
Metabolic and Structural Insights into Hydrogen Sulfide Mis-Regulation in Enterococcus faecalis

 , ,
, ,  and
and
Abstract
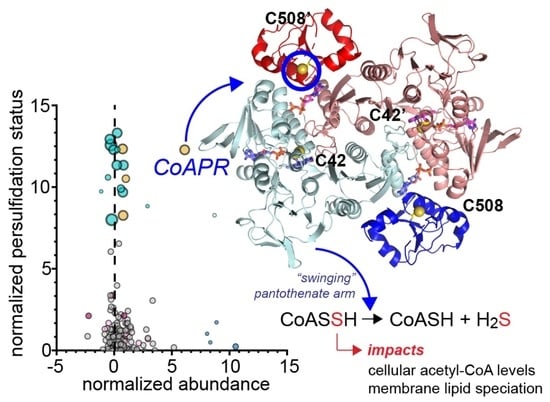
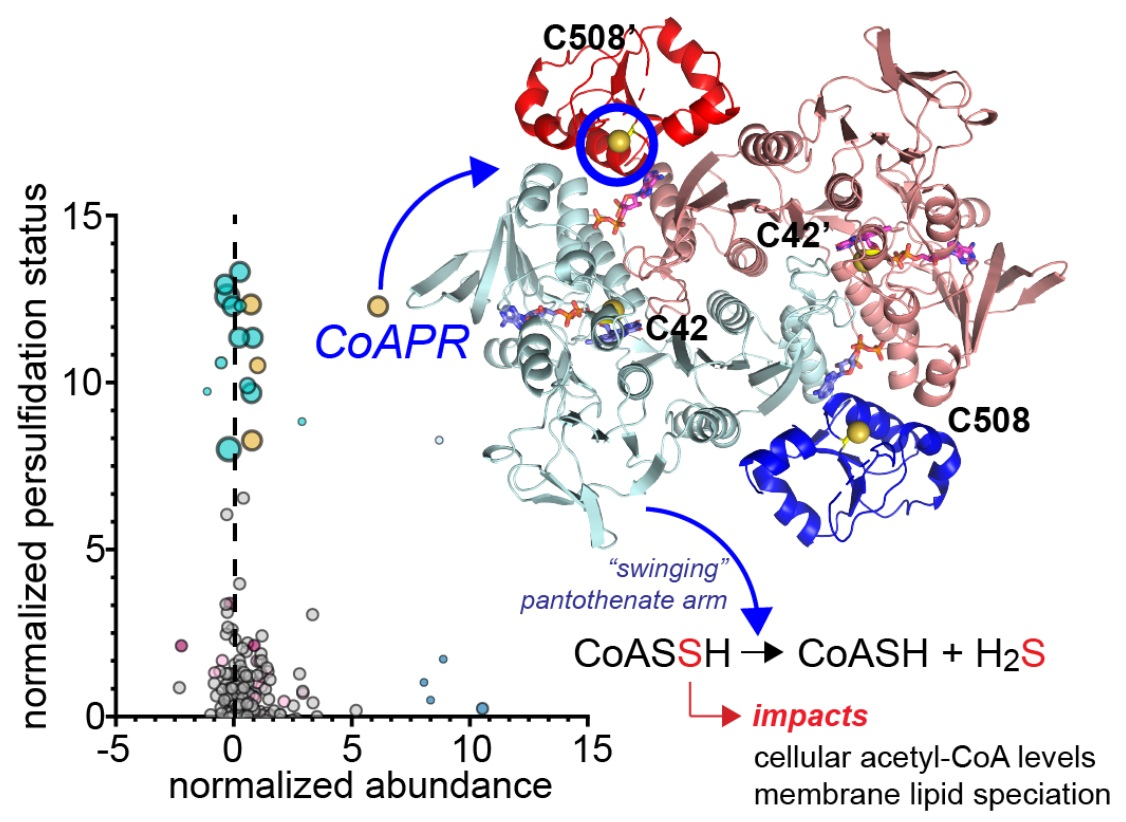
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Growth of Enterococcus faecalis
2.2. Proteomic Analysis of Wild Type E. faecalis before and after the Addition of Exogenous Na2S
2.3. Enrichment and Identification of S-Sulfurated Proteins in E. faecalis
2.4. LC-MS/MS Analysis of Proteome and S-Sulfurated Proteins
2.5. Extraction and Measurement of Cellular Fatty Acids
2.6. Quantitation of Acetyl-CoA
2.7. Cloning and Purification of Recombinant Ef Pta
2.8. Enzyme Assays of Ef Pta Activity
2.9. Statistical Rationale and Bioinformatics Analysis
2.10. Cloning and Purification of Recombinant EfCoAPR
2.11. X-ray Crystallography
2.12. Molecular Dynamics (MD) Simulations
3. Results
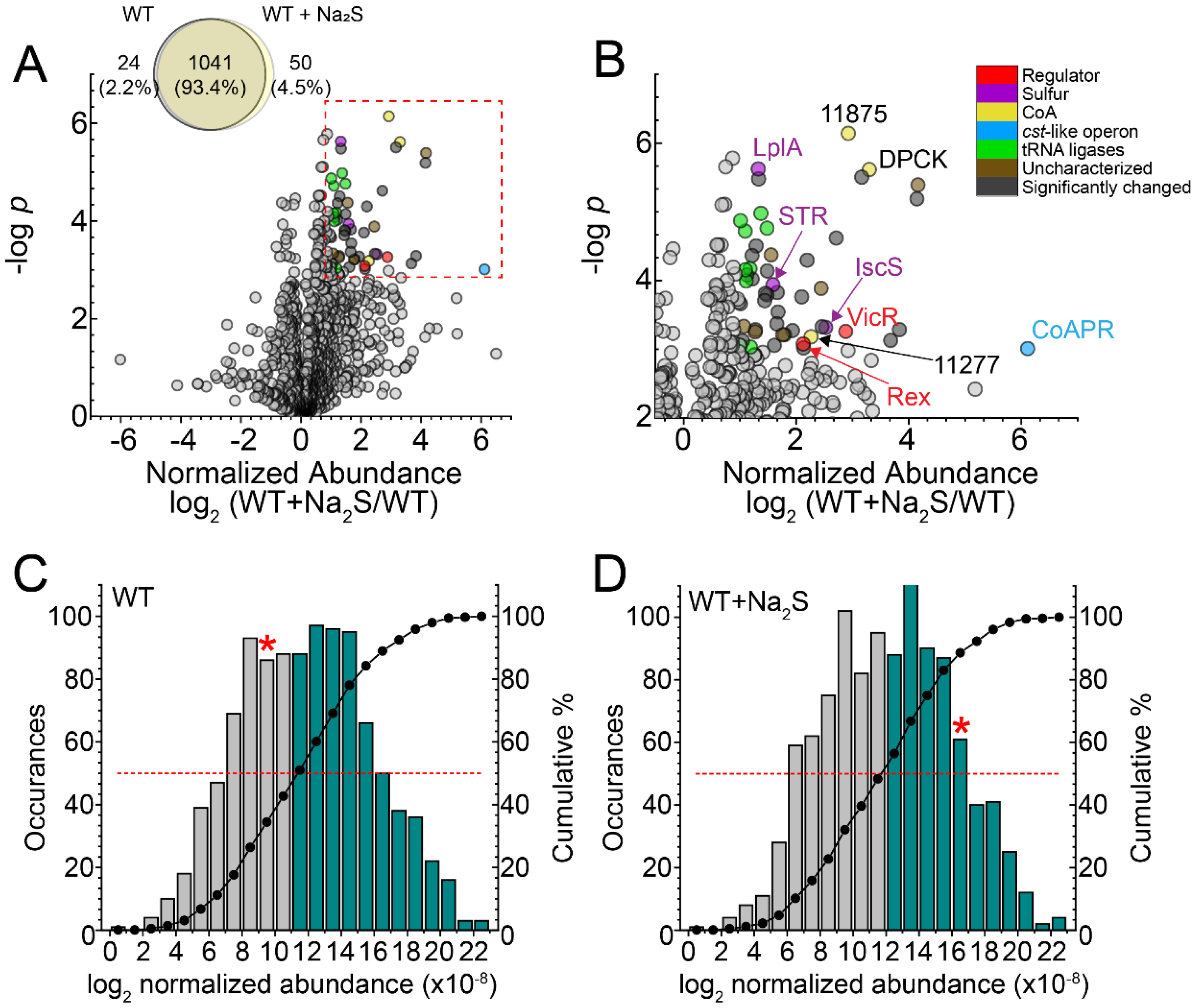
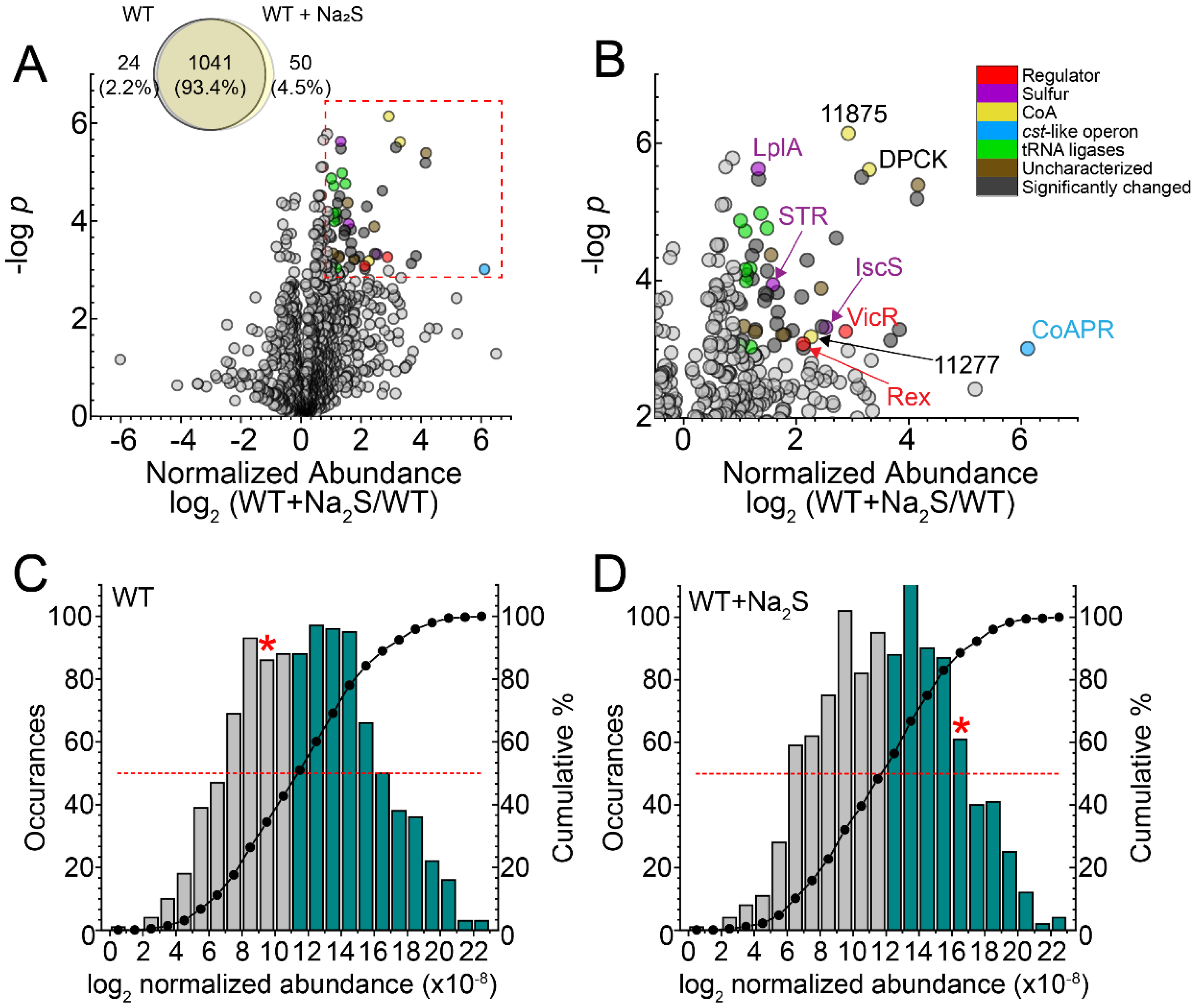
3.1. Global Proteomics Profiling of Wild-Type E. faecalis before and after Addition of Exogenous Na2S
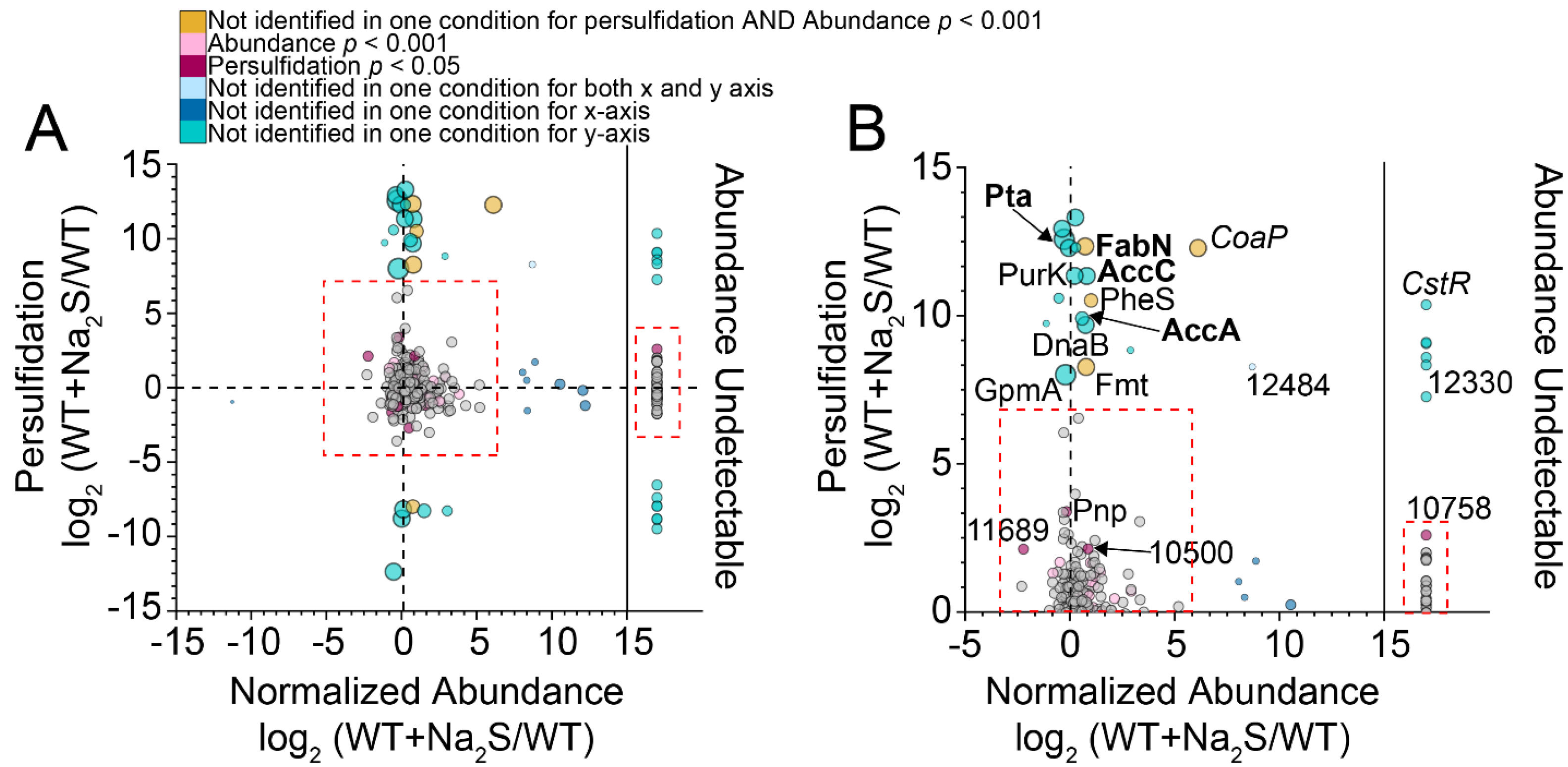
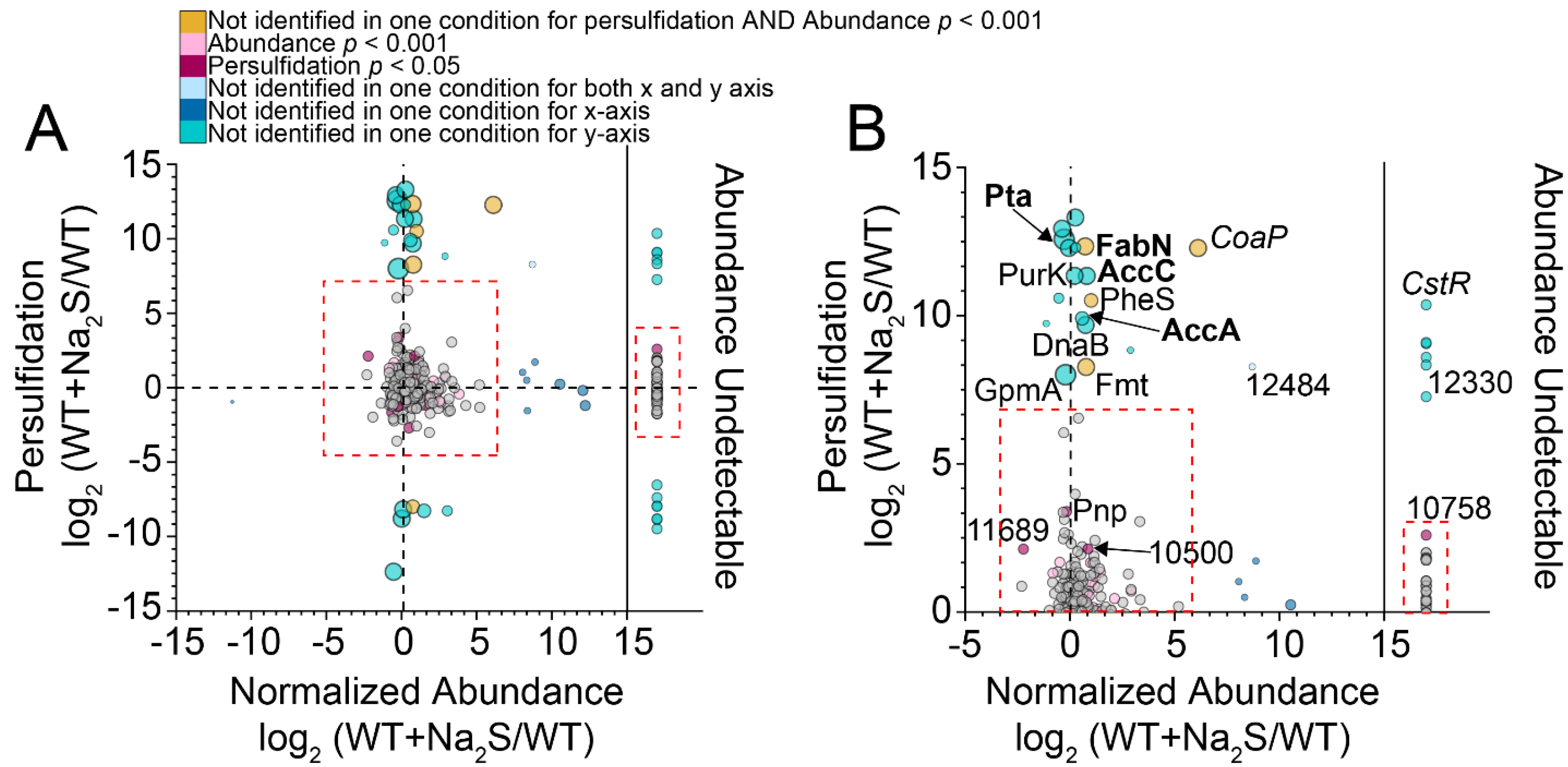
3.2. Proteome Persulfidation in E. faecalis
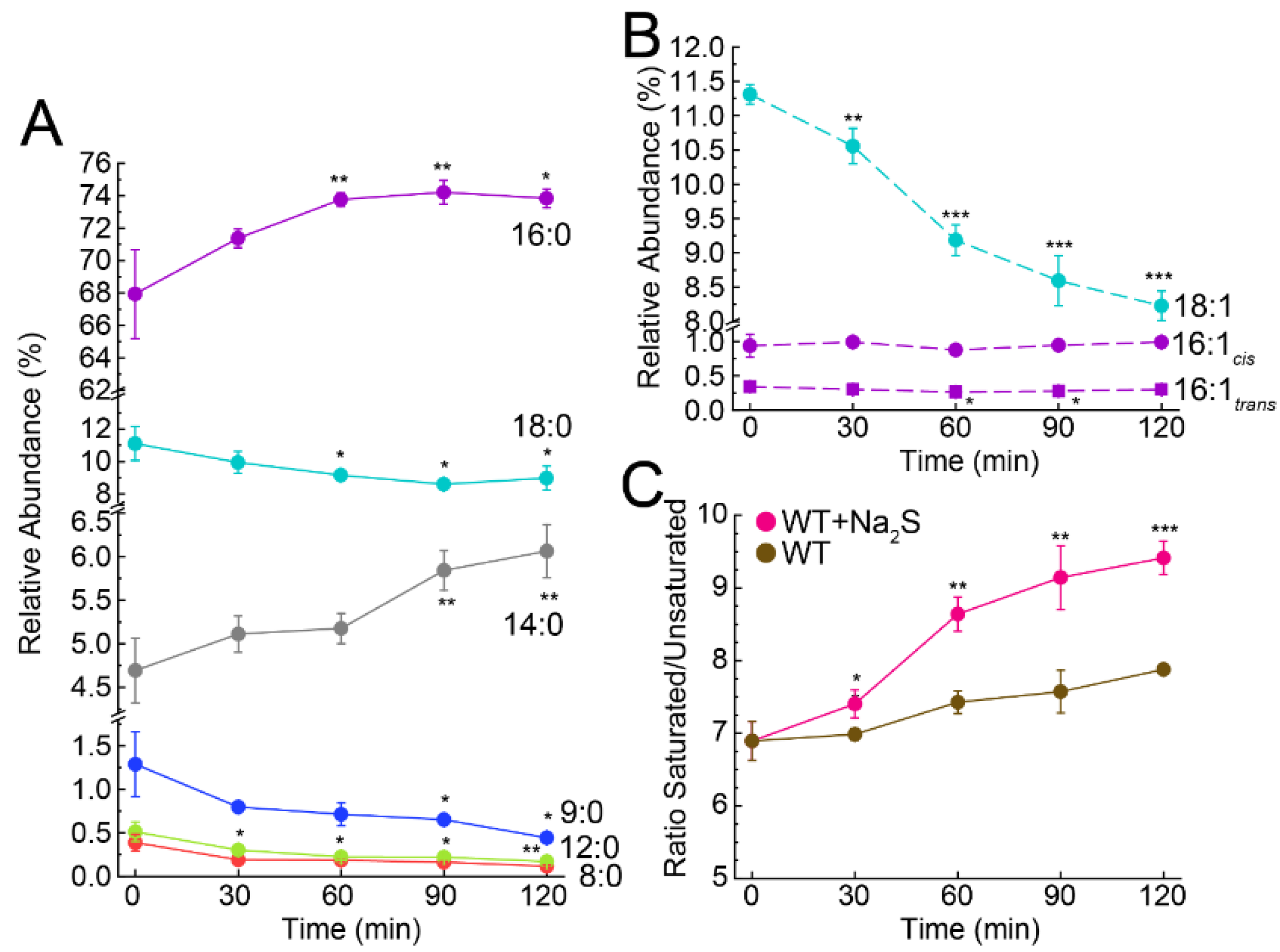
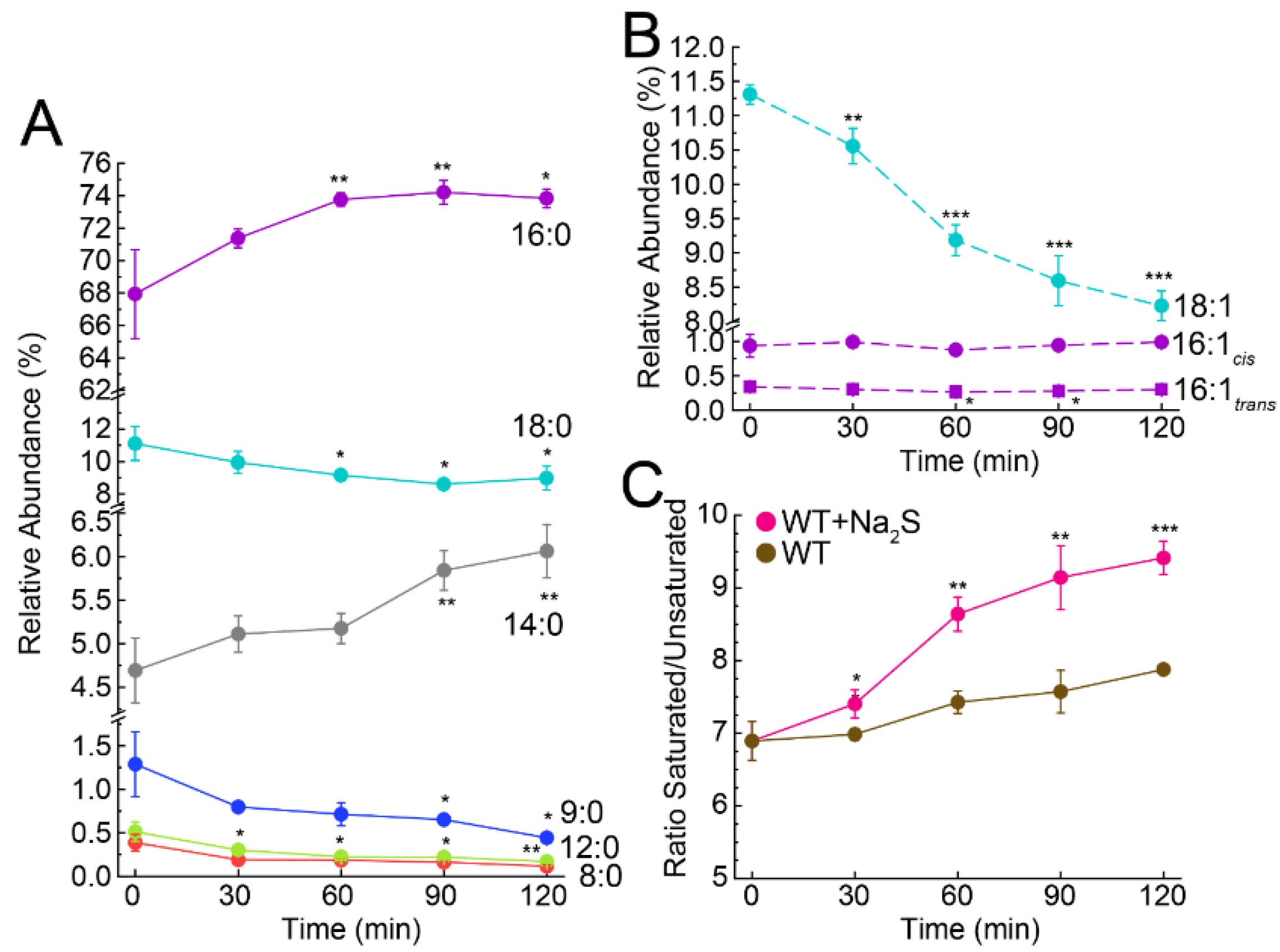
3.3. Exogenous Na2S Impacts Cellular Composition of Fatty Acids
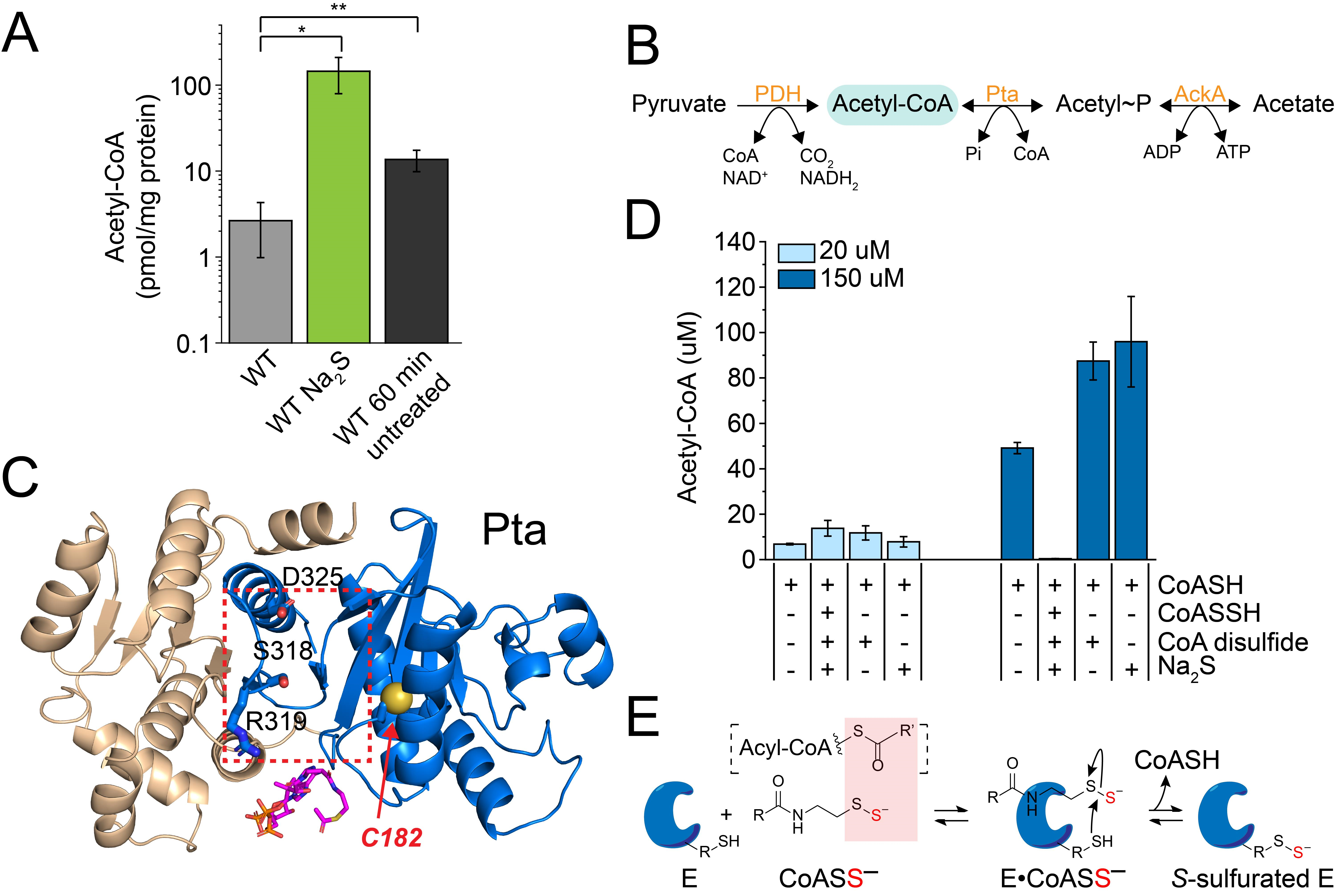
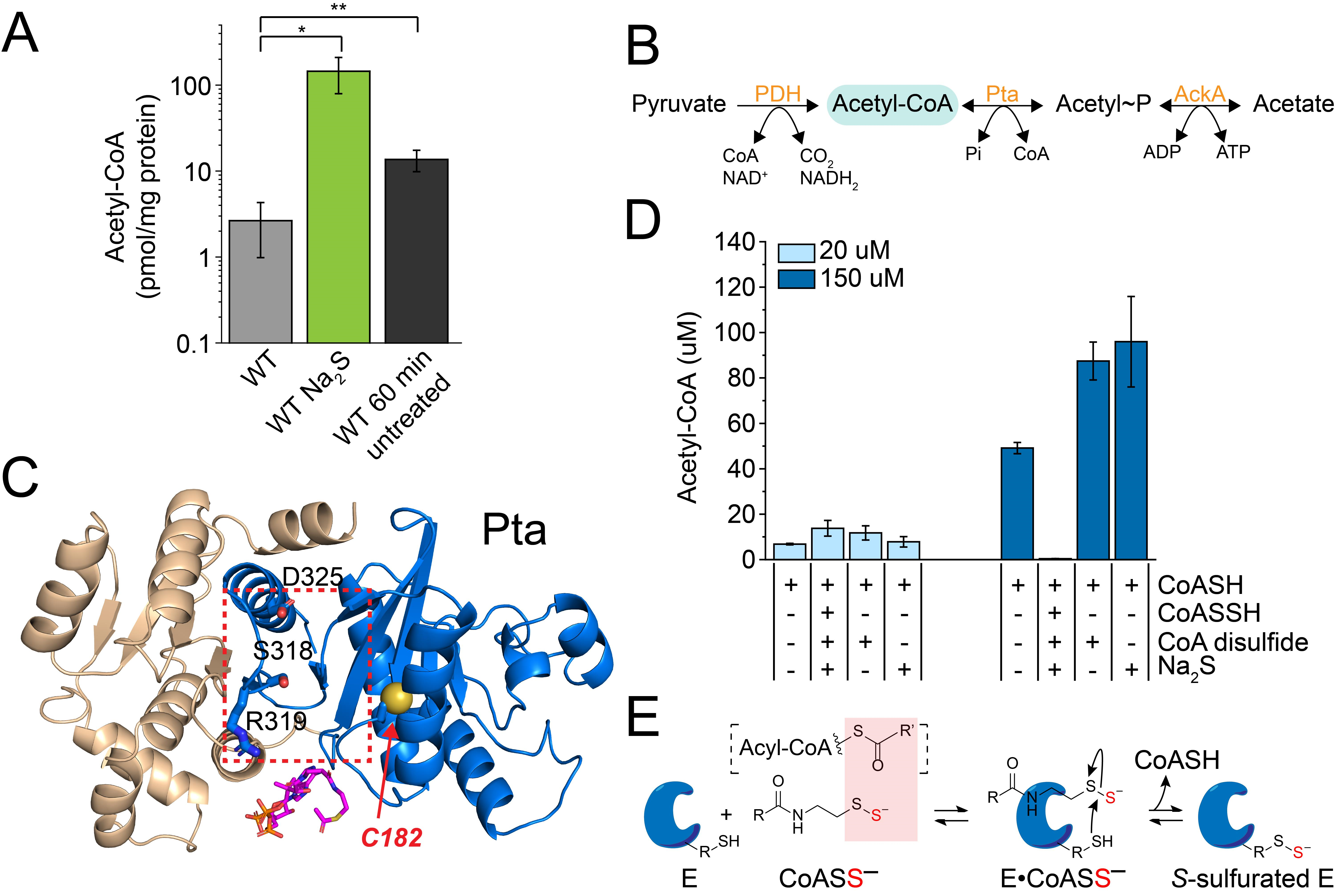
3.4. Exogenous Na2S Impacts Acetyl-CoA via Enzyme Inhibition of Phosphotransacetylase (Pta) by CoASSH
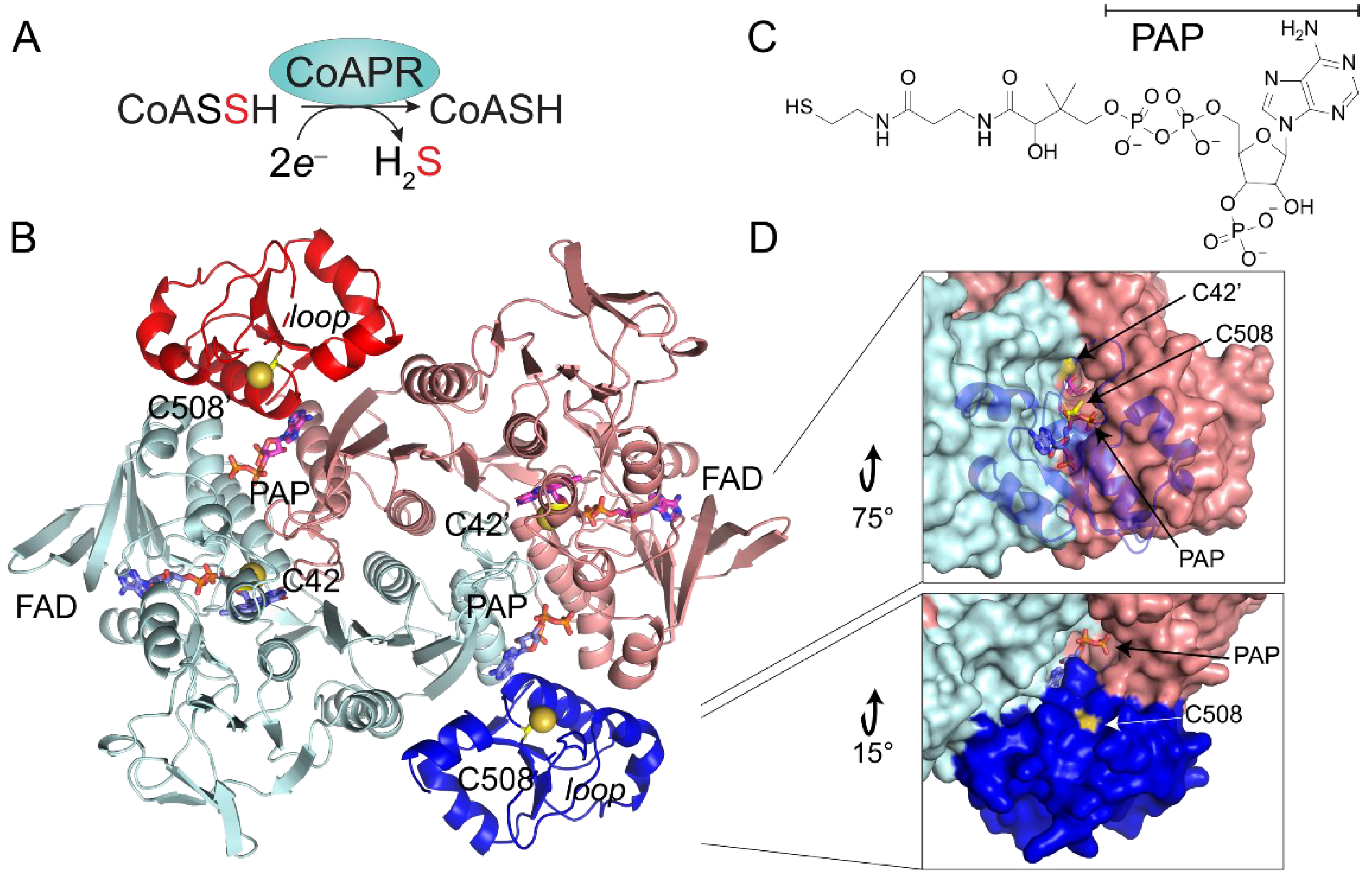
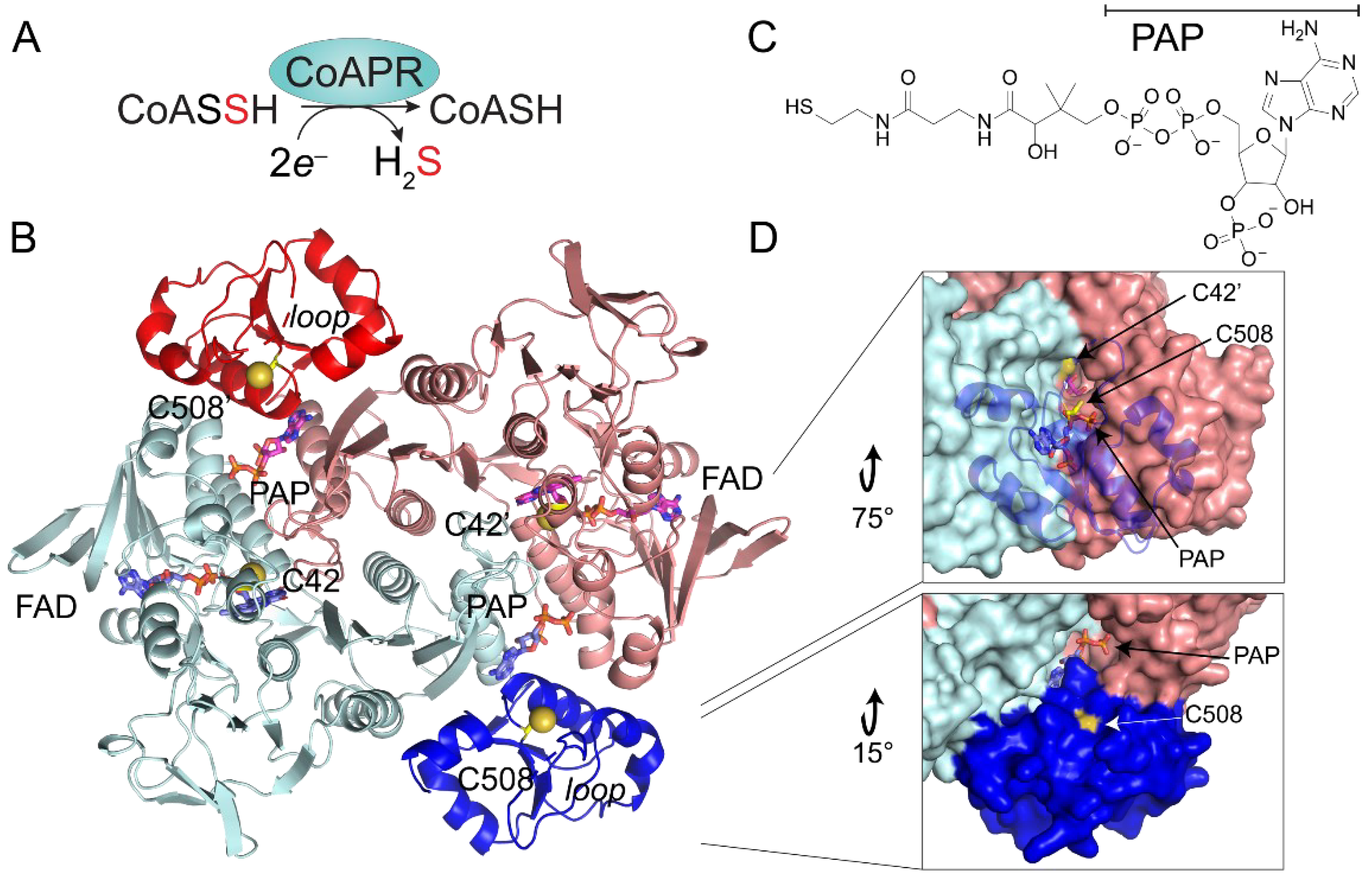
3.5. Crystallographic Structure of Ligand-Bound EfCoAPR
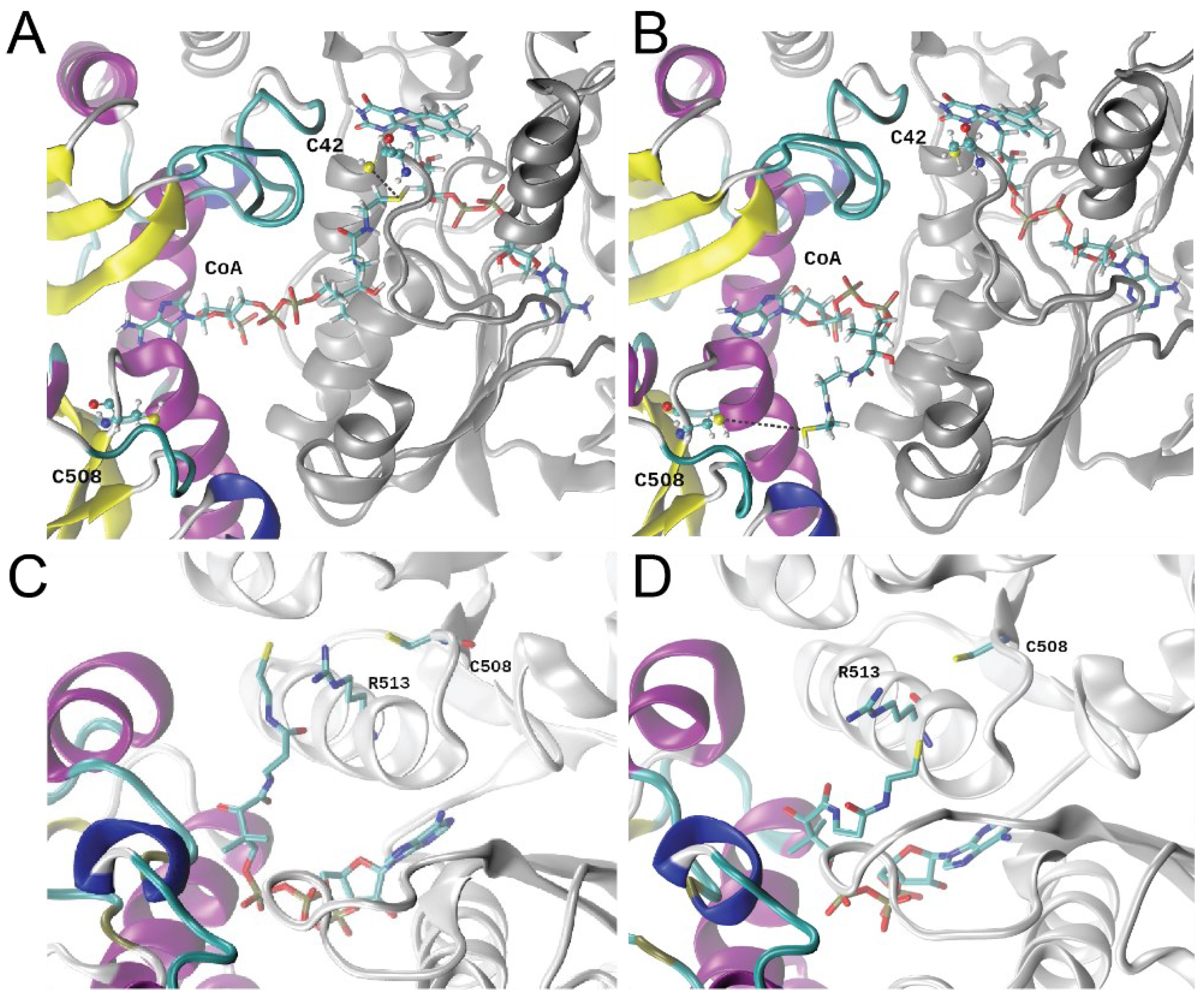
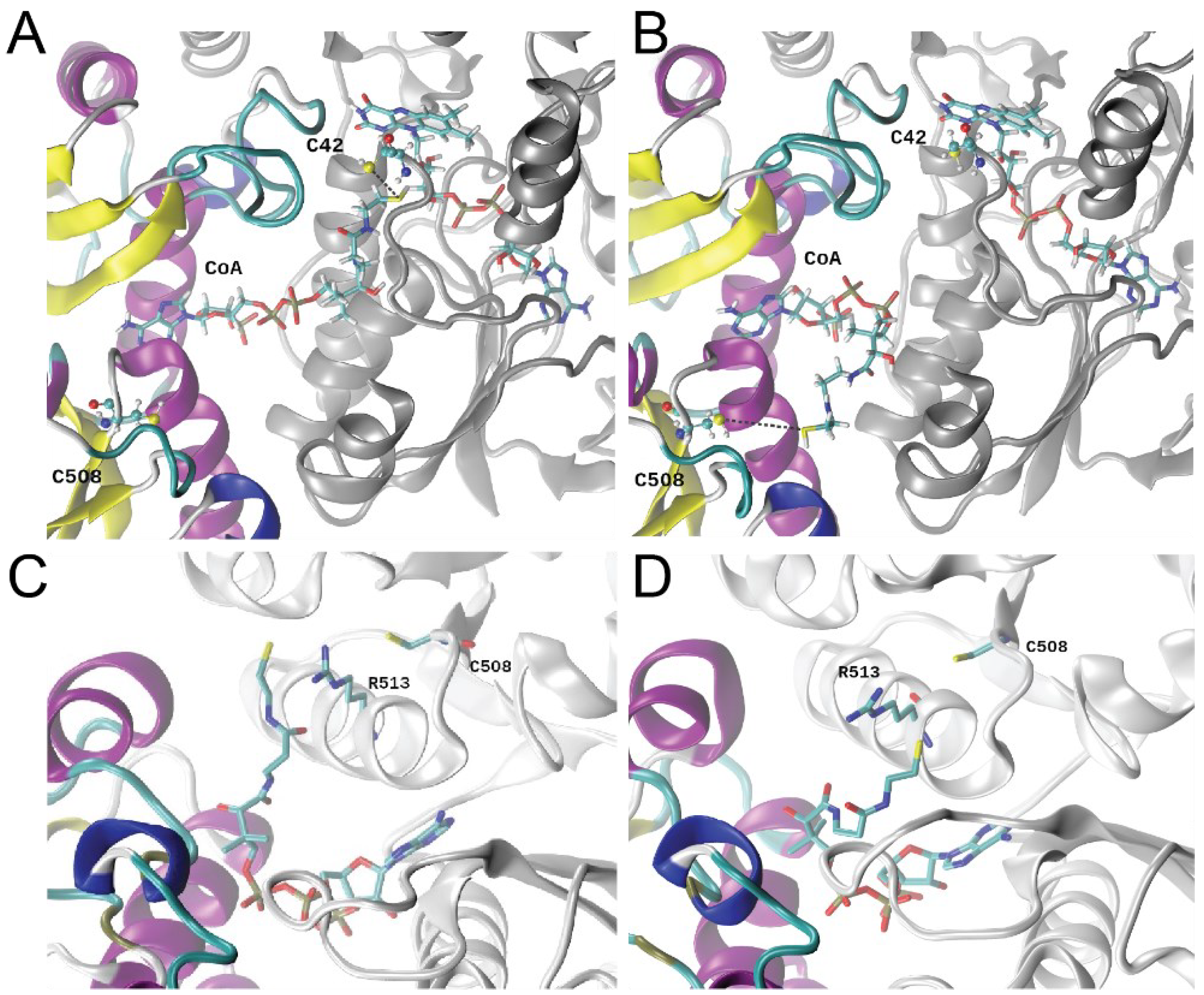
3.6. Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Toliver-Kinsky, T.; Cui, W.; Toro, G.; Lee, S.J.; Shatalin, K.; Nudler, E.; Szabo, C. H2S, a Bacterial Defense Mechanism against the Host Immune Response. Infect. Immun. 2019, 87, e00272-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mironov, A.; Seregina, T.; Nagornykh, M.; Luhachack, L.G.; Korolkova, N.; Lopes, L.E.; Kotova, V.; Zavilgelsky, G.; Shakulov, R.; Shatalin, K.; et al. Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, 6022–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhachack, L.; Rasouly, A.; Shamovsky, I.; Nudler, E. Transcription factor YcjW controls the emergency H2S production in E. coli. Nat. Commun. 2019, 10, 2868. [Google Scholar] [CrossRef]
- Saini, A.; Chinta, K.C.; Reddy, V.P.; Glasgow, J.N.; Stein, A.; Lamprecht, D.A.; Rahman, M.A.; Mackenzie, J.S.; Truebody, B.E.; Adamson, J.H.; et al. Hydrogen sulfide stimulates Mycobacterium tuberculosis respiration, growth and pathogenesis. Nat. Commun. 2020, 11, 557. [Google Scholar] [CrossRef]
- Shukla, P.; Khodade, V.S.; SharathChandra, M.; Chauhan, P.; Mishra, S.; Siddaramappa, S.; Pradeep, B.E.; Singh, A.; Chakrapani, H. “On demand” redox buffering by H2S contributes to antibiotic resistance revealed by a bacteria-specific H2S donor. Chem. Sci. 2017, 8, 4967–4972. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhang, Y.; Palmer, L.D.; Kehl-Fie, T.E.; Skaar, E.P.; Trinidad, J.C.; Giedroc, D.P. Hydrogen Sulfide and Reactive Sulfur Species Impact Proteome S-Sulfhydration and Global Virulence Regulation in Staphylococcus aureus. ACS Infect. Dis. 2017, 3, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [Green Version]
- Cuevasanta, E.; Zeida, A.; Carballal, S.; Wedmann, R.; Morzan, U.N.; Trujillo, M.; Radi, R.; Estrin, D.A.; Filipovic, M.R.; Alvarez, B. Insights into the mechanism of the reaction between hydrogen sulfide and peroxynitrite. Free Radic. Biol. Med. 2015, 80, 93–100. [Google Scholar] [CrossRef]
- Ono, K.; Kitamura, Y.; Zhang, T.; Tsutsuki, H.; Rahman, A.; Ihara, T.; Akaike, T.; Sawa, T. Cysteine Hydropersulfide Inactivates beta-Lactam Antibiotics with Formation of Ring-Opened Carbothioic S-Acids in Bacteria. ACS Chem. Biol. 2021, 16, 731–739. [Google Scholar] [CrossRef]
- Everett, S.A.; Wardman, P. Perthiols as antioxidants: Radical-scavenging and prooxidative mechanisms. Methods Enzymol. 1995, 251, 55–69. [Google Scholar] [PubMed]
- Shen, J.; Walsh, B.J.C.; Flores-Mireles, A.L.; Peng, H.; Zhang, Y.; Zhang, Y.; Trinidad, J.C.; Hultgren, S.J.; Giedroc, D.P. Hydrogen Sulfide Sensing through Reactive Sulfur Species (RSS) and Nitroxyl (HNO) in Enterococcus faecalis. ACS Chem. Biol. 2018, 13, 1610–1620. [Google Scholar] [CrossRef]
- Walsh, B.J.C.; Wang, J.; Edmonds, K.A.; Palmer, L.D.; Zhang, Y.; Trinidad, J.C.; Skaar, E.P.; Giedroc, D.P. The Response of Acinetobacter baumannii to Hydrogen Sulfide Reveals Two Independent Persulfide-Sensing Systems and a Connection to Biofilm Regulation. mBio 2020, 11, e01254-20. [Google Scholar] [CrossRef]
- Peng, H.; Shen, J.; Edmonds, K.A.; Luebke, J.L.; Hickey, A.K.; Palmer, L.D.; Chang, F.J.; Bruce, K.A.; Kehl-Fie, T.E.; Skaar, E.P.; et al. Sulfide homeostasis and nitroxyl intersect via formation of reactive sulfur species in Staphylococcus aureus. mSphere 2017, 2, e00082-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, D.H.; Eghbal, M.A.; Hindmarsh, W.; Roth, S.H.; O’Brien, P.J. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab. Rev. 2006, 38, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, Y.; Trinidad, J.C.; Giedroc, D.P. Thioredoxin Profiling of Multiple Thioredoxin-Like Proteins in Staphylococcus aureus. Front. Microbiol. 2018, 9, 2385. [Google Scholar] [CrossRef]
