
Oxidative Stress as a Potential Mechanism Underlying Membrane Hyperexcitability in Neurodegenerative Diseases

 ,
,  and
and 
Abstract
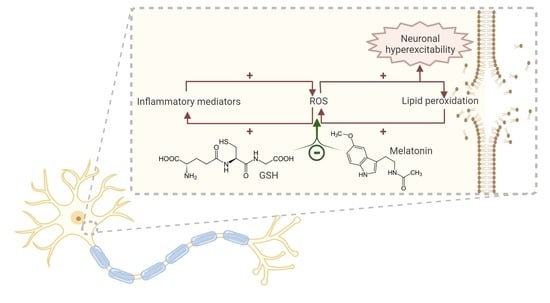
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pathogenesis of Neurodegenerative Disease: Oxidative Stress and Inflammation
2.1. Inflammation Triggers Neurodegenerative Diseases
2.2. Role of Oxidative Stress in Neurodegenerative Diseases
3. Synaptic Disbalance and Neuron Hyperexcitability in Neurodegenerative Diseases
3.1. Hyperexcitability in AD
3.2. Hyperexcitability in MS
3.3. Hyperexcitability in ALS
4. Impact of Oxidative Stress in Motor Cortex Neurons Hyperexcitability
4.1. Neuronal Excitability and Oxidative Stress
4.2. Neuronal Excitability and Time and Dose Dependent Effects of Oxidative Stress
4.3. Oxidative Stress, Synaptic Depression and Hyperexcitability
4.4. Temporal Course of Neuron Hyperexcitability
4.5. Peripheral Inflammation, Neuroinflammation and Hyperexcitability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirmani, B.F.; Shapiro, L.A.; Shetty, A.K. Neurological and Neurodegenerative Disorders: Novel Concepts and Treatment. Aging Dis. 2021, 12, 950. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, M.B.; Sanusi, K.O.; Ugusman, A.; Mohamed, W.; Kamal, H.; Ibrahim, N.H.; Khoo, C.S.; Kumar, J. Alzheimer’s Disease: An Update and Insights into Pathophysiology. Front. Aging Neurosci. 2022, 14, 742408. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Zapata, C.H.; Franco-Dáger, E.; Solano-Atehortúa, J.M.; Ahunca-Velásquex, L.F. Esclerosis Lateral Amiotrófica: Actualización. IATREIA 2016, 29, 194–205. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple Sclerosis—A Review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.W.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G. Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cell Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef]
- Kanai, K.; Shibuya, K.; Sato, Y.; Misawa, S.; Nasu, S.; Sekiguchi, Y.; Mitsuma, S.; Isose, S.; Fujimaki, Y.; Ohmori, S. Motor Axonal Excitability Properties Are Strong Predictors for Survival in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 734–738. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Özdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and Diagnostic Implications of Cortical Dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative Diseases and Oxidative Stress. Biomed. Pharmacother. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- BioRender. Available online: https://biorender.com (accessed on 30 June 2022).
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in Neurodegenerative Diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Bogár, F.; Fülöp, L.; Penke, B. Novel Therapeutic Target for Prevention of Neurodegenerative Diseases: Modulation of Neuroinflammation with Sig-1R Ligands. Biomolecules 2022, 12, 363. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.E.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 Knockout Prevents Neuron L.Loss in a Mouse Model of Alzheimer’s Disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Kocur, M.; Schneider, R.; Pulm, A.-K.; Bauer, J.; Kropp, S.; Gliem, M.; Ingwersen, J.; Goebels, N.; Alferink, J.; Prozorovski, T.; et al. IFNβ Secreted by Microglia Mediates Clearance of Myelin Debris in CNS Autoimmunity. Acta Neuropathol. Commun. 2015, 3, 20. [Google Scholar] [CrossRef]
- Lue, L.F.; Walker, D.G.; Rogers, J. Modeling Microglial Activation in Alzheimer’s Disease with Human Postmortem Microglial Cultures. Neurobiol. Aging 2001, 22, 945–956. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Béraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoji, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial Activation and Antioxidant Responses Induced by the Parkinson’s Disease Protein α-Synuclein. J. Neuroimmune Pharmacol. 2013, 8, 94–117. [Google Scholar] [CrossRef]
