2, 771–783. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Madamanchi, N.R.; Hakim, Z.S.; Rojas, M.; Runge, M.S. Thrombin and NAD(P)H oxidase-mediated regulation of CD44 and BMP4-Id pathway in VSMC, restenosis, and atherosclerosis. Circ. Res. 2006, 98, 1254–1263. [Google Scholar] [CrossRef]
- Li, Y.; Kracun, D.; Dustin, C.M.; El Massry, M.; Yuan, S.; Goossen, C.J.; DeVallance, E.R.; Sahoo, S.; St Hilaire, C.; Gurkar, A.U.; et al. Forestalling age-impaired angiogenesis and blood flow by targeting NOX: Interplay of NOX1, IL-6, and SASP in propagating cell senescence. Proc. Natl. Acad. Sci. USA 2021, 118, e2015666118. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Murdoch, C.E.; Brewer, A.C.; Ivetic, A.; Evans, P.; Shah, A.M.; Zhang, M. Endothelial NADPH oxidase 4 protects against angiotensin II-induced cardiac fibrosis and inflammation. ESC Heart Fail. 2021, 8, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Kant, S.; Reif, M.; Chen, K.; Pei, Y.; Angoff, R.; Sugamura, K.; Fitzgibbons, T.; Keaney, J.F., Jr. Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic. Biol. Med. 2015, 89, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schurmann, C.; Rezende, F.; Kruse, C.; Yasar, Y.; Lowe, O.; Fork, C.; van de Sluis, B.; Bremer, R.; Weissmann, N.; Shah, A.M.; et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur. Heart J. 2015, 36, 3447–3456. [Google Scholar] [CrossRef]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef]
- Bendall, J.K.; Rinze, R.; Adlam, D.; Tatham, A.L.; de Bono, J.; Wilson, N.; Volpi, E.; Channon, K.M. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: Studies in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007, 100, 1016–1025. [Google Scholar] [CrossRef]
- Douglas, G.; Bendall, J.K.; Crabtree, M.J.; Tatham, A.L.; Carter, E.E.; Hale, A.B.; Channon, K.M. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(-)/(-) mice. Cardiovasc Res. 2012, 94, 20–29. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef]
- Elbatreek, M.H.; Sadegh, S.; Anastasi, E.; Guney, E.; Nogales, C.; Kacprowski, T.; Hassan, A.A.; Teubner, A.; Huang, P.H.; Hsu, C.Y.; et al. NOX5-induced uncoupling of endothelial NO synthase is a causal mechanism and theragnostic target of an age-related hypertension endotype. PLoS Biol. 2020, 18, e3000885. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, K.; Kinoshita, H.; Kawashima, S.; Kawahito, S. Human Vascular Smooth Muscle Function and Oxidative Stress Induced by NADPH Oxidase with the Clinical Implications. Cells 2021, 10, 1947. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Gongora, M.C.; Harrison, D.G.; Lambeth, J.D.; Dikalov, S.; Griendling, K.K. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H673–H679. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; San Martin, A.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 480–487. [Google Scholar] [CrossRef]
- Bruder-Nascimento, T.; Chinnasamy, P.; Riascos-Bernal, D.F.; Cau, S.B.; Callera, G.E.; Touyz, R.M.; Tostes, R.C.; Sibinga, N.E. Angiotensin II induces Fat1 expression/activation and vascular smooth muscle cell migration via Nox1-dependent reactive oxygen species generation. J. Mol. Cell Cardiol. 2014, 66, 18–26. [Google Scholar] [CrossRef]
- Lozhkin, A.; Vendrov, A.E.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J. Mol. Cell Cardiol. 2017, 102, 10–21. [Google Scholar] [CrossRef]
- Xu, S.; Chamseddine, A.H.; Carrell, S.; Miller, F.J., Jr. Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol. 2014, 2, 642–650. [Google Scholar] [CrossRef]
- Camargo, L.L.; Montezano, A.C.; Hussain, M.; Wang, Y.; Zou, Z.; Rios, F.J.; Neves, K.B.; Alves-Lopes, R.; Awan, F.R.; Guzik, T.J.; et al. Central role of c-Src in NOX5- mediated redox signalling in vascular smooth muscle cells in human hypertension. Cardiovasc. Res. 2022, 118, 1359–1373. [Google Scholar] [CrossRef]
- Jay, D.B.; Papaharalambus, C.A.; Seidel-Rogol, B.; Dikalova, A.E.; Lassegue, B.; Griendling, K.K. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 329–335. [Google Scholar] [CrossRef]
- Montezano, A.C.; De Lucca Camargo, L.; Persson, P.; Rios, F.J.; Harvey, A.P.; Anagnostopoulou, A.; Palacios, R.; Gandara, A.C.P.; Alves-Lopes, R.; Neves, K.B.; et al. NADPH Oxidase 5 Is a Pro-Contractile Nox Isoform and a Point of Cross-Talk for Calcium and Redox Signaling-Implications in Vascular Function. J. Am. Heart Assoc. 2018, 7, e009388. [Google Scholar] [CrossRef] [PubMed]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Vagnozzi, R.J.; Johansen, A.K.; Maliken, B.D.; Prasad, V.; Boyer, J.G.; Brody, M.J.; Schips, T.; Kilian, K.K.; et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 2019, 4, e128722. [Google Scholar] [CrossRef] [PubMed]
- Meijles, D.N.; Pagano, P.J. Nox and Inflammation in the Vascular Adventitia. Hypertension 2016, 67, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Philip, J.L.; Razzaque, M.A.; Han, M.; Li, J.; Theccanat, T.; Xu, X.; Akhter, S.A. Regulation of mitochondrial oxidative stress by beta-arrestins in cultured human cardiac fibroblasts. Dis. Model. Mech. 2015, 8, 1579–1589. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Cross, A.R.; Jones, O.T.; Garcia, R.; Segal, A.W. The association of FAD with the cytochrome b-245 of human neutrophils. Biochem. J. 1982, 208, 759–763. [Google Scholar] [CrossRef]
- Volpp, B.D.; Nauseef, W.M.; Clark, R.A. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science 1988, 242, 1295–1297. [Google Scholar] [CrossRef]
- Nunoi, H.; Rotrosen, D.; Gallin, J.I.; Malech, H.L. Two forms of autosomal chronic granulomatous disease lack distinct neutrophil cytosol factors. Science 1988, 242, 1298–1301. [Google Scholar] [CrossRef]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z.G. NADPH Oxidases Are Essential for Macrophage Differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic. Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.U.; San Jose, G.; Pejenaute, A.; Landecho, M.F.; Diez, J.; Beloqui, O.; Fortuno, A.; Zalba, G. Association of phagocytic NADPH oxidase activity with hypertensive heart disease: A role for cardiotrophin-1? Hypertension 2014, 63, 468–474. [Google Scholar] [CrossRef]
- Emmerson, A.; Trevelin, S.C.; Mongue-Din, H.; Becker, P.D.; Ortiz, C.; Smyth, L.A.; Peng, Q.; Elgueta, R.; Sawyer, G.; Ivetic, A.; et al. Nox2 in regulatory T cells promotes angiotensin II-induced cardiovascular remodeling. J. Clin. Investig. 2018, 128, 3088–3101. [Google Scholar] [CrossRef]
- Matsushima, S.; Sadoshima, J. Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion. Antioxidants 2022, 11, 1069. [Google Scholar] [CrossRef]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O.; Hikoso, S.; Kashiwase, K.; Takeda, T.; Watanabe, T.; et al. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-kappa B. J. Biol. Chem. 2003, 278, 20770–20777. [Google Scholar] [CrossRef]
- Chen, K.; Pittman, R.N.; Popel, A.S. Nitric oxide in the vasculature: Where does it come from and where does it go? A quantitative perspective. Antioxid. Redox Signal. 2008, 10, 1185–1198. [Google Scholar] [CrossRef]
- Joyner, M.J.; Dietz, N.M. Nitric oxide and vasodilation in human limbs. J. Appl. Physiol. 1997, 83, 1785–1796. [Google Scholar] [CrossRef]
- Mongirdiene, A.; Skrodenis, L.; Varoneckaite, L.; Mierkyte, G.; Gerulis, J. Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies. Biomedicines 2022, 10, 602. [Google Scholar] [CrossRef] [PubMed]
- Leggat, J.; Bidault, G.; Vidal-Puig, A. Lipotoxicity: A driver of heart failure with preserved ejection fraction? Clin. Sci. 2021, 135, 2265–2283. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.; Kotter, S.; Grutzner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Hamdani, N.; Falcao-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Akers, S.R.; Trieu, L.; Ischiropoulos, H.; Doulias, P.T.; Tariq, A.; Vasim, I.; Koppula, M.R.; Syed, A.A.; Soto-Calderon, H.; et al. Heart Failure, Left Ventricular Remodeling, and Circulating Nitric Oxide Metabolites. J. Am. Heart Assoc. 2016, 5, e004133. [Google Scholar] [CrossRef] [PubMed]
- Zamani, P.; French, B.; Brandimarto, J.A.; Doulias, P.T.; Javaheri, A.; Chirinos, J.A.; Margulies, K.B.; Townsend, R.R.; Sweitzer, N.K.; Fang, J.C.; et al. Effect of Heart Failure With Preserved Ejection Fraction on Nitric Oxide Metabolites. Am. J. Cardiol. 2016, 118, 1855–1860. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Yoon, S.; Kim, M.; Lee, H.; Kang, G.; Bedi, K.; Margulies, K.B.; Jain, R.; Nam, K.I.; Kook, H.; Eom, G.H. S-Nitrosylation of Histone Deacetylase 2 by Neuronal Nitric Oxide Synthase as a Mechanism of Diastolic Dysfunction. Circulation 2021, 143, 1912–1925. [Google Scholar] [CrossRef]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef]
- Senni, M.; Greene, S.J.; Butler, J.; Fonarow, G.C.; Gheorghiade, M. Drug Development for Heart Failure With Preserved Ejection Fraction: What Pieces Are Missing From the Puzzle? Can. J. Cardiol. 2017, 33, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Massie, B.M.; Carson, P.E.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Zile, M.R.; Anderson, S.; Donovan, M.; Iverson, E.; Staiger, C.; et al. Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med. 2008, 359, 2456–2467. [Google Scholar] [CrossRef] [PubMed]
- Kitzman, D.W.; Hundley, W.G.; Brubaker, P.H.; Morgan, T.M.; Moore, J.B.; Stewart, K.P.; Little, W.C. A randomized double-blind trial of enalapril in older patients with heart failure and preserved ejection fraction: Effects on exercise tolerance and arterial distensibility. Circ. Heart Fail. 2010, 3, 477–485. [Google Scholar] [CrossRef]
- Cleland, J.G.; Tendera, M.; Adamus, J.; Freemantle, N.; Polonski, L.; Taylor, J.; Investigators, P.-C. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 2006, 27, 2338–2345. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef]
- Redfield, M.M.; Anstrom, K.J.; Levine, J.A.; Koepp, G.A.; Borlaug, B.A.; Chen, H.H.; LeWinter, M.M.; Joseph, S.M.; Shah, S.J.; Semigran, M.J.; et al. Isosorbide Mononitrate in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2015, 373, 2314–2324. [Google Scholar] [CrossRef]
- Borlaug, B.A.; Anstrom, K.J.; Lewis, G.D.; Shah, S.J.; Levine, J.A.; Koepp, G.A.; Givertz, M.M.; Felker, G.M.; LeWinter, M.M.; Mann, D.L.; et al. Effect of Inorganic Nitrite vs Placebo on Exercise Capacity Among Patients With Heart Failure With Preserved Ejection Fraction: The INDIE-HFpEF Randomized Clinical Trial. JAMA 2018, 320, 1764–1773. [Google Scholar] [CrossRef]
- Armstrong, P.W.; Lam, C.S.P.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; O'Connor, C.M.; Pieske, B.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; et al. Effect of Vericiguat vs Placebo on Quality of Life in Patients With Heart Failure and Preserved Ejection Fraction: The VITALITY-HFpEF Randomized Clinical Trial. JAMA 2020, 324, 1512–1521. [Google Scholar] [CrossRef]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef]
- Kalra, S. Sodium Glucose Co-Transporter-2 (SGLT2) Inhibitors: A Review of Their Basic and Clinical Pharmacology. Diabetes Ther. 2014, 5, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.E.; Windsor, S.L.; Borlaug, B.A.; Kitzman, D.W.; Shah, S.J.; Tang, F.; Khariton, Y.; Malik, A.O.; Khumri, T.; Umpierrez, G.; et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: A multicenter randomized trial. Nat. Med. 2021, 27, 1954–1960. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Tang, F.; Khariton, Y.; Husain, M.; Inzucchi, S.E.; McGuire, D.K.; Pitt, B.; Scirica, B.M.; Austin, B.; et al. Dapagliflozin Effects on Biomarkers, Symptoms, and Functional Status in Patients With Heart Failure With Reduced Ejection Fraction: The DEFINE-HF Trial. Circulation 2019, 140, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Waddingham, M.T.; Sonobe, T.; Tsuchimochi, H.; Edgley, A.J.; Sukumaran, V.; Chen, Y.C.; Hansra, S.S.; Schwenke, D.O.; Umetani, K.; Aoyama, K.; et al. Diastolic dysfunction is initiated by cardiomyocyte impairment ahead of endothelial dysfunction due to increased oxidative stress and inflammation in an experimental prediabetes model. J. Mol. Cell Cardiol. 2019, 137, 119–131. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Xue, M.; Li, T.; Wang, Y.; Chang, Y.; Cheng, Y.; Lu, Y.; Liu, X.; Xu, L.; Li, X.; Yu, X.; et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci. 2019, 133, 1705–1720. [Google Scholar] [CrossRef]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Chowdhury, F.I.; Nayan, S.I.; Rahman, M.M.; Khan, F.; Subhan, N.; et al. Canagliflozin attenuates isoprenaline-induced cardiac oxidative stress by stimulating multiple antioxidant and anti-inflammatory signaling pathways. Sci. Rep. 2020, 10, 14459. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Akoumianakis, I.; Badi, I.; Akawi, N.; Kotanidis, C.P.; Polkinghorne, M.; Stadiotti, I.; Sommariva, E.; Antonopoulos, A.S.; Carena, M.C.; et al. Effects of canagliflozin on human myocardial redox signalling: Clinical implications. Eur. Heart J. 2021, 42, 4947–4960. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Patients with HFpEF | Effect |
|---|---|
| Myocardium | • Trend towards increased NOX2 expression in cardiac macrophages [17] • Increased myocardial H2O2 levels [17,25] • Increased myocardial lipid peroxidation [25] |
| Serum | • Elevated levels of derivatives of reactive oxidative metabolites (DROMs) in patients with HFpEF with HF-related events [27,28] • Increased thiobarbituric acid reactive substances (TBARS, biomarker of lipid peroxidation) [19] |
| Peripheral Blood Monocytes | • Increased NOX1 and NOX4 mRNA levels correlate with diastolic dysfunction in patients with HFpEF [29] |
| Pre-clinical HFpEF Models | Effect |
| HFD †/L-NAME (mice) | • Increased myocardial Nox4 protein expression [30] |
| Unilateral nephrectomy and aldosterone infusion (mice) | • Increased myocardial oxidative stress (DHE fluorescence) [23,24] |
| Obese ZSF1/ZDF rats | • Increased cardiac macrophage Nox2 [17] • Increased myocardial H2O2 levels [17,25] • Increased myocardial lipid peroxidation [25] |
| DOCA/Western diet †† (Göttingen miniswine) | • Increased 8-isoprostane levels in plasma [20] |
| Streptozotocin, HFD †††, and renal artery embolization (Yorkshire x Landrace swine) | • Increased myocardial NADPH-stimulated superoxide production [26] |
| NOX Isoform | Cell Types | Cardiac | Vascular | |||
|---|---|---|---|---|---|---|
| Hypertrophy | Fibrosis | Inflammation | Hypertension | Atherosclerosis | ||
| NOX1 | CMs, ECs, VSMCs, Macrophages | + | + | + | + | + |
| NOX2 | CMs, ECs, VSMCs, Macrophages, Fibroblasts | + | + | + | + | + |
| NOX4 | CMs, VSMCs, Macrophages, Fibroblasts | + | + | + | + | |
| ECs | − | − | − | − | − | |
| NOX5 | CMs, ECs, VSMCs | + | + | + | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teuber, J.P.; Essandoh, K.; Hummel, S.L.; Madamanchi, N.R.; Brody, M.J. NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction. Antioxidants 2022, 11, 1822. https://doi.org/10.3390/antiox11091822
Teuber JP, Essandoh K, Hummel SL, Madamanchi NR, Brody MJ. NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction. Antioxidants. 2022; 11(9):1822. https://doi.org/10.3390/antiox11091822
Chicago/Turabian StyleTeuber, James P., Kobina Essandoh, Scott L. Hummel, Nageswara R. Madamanchi, and Matthew J. Brody. 2022. "NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction" Antioxidants 11, no. 9: 1822. https://doi.org/10.3390/antiox11091822
APA StyleTeuber, J. P., Essandoh, K., Hummel, S. L., Madamanchi, N. R., & Brody, M. J. (2022). NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction. Antioxidants, 11(9), 1822. https://doi.org/10.3390/antiox11091822