- Motta, J.P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; Da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef]
- Wallace, J.L.; Motta, J.P.; Buret, A.G. Hydrogen sulfide: An agent of stability at the microbiome-mucosa interface. Am. J. Physiol.-Gastrointest. Liver Physiol. 2018, 314, G143–G149. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Liang, F.; Shah Masood, W.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 S-sulfhydration, MAPK dependent anti-apoptosis and NF-kappaB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef]
- Pitcher, M.C.; Cummings, J.H. Hydrogen sulphide: A bacterial toxin in ulcerative colitis? Gut 1996, 39, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 2006, 4, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.Y.; Winston, J.H.; Shenoy, M.; Zhou, S.; Chen, J.D.; Pasricha, P.J. The endogenous hydrogen sulfide producing enzyme cystathionine-beta synthase contributes to visceral hypersensitivity in a rat model of irritable bowel syndrome. Mol. Pain 2009, 5, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.; Andrade-Oliveira, V.; McCulloch, J.A.; Hild, B.; Oh, J.H.; Perez-Chaparro, P.J.; Sim, C.K.; Lim, A.I.; Link, V.M.; Enamorado, M.; et al. Infection trains the host for microbiota-enhanced resistance to pathogens. Cell 2021, 184, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Walsh, B.J.C.; Giedroc, D.P. H2S and reactive sulfur signaling at the host-bacterial pathogen interface. J. Biol. Chem. 2020, 295, 13150–13168. [Google Scholar] [CrossRef]
- Grossoehme, N.; Kehl-Fie, T.E.; Ma, Z.; Adams, K.W.; Cowart, D.M.; Scott, R.A.; Skaar, E.P.; Giedroc, D.P. Control of Copper Resistance and Inorganic Sulfur Metabolism by Paralogous Regulators in Staphylococcus aureus. J. Biol. Chem. 2011, 286, 13522–13531. [Google Scholar] [CrossRef] [Green Version]
- Luebke, J.L.; Shen, J.; Bruce, K.E.; Kehl-Fie, T.E.; Peng, H.; Skaar, E.P.; Giedroc, D.P. The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol. 2014, 94, 1343–1360. [Google Scholar] [CrossRef] [Green Version]
- Fakhoury, J.N.; Zhang, Y.; Edmonds, K.A.; Bringas, M.; Luebke, J.L.; Gonzalez-Gutierrez, G.; Capdevila, D.A.; Giedroc, D.P. Functional asymmetry and chemical reactivity of CsoR family persulfide sensors. Nucleic. Acids Res. 2021, 49, 12556–12576. [Google Scholar] [CrossRef]
- Landry, A.P.; Moon, S.; Kim, H.; Yadav, P.K.; Guha, A.; Cho, U.S.; Banerjee, R. A Catalytic Trisulfide in Human Sulfide Quinone Oxidoreductase Catalyzes Coenzyme A Persulfide Synthesis and Inhibits Butyrate Oxidation. Cell Chem. Biol. 2019, 26, 1515–1525.e4. [Google Scholar] [CrossRef]
- Walsh, B.J.C.; Giedroc, D.P. Proteomics Profiling of S-sulfurated Proteins in Acinetobacter baumannii. Bio. Protoc. 2021, 11, e4000. [Google Scholar] [CrossRef]
- Smart, K.F.; Aggio, R.B.; Van Houtte, J.R.; Villas-Boas, S.G. Analytical platform for metabolome analysis of microbial cells using methyl chloroformate derivatization followed by gas chromatography-mass spectrometry. Nat. Prot. 2010, 5, 1709–1729. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Chu, D.B.; Marx, H.; Sauer, M.; Hann, S.; Koellensperger, G. LC-MS/MS-based analysis of coenzyme A and short-chain acyl-coenzyme A thioesters. Anal. Bioanal. Chem. 2015, 407, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Campos-Bermudez, V.A.; Bologna, F.P.; Andreo, C.S.; Drincovich, M.F. Functional dissection of Escherichia coli phosphotransacetylase structural domains and analysis of key compounds involved in activity regulation. FEBS J. 2010, 277, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Decker, K. Acetyl-Coenzyme A UV-Spectrophotometric Assay. In Methods of Enzymatic Analysis (Second English Edition); Elsevier Inc.: Amsterdam, The Netherlands; Verlag Chemie GmbH: Weinheim, Germany, 1974; Volume 4, pp. 1988–2044. [Google Scholar]
- O’Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef]
- D’Arcy, A.; Villard, F.; Marsh, M. An automated microseed matrix-screening method for protein crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 550–554. [Google Scholar] [CrossRef]
- Theveneau, P.; Baker, R.; Barrett, R.; Beteva, A.; Bowler, M.W.; Carpentier, P.; Caserotto, H.; de Sanctis, D.; Dobias, F.; Flot, D.; et al. The Upgrade Programme for the Structural Biology beam lines at the European Synchrotron Radiation Facility—High throughput sample evaluation and automation. J. Phys. Conf. Ser. 2013, 425, 012001. [Google Scholar] [CrossRef] [Green Version]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Wallen, J.R.; Mallett, T.C.; Boles, W.; Parsonage, D.; Furdui, C.M.; Karplus, P.A.; Claiborne, A. Crystal structure and catalytic properties of Bacillus anthracis CoADR-RHD: Implications for flavin-linked sulfur trafficking. Biochemistry 2009, 48, 9650–9667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 2.9.1–2.9.37. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L.G. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef]
- Vesic, D.; Kristich, C.J. A Rex family transcriptional repressor influences H2O2 accumulation by Enterococcus faecalis. J. Bacteriol. 2013, 195, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Yuille, H.M.; Blessie, V.; Gohring, N.; Igloi, Z.; Nishiguchi, K.; Nakayama, J.; Henderson, P.J.; Phillips-Jones, M.K. Expression, purification and activities of the entire family of intact membrane sensor kinases from Enterococcus faecalis. Mol. Membr. Biol. 2008, 25, 449–473. [Google Scholar] [CrossRef]
- Cronan, J.E.; Zhao, X.; Jiang, Y. Function, attachment and synthesis of lipoic acid in Escherichia coli. Adv. Microb. Physiol. 2005, 50, 103–146. [Google Scholar]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The Expanding Landscape of the Thiol Redox Proteome. Mol. Cell. Proteom. 2016, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Rock, C.O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef]
- Beld, J.; Lee, D.J.; Burkart, M.D. Fatty acid biosynthesis revisited: Structure elucidation and metabolic engineering. Mol. Biosyst. 2015, 11, 38–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronan, J.E., Jr.; Waldrop, G.L. Multi-subunit acetyl-CoA carboxylases. Prog. Lipid Res. 2002, 41, 407–435. [Google Scholar] [CrossRef]
- Wang, H.; Cronan, J.E. Functional replacement of the FabA and FabB proteins of Escherichia coli fatty acid synthesis by Enterococcus faecalis FabZ and FabF homologues. J. Biol. Chem. 2004, 279, 34489–34495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, A.K.; Duda, K.A.; Romero-Saavedra, F.; Engel, R.; Holst, O.; Huebner, J. Deletion of fabN in Enterococcus faecalis results in unsaturated fatty acid auxotrophy and decreased release of inflammatory cytokines. Innate Immun. 2016, 22, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.E.; Harp, J.R.; Fozo, E.M. Incorporation of exogenous fatty acids protects Enterococcus faecalis from membrane-damaging agents. Appl. Environ. Microbiol. 2014, 80, 6527–6538. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lawrence, S.H.; Luther, K.B.; Schindelin, H.; Ferry, J.G. Structural and functional studies suggest a catalytic mechanism for the phosphotransacetylase from Methanosarcina thermophila. J. Bacteriol. 2006, 188, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Brinsmade, S.R.; Escalante-Semerena, J.C. In vivo and in vitro analyses of single-amino acid variants of the Salmonella enterica phosphotransacetylase enzyme provide insights into the function of its N-terminal domain. J. Biol. Chem. 2007, 282, 12629–12640. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.S.; Jancarik, J.; Lou, Y.; Kuznetsova, K.; Yakunin, A.F.; Yokota, H.; Adams, P.; Kim, R.; Kim, S.H. Crystal structures of a phosphotransacetylase from Bacillus subtilis and its complex with acetyl phosphate. J. Struct. Funct. Genom. 2005, 6, 269–279. [Google Scholar] [CrossRef]
- Williamson, G.; Engel, P.C.; Mizzer, J.P.; Thorpe, C.; Massey, V. Evidence that the greening ligand in native butyryl-CoA dehydrogenase is a CoA persulfide. J. Biol. Chem. 1982, 257, 4314–4320. [Google Scholar] [CrossRef]
- Lee, K.H.; Humbarger, S.; Bahnvadia, R.; Sazinsky, M.H.; Crane, E.J., 3rd. Characterization of the mechanism of the NADH-dependent polysulfide reductase (Npsr) from Shewanella loihica PV-4: Formation of a productive NADH-enzyme complex and its role in the general mechanism of NADH and FAD-dependent enzymes. Biochim. Biophys. Acta 2014, 1844, 1708–1717. [Google Scholar] [CrossRef]
- Shabdar, S.; Anaclet, B.; Castineiras, A.G.; Desir, N.; Choe, N.; Crane, E.J., 3rd; Sazinsky, M.H. Structural and Kinetic Characterization of Hyperthermophilic NADH-Dependent Persulfide Reductase from Archaeoglobus fulgidus. Archaea 2021, 2021, 8817136. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.D.; Lukose, V.; Lee, K.H.; Lopez, K.; Sazinsky, M.H.; Crane, E.J., 3rd. Characterization of an NADH-dependent persulfide reductase from Shewanella loihica PV-4: Implications for the mechanism of sulfur respiration via FAD-dependent enzymes. Biochemistry 2011, 50, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Thiennimitr, P.; Winter, M.G.; Butler, B.P.; Huseby, D.L.; Crawford, R.W.; Russell, J.M.; Bevins, C.L.; Adams, L.G.; Tsolis, R.M.; et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 2010, 467, 426–429. [Google Scholar] [CrossRef]
- Korshunov, S.; Imlay, K.R.; Imlay, J.A. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 2016, 101, 62–77. [Google Scholar] [CrossRef] [Green Version]
- Jones-Carson, J.; Husain, M.; Liu, L.; Orlicky, D.J.; Vazquez-Torres, A. Cytochrome bd-Dependent Bioenergetics and Antinitrosative Defenses in Salmonella Pathogenesis. MBio 2016, 7, e02052-16. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Borisov, V.B.; Falabella, M.; Colaco, H.G.; Tinajero-Trejo, M.; Poole, R.K.; Vicente, J.B.; Sarti, P.; Giuffre, A. The Terminal Oxidase Cytochrome bd Promotes Sulfide-resistant Bacterial Respiration and Growth. Sci. Rep. 2016, 6, 23788. [Google Scholar] [CrossRef] [Green Version]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar] [CrossRef]
- Leekumjorn, S.; Cho, H.J.; Wu, Y.; Wright, N.T.; Sum, A.K.; Chan, C. The role of fatty acid unsaturation in minimizing biophysical changes on the structure and local effects of bilayer membranes. Biochim. Biophys. Acta 2009, 1788, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Brenner, R.R. Effect of unsaturated acids on membrane structure and enzyme kinetics. Prog. Lipid Res. 1984, 23, 69–96. [Google Scholar] [CrossRef]
- Shen, S.; Hang, X.; Zhuang, J.; Zhang, L.; Bi, H.; Zhang, L. A back-door Phenylalanine coordinates the stepwise hexameric loading of acyl carrier protein by the fatty acid biosynthesis enzyme beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ). Int. J. Biol. Macromol. 2019, 128, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Kimber, M.S.; Martin, F.; Lu, Y.; Houston, S.; Vedadi, M.; Dharamsi, A.; Fiebig, K.M.; Schmid, M.; Rock, C.O. The structure of (3R)-hydroxyacyl-acyl carrier protein dehydratase (FabZ) from Pseudomonas aeruginosa. J. Biol. Chem. 2004, 279, 52593–52602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.J.; White, S.W.; Rock, C.O. Domain swapping between Enterococcus faecalis FabN and FabZ proteins localizes the structural determinants for isomerase activity. J. Biol. Chem. 2005, 280, 30342–30348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xiao, J.; Xu, J.; Fu, T.; Cao, Z.; Zhu, L.; Chen, H.Z.; Shen, X.; Jiang, H.; Zhang, L. Crystal structure of FabZ-ACP complex reveals a dynamic seesaw-like catalytic mechanism of dehydratase in fatty acid biosynthesis. Cell Res. 2016, 26, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Broussard, T.C.; Kobe, M.J.; Pakhomova, S.; Neau, D.B.; Price, A.E.; Champion, T.S.; Waldrop, G.L. The three-dimensional structure of the biotin carboxylase-biotin carboxyl carrier protein complex of E. coli acetyl-CoA carboxylase. Structure 2013, 21, 650–657. [Google Scholar] [CrossRef] [Green Version]
- Broussard, T.C.; Price, A.E.; Laborde, S.M.; Waldrop, G.L. Complex formation and regulation of Escherichia coli acetyl-CoA carboxylase. Biochemistry 2013, 52, 3346–3357. [Google Scholar] [CrossRef]
- Bilder, P.; Lightle, S.; Bainbridge, G.; Ohren, J.; Finzel, B.; Sun, F.; Holley, S.; Al-Kassim, L.; Spessard, C.; Melnick, M.; et al. The structure of the carboxyltransferase component of acetyl-coA carboxylase reveals a zinc-binding motif unique to the bacterial enzyme. Biochemistry 2006, 45, 1712–1722. [Google Scholar] [CrossRef]
- Lee, T.H.; Hofferek, V.; Separovic, F.; Reid, G.E.; Aguilar, M.I. The role of bacterial lipid diversity and membrane properties in modulating antimicrobial peptide activity and drug resistance. Curr. Opin. Chem. Biol. 2019, 52, 85–92. [Google Scholar] [CrossRef]
- Harp, J.R.; Saito, H.E.; Bourdon, A.K.; Reyes, J.; Arias, C.A.; Campagna, S.R.; Fozo, E.M. Exogenous Fatty Acids Protect Enterococcus faecalis from Daptomycin-Induced Membrane Stress Independently of the Response Regulator LiaR. Appl. Environ. Microbiol. 2016, 82, 4410–4420. [Google Scholar] [CrossRef] [Green Version]
- Rashid, R.; Cazenave-Gassiot, A.; Gao, I.H.; Nair, Z.J.; Kumar, J.K.; Gao, L.; Kline, K.A.; Wenk, M.R. Comprehensive analysis of phospholipids and glycolipids in the opportunistic pathogen Enterococcus faecalis. PLoS ONE 2017, 12, e0175886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobocinska, J.; Roszczenko-Jasinska, P.; Ciesielska, A.; Kwiatkowska, K. Protein Palmitoylation and Its Role in Bacterial and Viral Infections. Front. Immunol. 2017, 8, 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, B.A.; Sutcliffe, I.C.; Harrington, D.J. Impact of lgt mutation on lipoprotein biosynthesis and in vitro phenotypes of Streptococcus agalactiae. Microbiology 2009, 155, 1451–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, S.W.; Charron, G.; Hang, H.C.; Galan, J.E. Subcellular targeting of Salmonella virulence proteins by host-mediated S-palmitoylation. Cell Host Microbe 2011, 10, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Spera, J.M.; Guaimas, F.; Corvi, M.M.; Ugalde, J.E. Brucella Hijacks Host-Mediated Palmitoylation To Stabilize and Localize PrpA to the Plasma Membrane. Infect. Immun. 2018, 86, e00402-18. [Google Scholar] [CrossRef] [Green Version]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar]
- Word, J.M.; Lovell, S.C.; LaBean, T.H.; Taylor, H.C.; Zalis, M.E.; Presley, B.K.; Richardson, J.S.; Richardson, D.C. Visualizing and quantifying molecular goodness-of-fit: Small-probe contact dots with explicit hydrogen atoms. J. Mol. Biol. 1999, 285, 1711–1733. [Google Scholar]
- Zhang, Y.M.; White, S.W.; Rock, C.O. Inhibiting bacterial fatty acid synthesis. J. Biol. Chem. 2006, 281, 17541–17544. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walsh, B.J.C.; Costa, S.S.; Edmonds, K.A.; Trinidad, J.C.; Issoglio, F.M.; Brito, J.A.; Giedroc, D.P. Metabolic and Structural Insights into Hydrogen Sulfide Mis-Regulation in Enterococcus faecalis. Antioxidants 2022, 11, 1607. https://doi.org/10.3390/antiox11081607
Walsh BJC, Costa SS, Edmonds KA, Trinidad JC, Issoglio FM, Brito JA, Giedroc DP. Metabolic and Structural Insights into Hydrogen Sulfide Mis-Regulation in Enterococcus faecalis. Antioxidants. 2022; 11(8):1607. https://doi.org/10.3390/antiox11081607
Chicago/Turabian StyleWalsh, Brenna J. C., Sofia Soares Costa, Katherine A. Edmonds, Jonathan C. Trinidad, Federico M. Issoglio, José A. Brito, and David P. Giedroc. 2022. "Metabolic and Structural Insights into Hydrogen Sulfide Mis-Regulation in Enterococcus faecalis" Antioxidants 11, no. 8: 1607. https://doi.org/10.3390/antiox11081607
APA StyleWalsh, B. J. C., Costa, S. S., Edmonds, K. A., Trinidad, J. C., Issoglio, F. M., Brito, J. A., & Giedroc, D. P. (2022). Metabolic and Structural Insights into Hydrogen Sulfide Mis-Regulation in Enterococcus faecalis. Antioxidants, 11(8), 1607. https://doi.org/10.3390/antiox11081607