- Komine, O.; Yamanaka, K. Neuroinflammation in Motor Neuron Disease. Nagoya J. Med. Sci. 2015, 77, 537–549. [Google Scholar]
- Bogie, J.F.J.; Stinissen, P.; Hendriks, J.J.A. Macrophage Subsets and Microglia in Multiple Sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte Dysfunction in Alzheimer Disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Kashon, M.L.; Ross, G.W.; O’Callaghan, J.P.; Miller, D.B.; Petrovitch, H.; Burchfiel, C.M.; Sharp, D.S.; Markesbery, W.R.; Davis, D.G.; Hardman, J.; et al. Associations of Cortical Astrogliosis with Cognitive Performance and Dementia Status. J. Alzheimers Dis. 2004, 6, 581–595. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s Disease. Park. Relat. Disord. 2012, 18 (Suppl. 1), S210–S212. [Google Scholar] [CrossRef]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic Expression of Parkinson’s Disease-Related A53T Alpha-Synuclein Causes Neurodegeneration in Mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 Domain of Bcl-XL Rescues Astrocyte Degeneration in Amyotrophic Lateral Sclerosis by Modulating Intracellular Calcium Signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef]
- Perriard, G.; Mathias, A.; Enz, L.; Canales, M.; Schluep, M.; Gentner, M.; Schaeren-Wiemers, N.; Du Pasquier, R.A. Interleukin-22 Is Increased in Multiple Sclerosis Patients and Targets Astrocytes. J. Neuroinflammation 2015, 12, 119. [Google Scholar] [CrossRef]
- Brosnan, C.F.; Raine, C.S. The Astrocyte in Multiple Sclerosis Revisited. Glia 2013, 61, 453–465. [Google Scholar] [CrossRef]
- Arroyo, D.S.; Soria, J.A.; Gaviglio, E.A.; Rodriguez-Galan, M.C.; Iribarren, P. Toll-like Receptors Are Key Players in Neurodegeneration. Int. Immunopharmacol. 2011, 11, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Owens, T. Toll-like Receptors in Neurodegeneration. Curr. Top. Microbiol. Immunol. 2009, 336, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Griffioen, K.J.; Lathia, J.D.; Tang, S.-C.; Mattson, M.P.; Arumugam, T.V. Toll-like Receptors in Neurodegeneration. Brain Res. Rev. 2009, 59, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Marta, M.; Meier, U.C.; Lobell, A. Regulation of Autoimmune Encephalomyelitis by Toll-like Receptors. Autoimmun. Rev. 2009, 8, 506–509. [Google Scholar] [CrossRef]
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of Toll-like Receptor Signalling in Abeta Uptake and Clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef]
- Prinz, M.; Garbe, F.; Schmidt, H.; Mildner, A.; Gutcher, I.; Wolter, K.; Piesche, M.; Schroers, R.; Weiss, E.; Kirschning, C.J.; et al. Innate Immunity Mediated by TLR9 Modulates Pathogenicity in an Animal Model of Multiple Sclerosis. J. Clin. Investig. 2006, 116, 456–464. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, Oxidative Stress and Neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef]
- Urano, S.; Asai, Y.; Makabe, S.; Matsuo, M.; Izumiyama, N.; Ohtsubo, K.; Endo, T. Oxidative Injury of Synapse and Alteration of Antioxidative Defense Systems in Rats, and Its Prevention by Vitamin E. Eur. J. Biochem. 1997, 245, 64–70. [Google Scholar] [CrossRef]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv. Ther. 2019, 37, 113–139. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Padurariu, M.; Ciobica, A.; Lefter, R.; Serban, I.L.; Stefanescu, C.; Chirita, R. The Oxidative Stress Hypothesis in Alzheimer’s Disease. Psychiatr. Danub. 2013, 25, 401–409. [Google Scholar]
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. [Google Scholar] [CrossRef]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and Regulated α-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by a Disintegrin Metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid Induction of Alzheimer Aβ Amyloid Formation by Zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Ghilardi, J.R.; Rogers, S.; DeMaster, E.; Allen, C.J.; Stimson, E.R.; Maggio, J.E. Aluminum, Iron, and Zinc Ions Promote Aggregation of Physiological Concentrations of Β-amyloid Peptide. J. Neurochem. 1993, 61, 1171–1174. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative Stress, Aging, and Diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef]
- Tong, H.; Zhang, X.; Meng, X.; Lu, L.; Mai, D.; Qu, S. Simvastatin Inhibits Activation of NADPH Oxidase/P38 MAPK Pathway and Enhances Expression of Antioxidant Protein in Parkinson Disease Models. Front. Mol. Neurosci. 2018, 11, 165. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Hare, D.J.; Double, K.L. Iron and Dopamine: A Toxic Couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef]
- Hare, D.J.; Kysenius, K.; Paul, B.; Knauer, B.; Hutchinson, R.W.; O’Connor, C.; Fryer, F.; Hennessey, T.P.; Bush, A.I.; Crouch, P.J. Imaging Metals in Brain Tissue by Laser Ablation-Inductively Coupled Plasma-Mass Spectrometry (LA-ICP-MS). JoVE (J. Vis. Exp.) 2017, 119, e55042. [Google Scholar] [CrossRef]
- Duce, J.A.; Wong, B.X.; Durham, H.; Devedjian, J.-C.; Smith, D.P.; Devos, D. Post Translational Changes to α-Synuclein Control Iron and Dopamine Trafficking; a Concept for Neuron Vulnerability in Parkinson’s Disease. Mol. Neurodegener. 2017, 12, 45. [Google Scholar] [CrossRef]
- Park, H.R.; Yang, E.J. Oxidative Stress as a Therapeutic Target in Amyotrophic Lateral Sclerosis: Opportunities and Limitations. Diagnostics 2021, 11, 1546. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Foran, E.; Trotti, D. Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2009, 11, 1587–1602. [Google Scholar] [CrossRef]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate Transporters Are Oxidant-Vulnerable: A Molecular Link between Oxidative and Excitotoxic Neurodegeneration? Trends Pharm. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef]
- Rao, S.D.; Yin, H.Z.; Weiss, J.H. Disruption of Glial Glutamate Transport by Reactive Oxygen Species Produced in Motor Neurons. J. Neurosci. 2003, 23, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.R.; Shaw, P.J. Update on the Glutamatergic Neurotransmitter System and the Role of Excitotoxicity in Amyotrophic Lateral Sclerosis. Muscle Nerve 2002, 26, 438–458. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.D.; Weiss, J.H. Excitotoxic and Oxidative Cross-Talk between Motor Neurons and Glia in ALS Pathogenesis. Trends Neurosci. 2004, 27, 17–23. [Google Scholar] [CrossRef]
- Hirano, A.; Nakano, I.; Kurland, L.T.; Mulder, D.W.; Holley, P.W.; Saccomanno, G. Fine Structural Study of Neurofibrillary Changes in a Family with Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 471–480. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative Stress in Multiple Sclerosis: Central and Peripheral Mode of Action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Fischer, M.T.; Wimmer, I.; Höftberger, R.; Gerlach, S.; Haider, L.; Zrzavy, T.; Hametner, S.; Mahad, D.; Binder, C.J.; Krumbholz, M. Disease-Specific Molecular Events in Cortical Multiple Sclerosis Lesions. Brain 2013, 136, 1799–1815. [Google Scholar] [CrossRef]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Höftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative Damage in Multiple Sclerosis Lesions. Brain 2011, 134, 1914–1924. [Google Scholar] [CrossRef]
- Ibitoye, R.; Kemp, K.; Rice, C.; Hares, K.; Scolding, N.; Wilkins, A. Oxidative Stress-Related Biomarkers in Multiple Sclerosis: A Review. Biomark. Med. 2016, 10, 889–902. [Google Scholar] [CrossRef]
- Olla, S.; Steri, M.; Formato, A.; Whalen, M.B.; Corbisiero, S.; Agresti, C. Combining Human Genetics of Multiple Sclerosis with Oxidative Stress Phenotype for Drug Repositioning. Pharmaceutics 2021, 13, 2064. [Google Scholar] [CrossRef]
- Corthay, A. A Three-cell Model for Activation of Naive T Helper Cells. Scand. J. Immunol. 2006, 64, 93–96. [Google Scholar] [CrossRef]
- Zhang, Q.; Fujino, M.; Xu, J.; Li, X. The Role and Potential Therapeutic Application of Myeloid-Derived Suppressor Cells in Allo-and Autoimmunity. Mediat. Inflamm. 2015, 2015, 421927. [Google Scholar] [CrossRef]
- Imaizumi, S.; Kondo, T.; Deli, M.A.; Gobbel, G.; Joó, F.; Epstein, C.J.; Yoshimoto, T.; Chan, P.H. The Influence of Oxygen Free Radicals on the Permeability of the Monolayer of Cultured Brain Endothelial Cells. Neurochem. Int. 1996, 29, 205–211. [Google Scholar] [CrossRef]
- Ju, N.; Li, Y.; Liu, F.; Jiang, H.; Macknik, S.L.; Martinez-Conde, S.; Tang, S. Spatiotemporal Functional Organization of Excitatory Synaptic Inputs onto Macaque V1 Neurons. Nat. Commun. 2020, 11, 697. [Google Scholar] [CrossRef]
- Iascone, D.M.; Li, Y.; Sümbül, U.; Doron, M.; Chen, H.; Andreu, V.; Goudy, F.; Blockus, H.; Abbott, L.F.; Segev, I.; et al. Whole-Neuron Synaptic Mapping Reveals Spatially Precise Excitatory/Inhibitory Balance Limiting Dendritic and Somatic Spiking. Neuron 2020, 106, 566–578.e8. [Google Scholar] [CrossRef]
- Ren, S.-Q.; Yao, W.; Yan, J.-Z.; Jin, C.; Yin, J.-J.; Yuan, J.; Yu, S.; Cheng, Z. Amyloid β Causes Excitation/Inhibition Imbalance through Dopamine Receptor 1-Dependent Disruption of Fast-Spiking GABAergic Input in Anterior Cingulate Cortex. Sci. Rep. 2018, 8, 302. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Malinovskaya, N.A.; Komleva, Y.K.; Gorina, Y.V.; Shuvaev, A.N.; Olovyannikova, R.Y.; Belozor, O.S.; Belova, O.A.; Higashida, H.; Salmina, A.B. Excitation/Inhibition Imbalance and Impaired Neurogenesis in Neurodevelopmental and Neurodegenerative Disorders. Rev. Neurosci. 2019, 30, 807–820. [Google Scholar] [CrossRef]
- Ghosh, I.; Liu, C.S.; Swardfager, W.; Lanctôt, K.L.; Anderson, N.D. The Potential Roles of Excitatory-Inhibitory Imbalances and the Repressor Element-1 Silencing Transcription Factor in Aging and Aging-Associated Diseases. Mol. Cell Neurosci. 2021, 117, 103683. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network Abnormalities and Interneuron Dysfunction in Alzheimer Disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Yu, T.; Liu, X.; Wu, J.; Wang, Q. Electrophysiological Biomarkers of Epileptogenicity in Alzheimer’s Disease. Front. Hum. Neurosci. 2021, 15, 747077. [Google Scholar] [CrossRef]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and Impact of Subclinical Epileptiform Activity in Alzheimer’s Disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Sarkis, R.A.; Dickerson, B.C.; Cole, A.J.; Chemali, Z.N. Clinical and Neurophysiologic Characteristics of Unprovoked Seizures in Patients Diagnosed with Dementia. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 56–61. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- Sperling, R.A.; Dickerson, B.C.; Pihlajamaki, M.; Vannini, P.; LaViolette, P.S.; Vitolo, O.V.; Hedden, T.; Becker, J.A.; Rentz, D.M.; Selkoe, D.J.; et al. Functional Alterations in Memory Networks in Early Alzheimer’s Disease. Neuromolecular Med. 2010, 12, 27–43. [Google Scholar] [CrossRef]
- Targa Dias Anastacio, H.; Matosin, N.; Ooi, L. Neuronal Hyperexcitability in Alzheimer’s Disease: What Are the Drivers behind This Aberrant Phenotype? Transl. Psychiatry 2022, 12, 1–14. [Google Scholar] [CrossRef]
- Lerdkrai, C.; Asavapanumas, N.; Brawek, B.; Kovalchuk, Y.; Mojtahedi, N.; Olmedillas del Moral, M.; Garaschuk, O. Intracellular Ca2+ Stores Control in Vivo Neuronal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1279–E1288. [Google Scholar] [CrossRef]
- Busche, M.A.; Konnerth, A. Impairments of Neural Circuit Function in Alzheimer’s Disease. Philos. Trans. R. Soc. B: Biol. Sci. 2016, 371, 20150429. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical Role of Soluble Amyloid-β for Early Hippocampal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef]
- Leal, S.L.; Landau, S.M.; Bell, R.K.; Jagust, W.J. Hippocampal Activation Is Associated with Longitudinal Amyloid Accumulation and Cognitive Decline. eLife 2017, 6, e22978. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A. Amyloid β-Induced Neuronal Hyperexcitability Triggers Progressive Epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.A.; Gebhardt, F.M.; Mitrovic, A.D.; Vandenberg, R.J.; Dodd, P.R. Glutamate Transporter Variants Reduce Glutamate Uptake in Alzheimer’s Disease. Neurobiol. Aging 2011, 32, 553.e1–553.e11. [Google Scholar] [CrossRef] [PubMed]
- le Prince, G.; Delaere, P.; Fages, C.; Lefrançois, T.; Touret, M.; Salanon, M.; Tardy, M. Glutamine Synthetase (GS) Expression Is Reduced in Senile Dementia of the Alzheimer Type. Neurochem. Res. 1995, 20, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.-E.; Zhang, W.-M.; Yang, Z.Z.; Zhu, H.-M.; Yan, K.; Li, S.; Bagnard, D.; Dawe, G.S.; Ma, Q.-H.; Xiao, Z.-C. Amyloid Precursor Protein at Node of Ranvier Modulates Nodal Formation. Cell Adh. Migr. 2014, 8, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.-J.; Yang, G.; Wu, W.; Li, Q.-F.; Xu, D.-E.; Ntim, M.; Jiang, C.-Y.; Liu, J.-C.; Zhang, Y.; Wang, Y.-Z.; et al. Reducing Nav1.6 Expression Attenuates the Pathogenesis of Alzheimer’s Disease by Suppressing BACE1 Transcription. Aging Cell 2022, 21, e13593. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Palmieri, L.; Spera, I.; Porcelli, V.; Palmieri, F.; Fabis-Pedrini, M.J.; Kean, R.B.; Barkhouse, D.A.; Curtis, M.T.; Hooper, D.C. Oxidative Stress and Reduced Glutamine Synthetase Activity in the Absence of Inflammation in the Cortex of Mice with Experimental Allergic Encephalomyelitis. Neuroscience 2011, 185, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Muzio, L.; De Chiara, V.; Grasselli, G.; Musella, A.; Musumeci, G.; Mandolesi, G.; De Ceglia, R.; Maida, S.; Biffi, E.; et al. Impaired Striatal GABA Transmission in Experimental Autoimmune Encephalomyelitis. Brain Behav. Immun. 2011, 25, 947–956. [Google Scholar] [CrossRef]
- Centonze, D.; Muzio, L.; Rossi, S.; Cavasinni, F.; De Chiara, V.; Bergami, A.; Musella, A.; D’Amelio, M.; Cavallucci, V.; Martorana, A.; et al. Inflammation Triggers Synaptic Alteration and Degeneration in Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef]
- Bassi, M.S.; Mori, F.; Buttari, F.; Marfia, G.A.; Sancesario, A.; Centonze, D.; Iezzi, E. Neurophysiology of Synaptic Functioning in Multiple Sclerosis. Clin. Neurophysiol. 2017, 128, 1148–1157. [Google Scholar] [CrossRef]
- Reyes-Leiva, D.; Dols-Icardo, O.; Sirisi, S.; Cortés-Vicente, E.; Turon-Sans, J.; de Luna, N.; Blesa, R.; Belbin, O.; Montal, V.; Alcolea, D.; et al. Pathophysiological Underpinnings of Extra-Motor Neurodegeneration in Amyotrophic Lateral Sclerosis: New Insights From Biomarker Studies. Front. Neurol. 2021, 12, 750543. [Google Scholar] [CrossRef]
- Vucic, S.; Pavey, N.; Haidar, M.; Turner, B.J.; Kiernan, M.C. Cortical Hyperexcitability: Diagnostic and Pathogenic Biomarker of ALS. Neurosci. Lett. 2021, 759, 136039. [Google Scholar] [CrossRef]
- Van den Bos, M.A.J.; Higashihara, M.; Geevasinga, N.; Menon, P.; Kiernan, M.C.; Vucic, S. Imbalance of Cortical Facilitatory and Inhibitory Circuits Underlies Hyperexcitability in ALS. Neurology 2018, 91, e1669–e1676. [Google Scholar] [CrossRef]
- Menon, P.; Geevasinga, N.; van den Bos, M.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical Hyperexcitability and Disease Spread in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2017, 24, 816–824. [Google Scholar] [CrossRef]
- Fogarty, M.J. Driven to Decay: Excitability and Synaptic Abnormalities in Amyotrophic Lateral Sclerosis. Brain Res. Bull. 2018, 140, 318–333. [Google Scholar] [CrossRef]
- Huynh, W.; Simon, N.G.; Grosskreutz, J.; Turner, M.R.; Vucic, S.; Kiernan, M.C. Assessment of the Upper Motor Neuron in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2016, 127, 2643–2660. [Google Scholar] [CrossRef]
- van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef]
- Mills, K.R.; Nithi, K.A. Corticomotor Threshold Is Reduced in Early Sporadic Amyotrophic Lateral Sclerosis. Muscle Nerve 1997, 20, 1137–1141. [Google Scholar] [CrossRef]
- Zanette, G.; Tamburin, S.; Manganotti, P.; Refatti, N.; Forgione, A.; Rizzuto, N. Different Mechanisms Contribute to Motor Cortex Hyperexcitability in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2002, 113, 1688–1697. [Google Scholar] [CrossRef]
- Boston-Howes, W.; Gibb, S.L.; Williams, E.O.; Pasinelli, P.; Brown, R.H.J.; Trotti, D. Caspase-3 Cleaves and Inactivates the Glutamate Transporter EAAT2. J. Biol. Chem. 2006, 281, 14076–14084. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte Glutamate Transport: Review of Properties, Regulation, and Physiological Functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Foerster, B.R.; Callaghan, B.C.; Petrou, M.; Edden, R.A.E.; Chenevert, T.L.; Feldman, E.L. Decreased Motor Cortex -Aminobutyric Acid in Amyotrophic Lateral Sclerosis. Neurology 2012, 78, 1596–1600. [Google Scholar] [CrossRef]
- Petri, S.; Krampfl, K.; Hashemi, F.; Grothe, C.; Hori, A.; Dengler, R.; Bufler, J. Distribution of GABAA Receptor MRNA in the Motor Cortex of ALS Patients. J. Neuropathol. Exp. Neurol. 2003, 62, 1041–1051. [Google Scholar] [CrossRef]
- Nieto-Gonzalez, J.L.; Moser, J.; Lauritzen, M.; Schmitt-John, T.; Jensen, K. Reduced GABAergic Inhibition Explains Cortical Hyperexcitability in the Wobbler Mouse Model of ALS. Cereb. Cortex 2011, 21, 625–635. [Google Scholar] [CrossRef]
- Pieri, M.; Carunchio, I.; Curcio, L.; Mercuri, N.B.; Zona, C. Increased Persistent Sodium Current Determines Cortical Hyperexcitability in a Genetic Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2009, 215, 368–379. [Google Scholar] [CrossRef]
- Fogarty, M.J.; Noakes, P.G.; Bellingham, M.C. Motor Cortex Layer V Pyramidal Neurons Exhibit Dendritic Regression, Spine Loss, and Increased Synaptic Excitation in the Presymptomatic HSOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2015, 35, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical Synaptic and Dendritic Spine Abnormalities in a Presymptomatic TDP-43 Model of Amyotrophic Lateral Sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Kiernan, M.C. Upregulation of Persistent Sodium Conductances in Familial ALS. J. Neurol. Neurosurg. Psychiatry 2010, 81, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Pardillo-Díaz, R.; Carrascal, L.; Ayala, A.; Nunez-Abades, P. Oxidative Stress Induced by Cumene Hydroperoxide Evokes Changes in Neuronal Excitability of Rat Motor Cortex Neurons. Neuroscience 2015, 289, 85–98. [Google Scholar] [CrossRef]
- Nakaya, H.; Takeda, Y.; Tohse, N.; Kanno, M. Mechanism of the Membrane Depolarization Induced by Oxidative Stress in Guinea-Pig Ventricular Cells. J. Mol. Cell Cardiol. 1992, 24, 523–534. [Google Scholar] [CrossRef]
- Nani, F.; Cifra, A.; Nistri, A. Transient Oxidative Stress Evokes Early Changes in the Functional Properties of Neonatal Rat Hypoglossal Motoneurons in Vitro. Eur. J. Neurosci. 2010, 31, 951–966. [Google Scholar] [CrossRef]
- Bhatnagar, A. Electrophysiological Effects of 4-Hydroxynonenal, an Aldehydic Product of Lipid Peroxidation, on Isolated Rat Ventricular Myocytes. Circ. Res. 1995, 76, 293–304. [Google Scholar] [CrossRef]
- Jovanovic, Z.; Jovanovic, S. Comparison of the Effects of Cumene Hydroperoxide and Hydrogen Peroxide on Retzius Nerve Cells of the Leech Haemopis Sanguisuga. Exp. Anim. 2013, 62, 9–17. [Google Scholar] [CrossRef][Green Version]
- Angelova, P.; Müller, W. Oxidative Modulation of the Transient Potassium Current IA by Intracellular Arachidonic Acid in Rat CA1 Pyramidal Neurons. Eur. J. Neurosci. 2006, 23, 2375–2384. [Google Scholar] [CrossRef]
- Lamanauskas, N.; Nistri, A. Riluzole Blocks Persistent Na+ and Ca2+ Currents and Modulates Release of Glutamate via Presynaptic NMDA Receptors on Neonatal Rat Hypoglossal Motoneurons in Vitro. Eur. J. Neurosci. 2008, 27, 2501–2514. [Google Scholar] [CrossRef]
- Pouokam, E.; Rehn, M.; Diener, M. Effects of H2O2 at Rat Myenteric Neurones in Culture. Eur. J. Pharm. 2009, 615, 40–49. [Google Scholar] [CrossRef]
- Huang, W.F.; Ouyang, S.; Zhang, H. The Characteristics and Oxidative Modulation of Large-Conductance Calcium-Activated Potassium Channels in Guinea-Pig Colon Smooth Muscle Cells. Sheng Li Xue Bao 2009, 61, 285–291. [Google Scholar]
- Pardillo-Díaz, R.; Carrascal, L.; Muñoz, M.F.; Ayala, A.; Nunez-Abades, P. Time and Dose Dependent Effects of Oxidative Stress Induced by Cumene Hydroperoxide in Neuronal Excitability of Rat Motor Cortex Neurons. Neurotoxicology 2016, 53, 201–214. [Google Scholar] [CrossRef]
- Robinson, R.B.; Siegelbaum, S.A. Hyperpolarization-Activated Cation Currents: From Molecules to Physiological Function. Annu. Rev. Physiol. 2003, 65, 453–480. [Google Scholar] [CrossRef]
- Vimard, F.; Saucet, M.; Nicole, O.; Feuilloley, M.; Duval, D. Toxicity Induced by Cumene Hydroperoxide in PC12 Cells: Protective Role of Thiol Donors. J. Biochem. Mol. Toxicol. 2011, 25, 205–215. [Google Scholar] [CrossRef]
- Oh, J.M.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Oxidative Stress Impairs Autophagic Flux in Prion Protein-Deficient Hippocampal Cells. Autophagy 2012, 8, 1448–1461. [Google Scholar] [CrossRef]
- Carrascal, L.; Nunez-Abades, P.; Ayala, A.; Cano, M. Role of Melatonin in the Inflammatory Process and Its Therapeutic Potential. Curr. Pharm. Des. 2018, 24, 1563–1588. [Google Scholar] [CrossRef]
- Pardillo-Diaz, R.; Carrascal, L.; Barrionuevo, G.; Nunez-Abades, P. Oxidative Stress Induced by Cumene Hydroperoxide Produces Synaptic Depression and Transient Hyperexcitability in Rat Primary Motor Cortex Neurons. Mol. Cell. Neurosci. 2017, 82, 204–217. [Google Scholar] [CrossRef]
- Maekawa, S.; Al-Sarraj, S.; Kibble, M.; Landau, S.; Parnavelas, J.; Cotter, D.; Everall, I.; Leigh, P.N. Cortical Selective Vulnerability in Motor Neuron Disease: A Morphometric Study. Brain 2004, 127, 1237–1251. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Chapter 4-Reactive Oxygen Species, Oxidative Damage and Cell Death. In Immunity and Inflammation in Health and Disease; Chatterjee, S., Jungraithmayr, W., Bagchi, D.B.T.-I., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 45–55. ISBN 978-0-12-805417-8. [Google Scholar]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) Modified Proteins in Metabolic Diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef]
- Deepashree, S.; Niveditha, S.; Shivanandappa, T.; Ramesh, S.R. Oxidative Stress Resistance as a Factor in Aging: Evidence from an Extended Longevity Phenotype of Drosophila Melanogaster. Biogerontology 2019, 20, 497–513. [Google Scholar] [CrossRef]
- Guevara, R.; Gianotti, M.; Oliver, J.; