
COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Mitochondria Isolation
2.3. High Resolution Respirometry and ROS Production Evaluation
2.4. Western Blot Analysis
2.5. Mitochondrial Membrane Potential (ΔΨm) Measurement
2.6. Transmission Electron Microscopy (TEM)
2.7. RNA Isolation, and Quantitative RT-PCR
2.8. Immunofluorescence
2.9. Statistics
3. Results
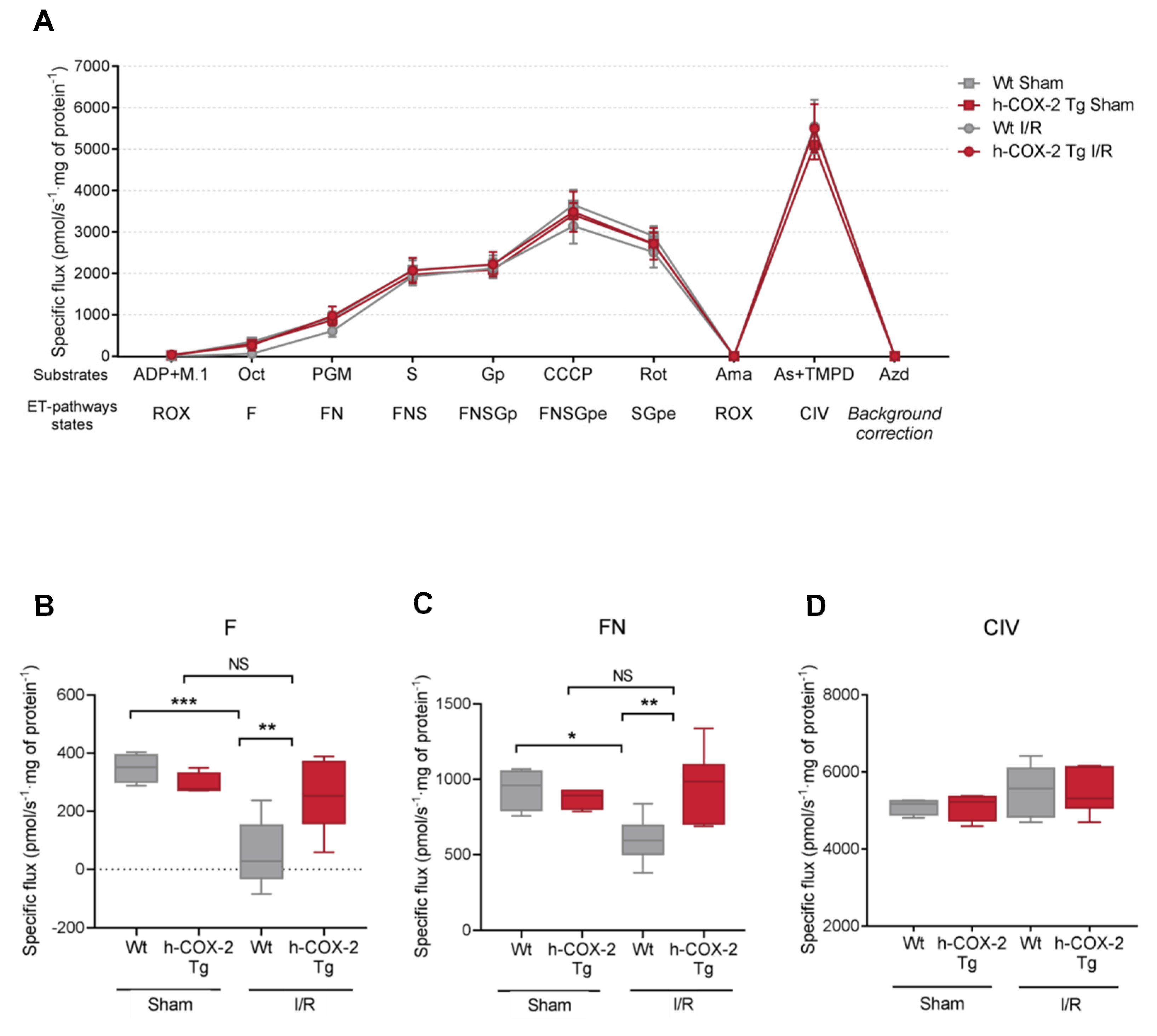
3.1. Mitochondrial Respiration Is Higher When COX-2 Is Overexpressed after I/R
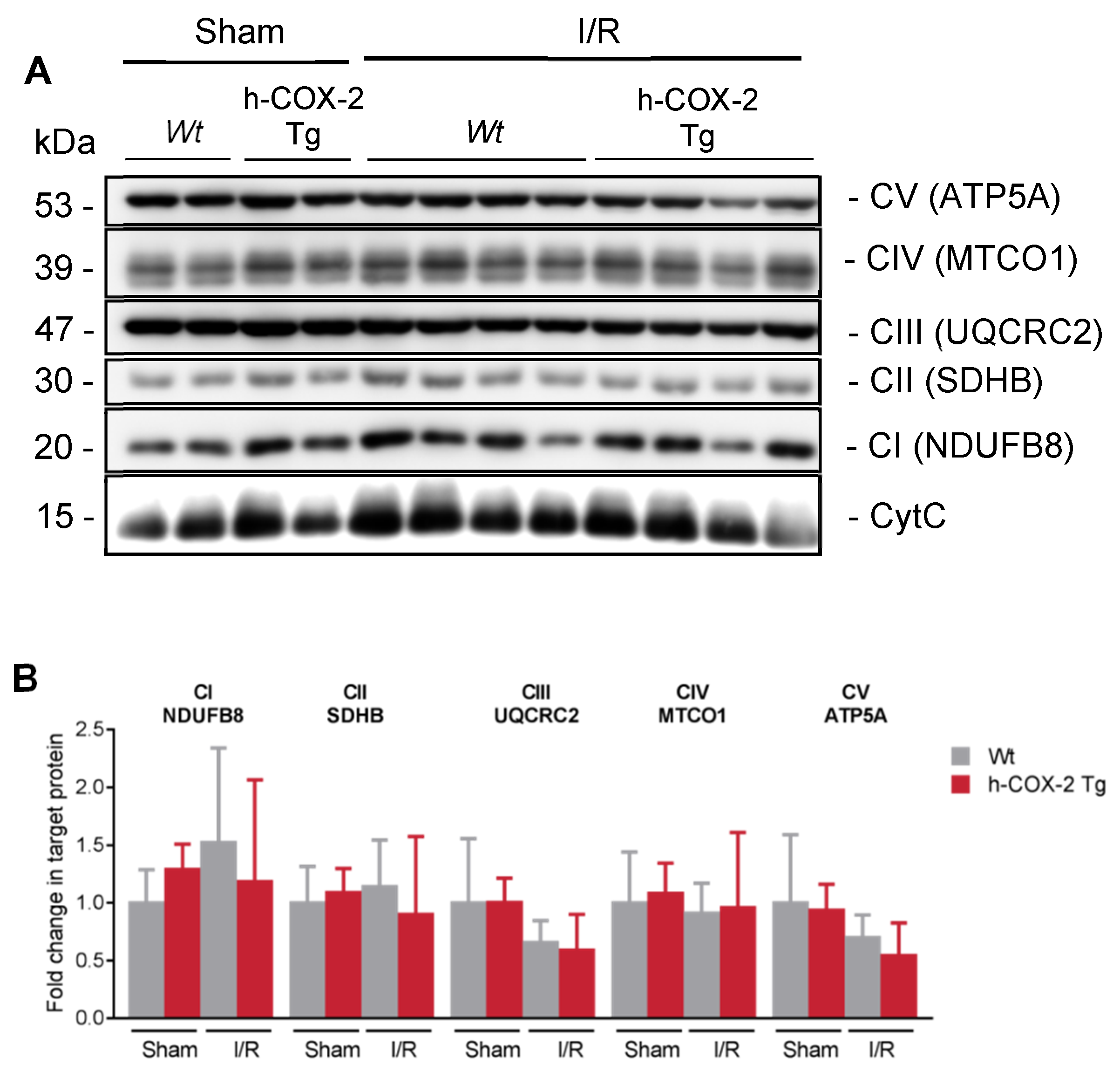
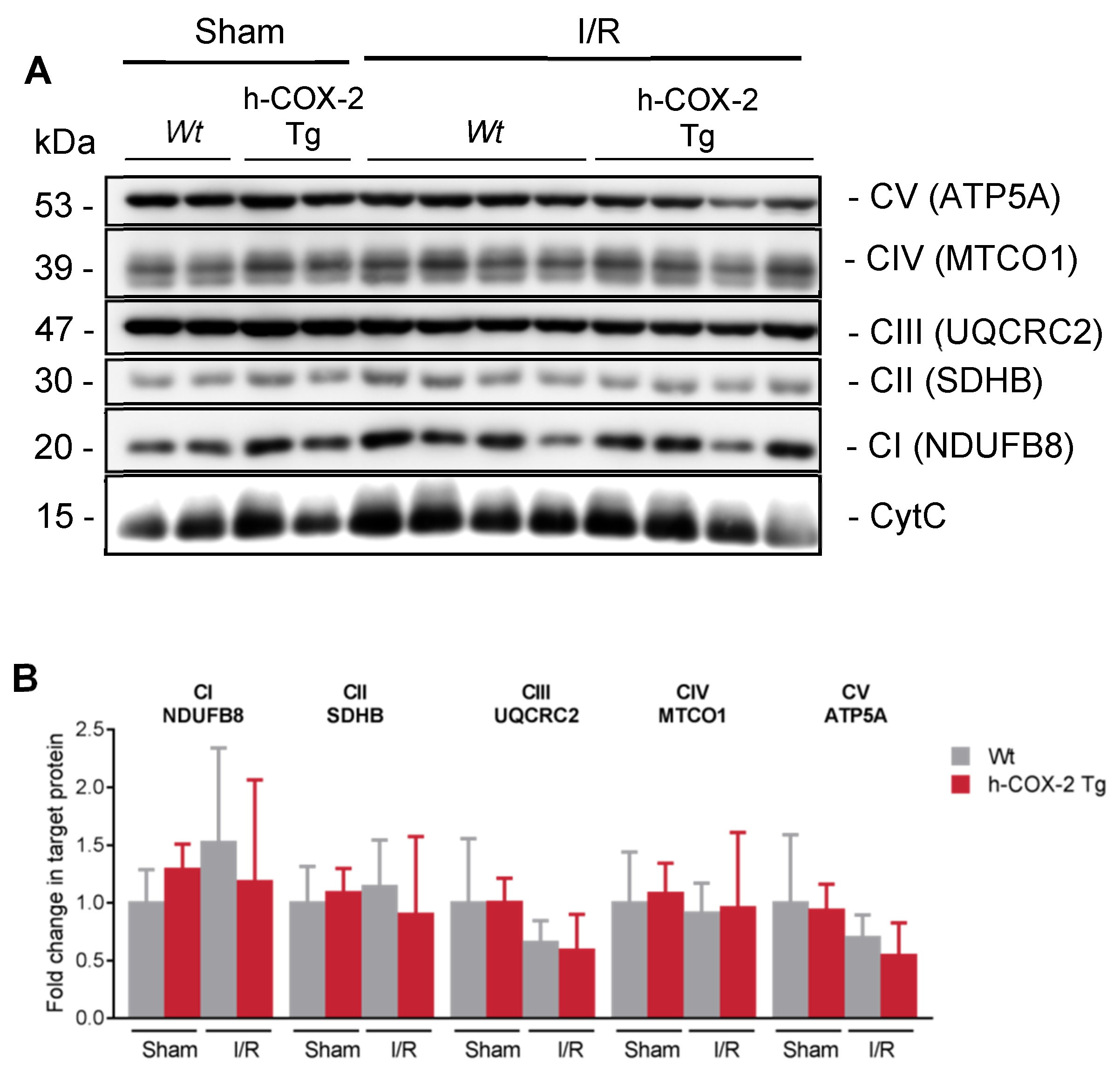
3.2. Higher Mitochondrial Activity Is Not Due to an Increased Presence of ETC Complexes or to a Better Association in Supercomplexes
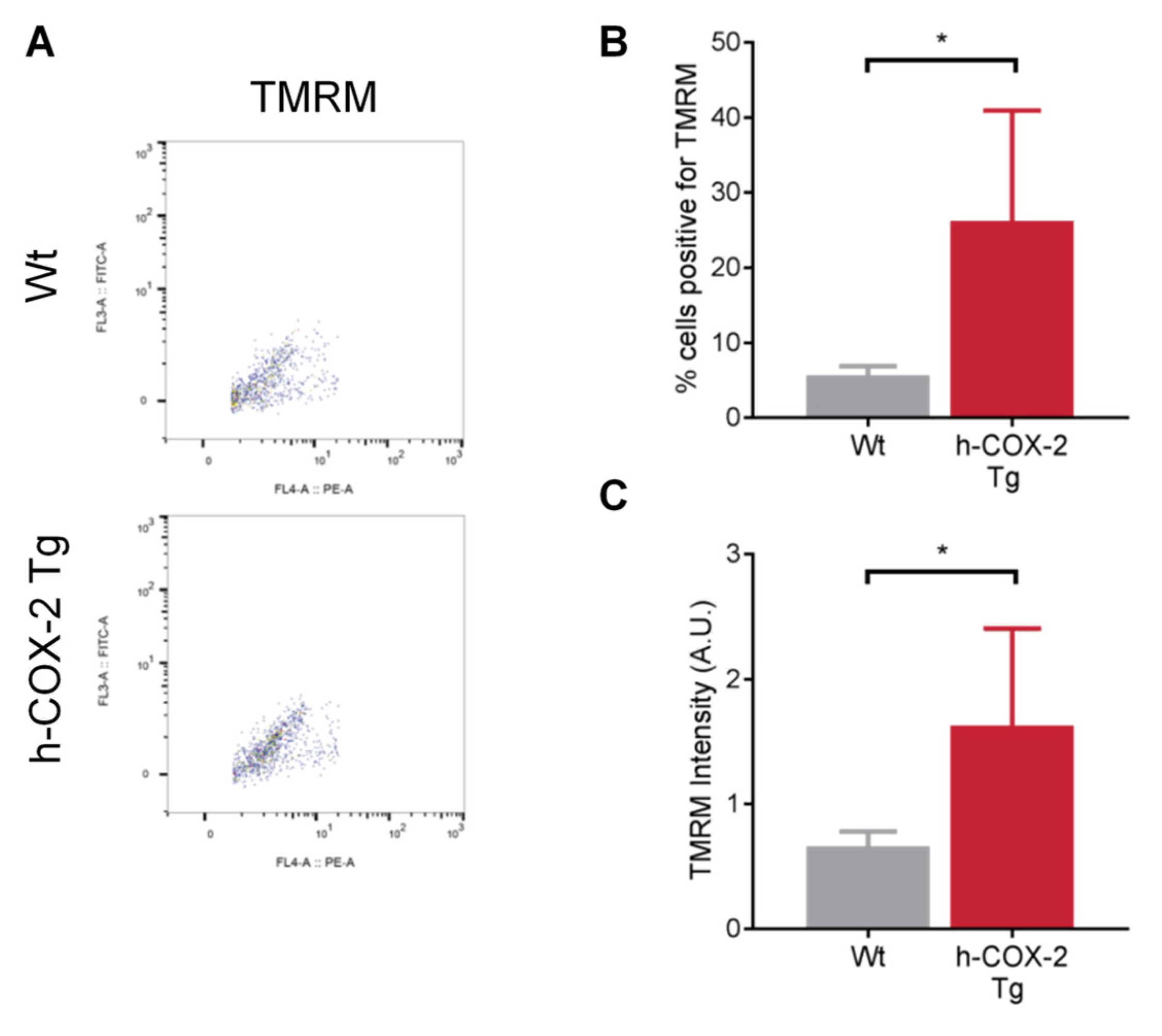
3.3. h-COX-2 Tg-Derived Mitochondria Have an Intact Mitochondrial Membrane Potential (ΔΨm) after I/R Compared to Wt
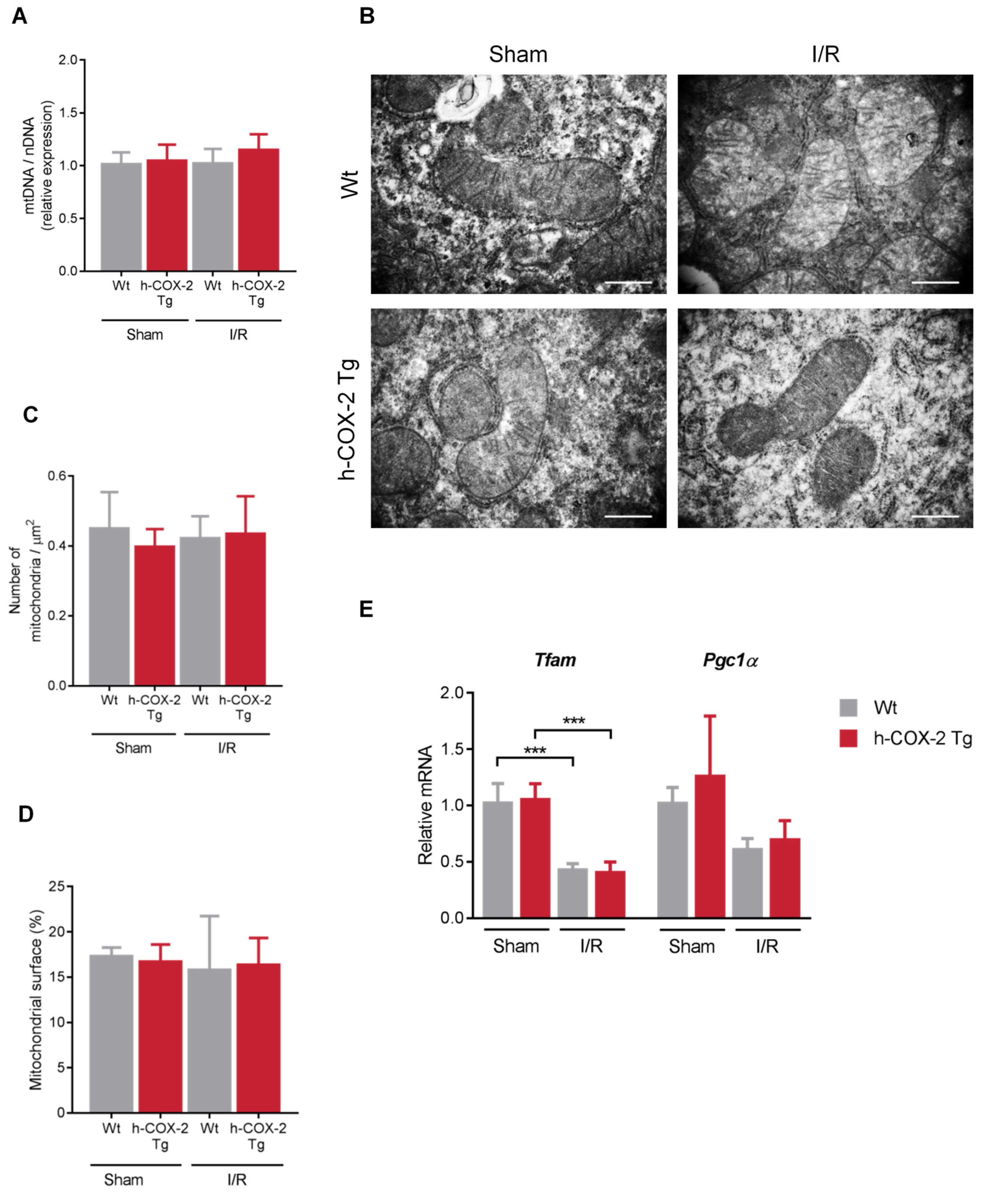
3.4. Mitochondrial Aspect and Functionality Are Not Altered Because of the Overexpression of COX-2 after I/R
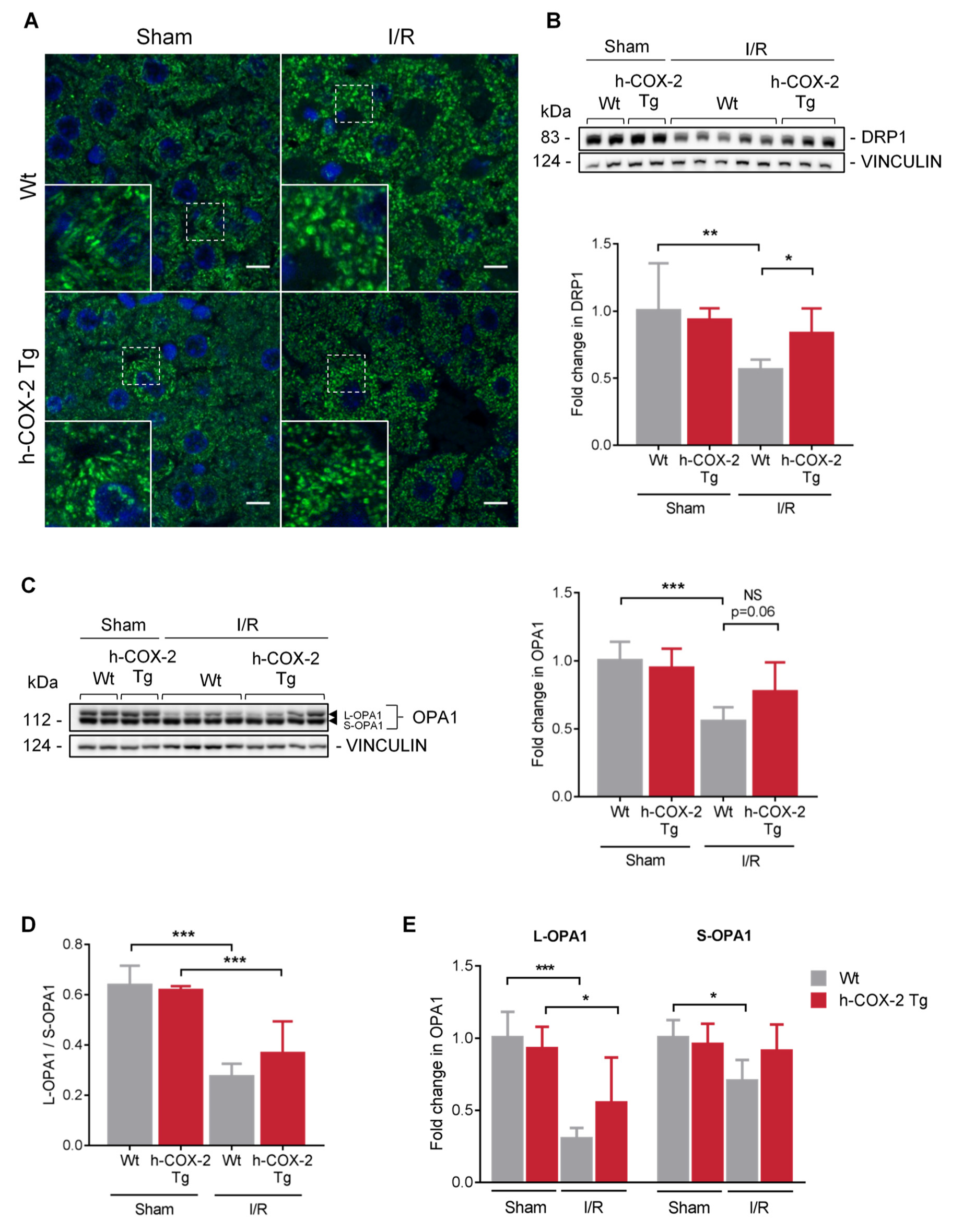
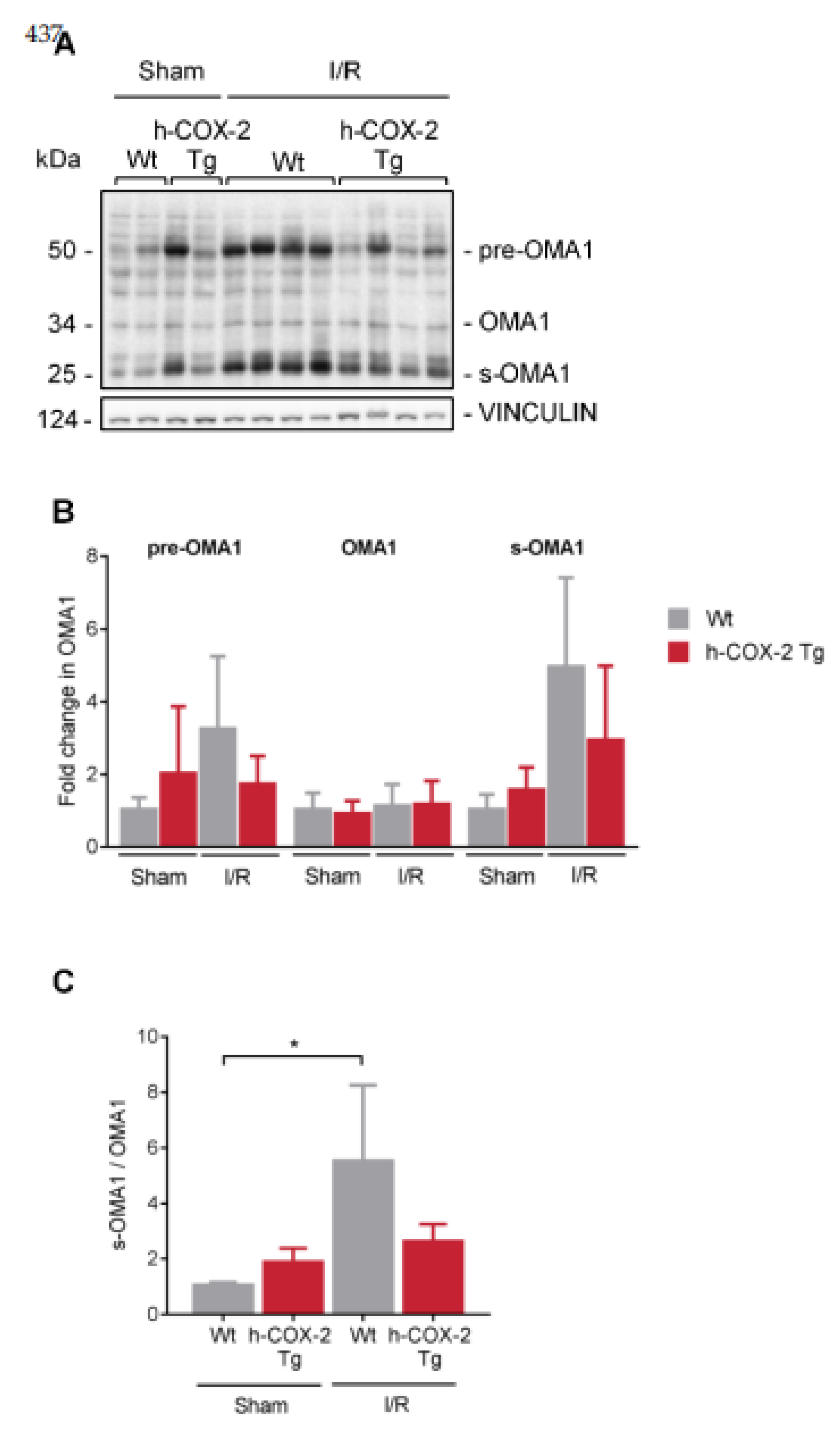
3.5. OPA1 Processing Is Altered through OMA1 Activity in Wt Mice after I/R
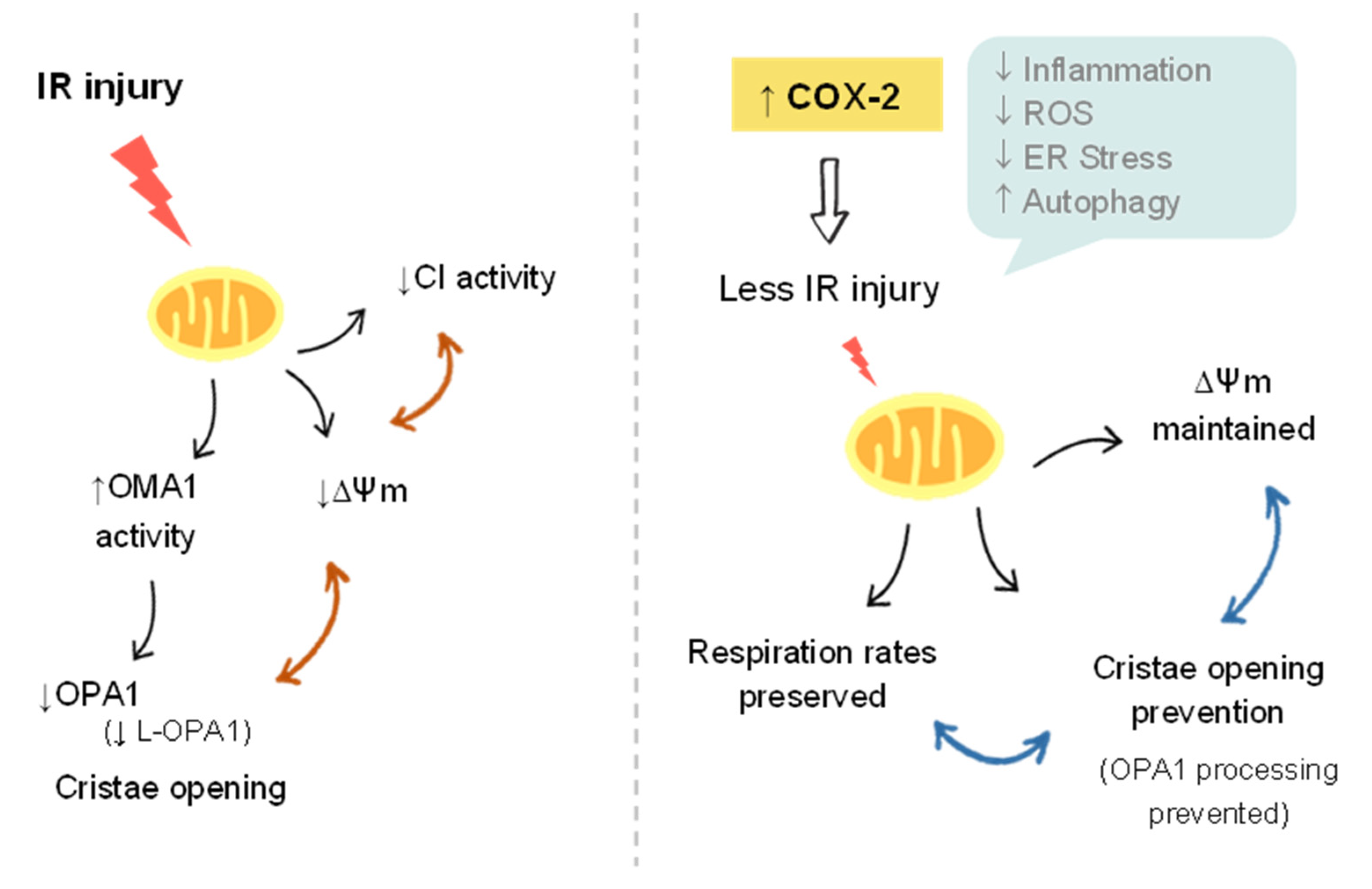
4. Discussion
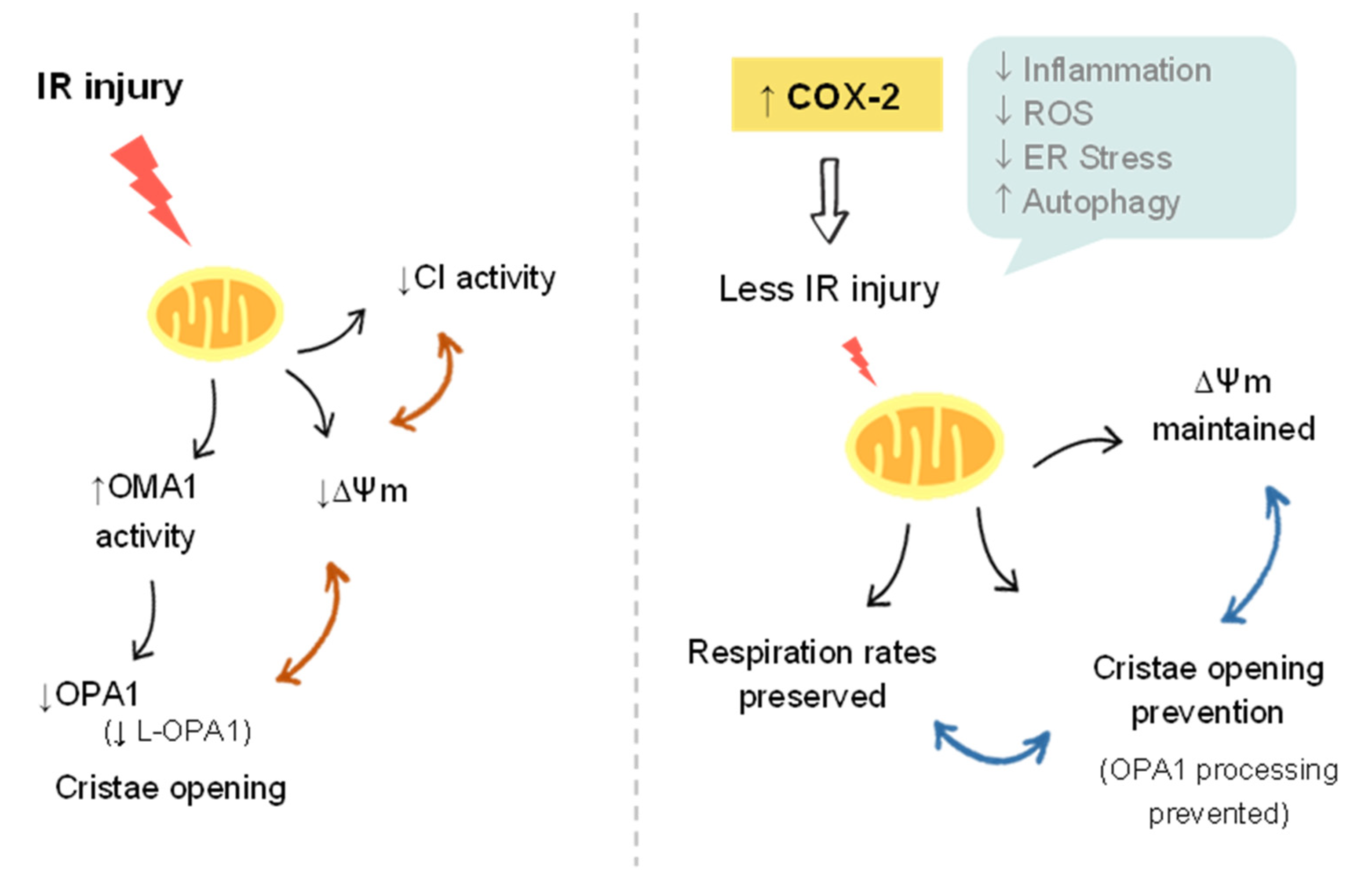
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barahman, M.; Asp, P.; Roy-Chowdhury, N.; Kinkhabwala, M.; Roy-Chowdhury, J.; Kabarriti, R.; Guha, C. Hepatocyte Transplantation: Quo Vadis? Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Molecular Mechanisms of Hepatic Ischemia-Reperfusion Injury and Preconditioning. Am. J. Physiol. Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef] [PubMed]
- Coito, J.; Duarte, S.; Moore, C.; Busuttil, R.W.; Hamada, A.T.; Tsuchihashi, S.; Avanesyan, A. Injury Recruitment in Hepatic Ischemia/Reperfusion Immune Responses and Impairs Neutrophil Cyclooxygenase-2 Deficiency Enhances Th2. J. Immunol. Ref. 2008, 180, 1843–1853. [Google Scholar] [CrossRef]
- Demiryilmaz, I.; Turan, M.I.; Kisaoglu, A.; Gulapoglu, M.; Yilmaz, I.; Suleyman, H. Protective Effect of Nimesulide against Hepatic Ischemia/Reperfusion Injury in Rats: Effects on Oxidant/Antioxidants, DNA Mutation and COX-1/COX-2 Levels. Pharmacol. Rep. 2014, 66, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Schumacker, P.T. Oxygen Sensing by Mitochondria at Complex III: The Paradox of Increased Reactive Oxygen Species during Hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Klinman, J.P. How Do Enzymes Activate Oxygen without Inactivating Themselves? Acc. Chem. Res. 2007, 40, 325–333. [Google Scholar] [CrossRef]
- Selzner, N. Protective Strategies against Ischemic Injury of the Liver. Gastroenterology 2003, 125, 917–936. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Peralta, C.; Rosello-Catafau, J.; Palmeira, C.M. Prevention of I/R Injury in Fatty Livers by Ischemic Preconditioning Is Associated with Increased Mitochondrial Tolerance: The Key Role of ATPsynthase and Mitochondrial Permeability Transition. Transpl. Int. 2009, 22, 1081–1090. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial Reactive Oxygen Species: A Double Edged Sword in Ischemia/Reperfusion vs Preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 Processing in Cell Death and Disease—The Long and Short of It. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and Dysfunctions of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 Requires Mitofusin 1 to Promote Mitochondrial Fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Pagliuso, A.; Cossart, P.; Stavru, F. The Ever-Growing Complexity of the Mitochondrial Fission Machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 Regulates Mitochondrial Dynamics through Inhibition of the Fusion Machinery. EMBO J. 2019, 38, 1–21. [Google Scholar] [CrossRef]
- Griparic, L.; Kanazawa, T.; Van Der Bliek, A.M. Regulation of the Mitochondrial Dynamin-like Protein Opa1 by Proteolytic Cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 Processing Controls Mitochondrial Fusion and Is Regulated by MRNA Splicing, Membrane Potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Stiburek, L.; Cesnekova, J.; Kostkova, O.; Fornuskova, D.; Vinsova, K.; Wenchich, L.; Houstek, J.; Zeman, J. YME1L Controls the Accumulation of Respiratory Chain Subunits and Is Required for Apoptotic Resistance, Cristae Morphogenesis, and Cell Proliferation. Mol. Biol. Cell 2012, 23, 1010–1023. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. Bioenerg. Commun. 2020, 2020. [Google Scholar] [CrossRef]
- Davila, M.P.; Muñoz, P.M.; Tapia, J.A.; Ferrusola, C.O.; Da Silva, C.C.B.; Peña, F.J. Inhibition of Mitochondrial Complex I Leads to Decreased Motility and Membrane Integrity Related to Increased Hydrogen Peroxide and Reduced ATP Production, While the Inhibition of Glycolysis Has Less Impact on Sperm Motility. PLoS ONE 2015, 10, e0138777. [Google Scholar] [CrossRef]
- Callejas, N.A.; Boscá, L.; Williams, C.S.; DuBois, R.N.; Martín-Sanz, P. Regulation of Cyclooxygenase 2 Expression in Hepatocytes by CCAAT/Enhancer-Binding Proteins. Gastroenterology 2000, 119, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sanz, P.; Callejas, N.A.; Casado, M.; Díaz-Guerra, M.J.M.; Boscá, L. Expression of Cyclooxygenase-2 in Foetal Rat Hepatocytes Stimulated with Lipopolysaccharide and pro-Inflammatory Cytokines. Br. J. Pharmacol. 1998, 125, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Casado, M.; Mollá, B.; Roy, R.; Fernández-Martínez, A.; Cucarella, C.; Mayoral, R.; Boscá, L.; Martín-Sanz, P. Protection against Fas-Induced Liver Apoptosis in Transgenic Mice Expressing Cyclooxygenase 2 in Hepatocytes. Hepatology 2007, 45, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.M.; Brand, M.D.; Krauss, S.; Meisel, C.; Vergin, H.; Burmester, G.-R.; Buttgereit, F. Nonsteroidal Antiinflammatory Drugs and a Selective Cyclooxygenase 2 Inhibitor Uncouple Mitochondria in Intact Cells. Arthritis Rheum. 2003, 48, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chen, H.; Wang, C.; Xu, H.; Liu, F.; Guo, M.; Wang, Q.; Shi, X. Flurbiprofen, a Cyclooxygenase Inhibitor, Protects Mice from Hepatic Ischemia/Reperfusion Injury by Inhibiting GSK-3β Signaling and Mitochondrial Permeability Transition. Mol. Med. 2012, 18, 1128–1135. [Google Scholar] [CrossRef]
- Tolba, R.H.; Fet, N.; Yonezawa, K.; Taura, K.; Nakajima, A.; Hata, K.; Okamura, Y.; Uchinami, H.; Klinge, U.; Minor, T.; et al. Role of Preferential Cyclooxygenase-2 Inhibition by Meloxicam in Ischemia/Reperfusion Injury of the Rat Liver. Eur. Surg. Res. 2014, 53, 11–24. [Google Scholar] [CrossRef]
- Motiño, O.; Francés, D.E.; Casanova, N.; Fuertes-Agudo, M.; Cucarella, C.; Flores, J.M.; Vallejo-Cremades, M.T.; Olmedilla, L.; Pérez Peña, J.; Bañares, R.; et al. Protective Role of Hepatocyte Cyclooxygenase-2 Expression against Liver Ischemia–Reperfusion Injury in Mice. Hepatology 2019, 70, 650–665. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle Isolation: Functional Mitochondria from Mouse Liver, Muscle and Cultured Filroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. Methods Mol. Biol. 2018, 1782, 31–70. [Google Scholar]
- Lam, J.; Katti, P.; Biete, M.; Mungai, M.; AshShareef, S.; Neikirk, K.; Garza Lopez, E.; Vue, Z.; Christensen, T.A.; Beasley, H.K.; et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells 2021, 10, 2177. [Google Scholar] [CrossRef]
- van Zutphen, T.; Ciapaite, J.; Bloks, V.W.; Ackereley, C.; Gerding, A.; Jurdzinski, A.; de Moraes, R.A.; Zhang, L.; Wolters, J.C.; Bischoff, R.; et al. Malnutrition-Associated Liver Steatosis and ATP Depletion Is Caused by Peroxisomal and Mitochondrial Dysfunction. J. Hepatol. 2016, 65, 1198–1208. [Google Scholar] [CrossRef]
- Schägger, H. Respiratory Chain Supercomplexes. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2001, 52, 119–128. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin Alleviates Liver Ischemia-Reperfusion Injury by Inhibiting Excessive Mitochondrial Fission, Promoting Mitochondrial Biogenesis and Decreasing Oxidative Stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Manjarrés-Raza, I.; Vicente-Gutiérrez, C.; Corrado, M.; Bolaños, J.P.; Scorrano, L. Opa1 Relies on Cristae Preservation and ATP Synthase to Curtail Reactive Oxygen Species Accumulation in Mitochondria. Redox Biol. 2021, 41, 101944. [Google Scholar] [CrossRef]
- Labbé, K.; Murley, A.; Nunnari, J. Determinants and Functions of Mitochondrial Behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391. [Google Scholar] [CrossRef]
- Zhou, Y.; Long, Q.; Wu, H.; Li, W.; Qi, J.; Wu, Y.; Xiang, G.; Tang, H.; Yang, L.; Chen, K.; et al. Topology-Dependent, Bifurcated Mitochondrial Quality Control under Starvation. Autophagy 2020, 16, 562–574. [Google Scholar] [CrossRef]
- Liu, X.; Hajnó, G. Altered Fusion Dynamics Underlie Unique Morphological Changes in Mitochondria during Hypoxia-Reoxygenation Stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial Energetics in the Kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Ong, S.-B.; Subrayan, S.; Lim, Y.S.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart Against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- Bouche, L.; Kamel, R.; Tamareille, S.; Garcia, G.; Villedieu, C.; Pillot, B.; Gueguen, N.; Chehaitly, A.; Manuel Chao de la Barca, J.; Beaumont, J.; et al. DRP1 Haploinsufficiency Attenuates Cardiac Ischemia/Reperfusion Injuries. PLoS ONE 2021, 16, e0248554. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Tummers, B.; Green, D.R. Crashing the Computer: Apoptosis vs. Necroptosis in Neuroinflammation. Cell Death Differ. 2019, 26, 41–52. [Google Scholar] [CrossRef]
- Krause, J.; Löser, A.; Lemoine, M.D.; Christ, T.; Scherschel, K.; Meyer, C.; Blankenberg, S.; Zeller, T.; Eschenhagen, T.; Stenzig, J. Rat Atrial Engineered Heart Tissue: A New In Vitro Model to Study Atrial Biology. Basic Res. Cardiol. 2018, 113, 41. [Google Scholar] [CrossRef]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vázquez, J.; Enríquez, J.A.; Gerle, C.; et al. The Cristae Modulator Optic Atrophy 1 Requires Mitochondrial ATP Synthase Oligomers to Safeguard Mitochondrial Function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef]
- Jiang, S.J.; Li, W.; An, W. Adenoviral Gene Transfer of Hepatic Stimulator Substance Confers Resistance against Hepatic Ischemia-Reperfusion Injury by Improving Mitochondrial Function. Hum. Gene Ther. 2013, 24, 443–456. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 Processing and Mitochondrial Fusion by M-AAA Protease Isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; Van Der Bliek, A.M. Inducible Proteolytic Inactivation of OPA1 Mediated by the OMA1 Protease in Mammalian Cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Quirós, P.M.; Ramsay, A.J.; Sala, D.; Fernández-Vizarra, E.; Rodríguez, F.; Peinado, J.R.; Fernández-García, M.S.; Vega, J.A.; Enríquez, J.A.; Zorzano, A.; et al. Loss of Mitochondrial Protease OMA1 Alters Processing of the GTPase OPA1 and Causes Obesity and Defective Thermogenesis in Mice. EMBO J. 2012, 31, 2117–2133. [Google Scholar] [CrossRef]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schägger, H. Supercomplexes and Subcomplexes of Mitochondrial Oxidative Phosphorylation. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1066–1072. [Google Scholar] [CrossRef]
- García-Poyatos, C.; Cogliati, S.; Calvo, E.; Hernansanz-Agustín, P.; Lagarrigue, S.; Magni, R.; Botos, M.; Langa, X.; Amati, F.; Vázquez, J.; et al. Scaf1 Promotes Respiratory Supercomplexes and Metabolic Efficiency in Zebrafish. EMBO Rep. 2020, 21, e50287. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Peralta, C.; Bartrons, R.; Serafin, A.; Blázquez, C.; Guzmán, M.; Prats, N.; Xaus, C.; Cutillas, B.; Gelpí, E.; Roselló-Catafau, J. Adenosine Monophosphate-Activated Protein Kinase Mediates the Protective Effects of Ischemic Preconditioning on Hepatic Ischemia-Reperfusion Injury in the Rat. Hepatology 2001, 34, 1164–1173. [Google Scholar] [CrossRef]
- Francés, D.E.; Motiño, O.; Agrá, N.; González-Rodríguez, Á.; Fernández-Álvarez, A.; Cucarella, C.; Mayoral, R.; Castro-Sánchez, L.; García-Casarrubios, E.; Boscá, L.; et al. Hepatic Cyclooxygenase-2 Expression Protects against Diet-Induced Steatosis, Obesity, and Insulin Resistance. Diabetes 2015, 64, 1522–1531. [Google Scholar] [CrossRef]
- Lai, Y.; Lin, P.; Chen, M.; Zhang, Y.; Chen, J.; Zheng, M.; Liu, J.; Du, H.; Chen, R.; Pan, X.; et al. Restoration of L-OPA1 Alleviates Acute Ischemic Stroke Injury in Rats via Inhibiting Neuronal Apoptosis and Preserving Mitochondrial Function. Redox Biol. 2020, 34, 101503. [Google Scholar] [CrossRef]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial Rhomboid PARL Regulates Cytochrome c Release during Apoptosis via OPA1-Dependent Cristae Remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuertes-Agudo, M.; Luque-Tévar, M.; Cucarella, C.; Brea, R.; Boscá, L.; Quintana-Cabrera, R.; Martín-Sanz, P.; Casado, M. COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury. Antioxidants 2022, 11, 1724. https://doi.org/10.3390/antiox11091724
Fuertes-Agudo M, Luque-Tévar M, Cucarella C, Brea R, Boscá L, Quintana-Cabrera R, Martín-Sanz P, Casado M. COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury. Antioxidants. 2022; 11(9):1724. https://doi.org/10.3390/antiox11091724
Chicago/Turabian StyleFuertes-Agudo, Marina, María Luque-Tévar, Carme Cucarella, Rocío Brea, Lisardo Boscá, Rubén Quintana-Cabrera, Paloma Martín-Sanz, and Marta Casado. 2022. "COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury" Antioxidants 11, no. 9: 1724. https://doi.org/10.3390/antiox11091724
APA StyleFuertes-Agudo, M., Luque-Tévar, M., Cucarella, C., Brea, R., Boscá, L., Quintana-Cabrera, R., Martín-Sanz, P., & Casado, M. (2022). COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury. Antioxidants, 11(9), 1724. https://doi.org/10.3390/antiox11091724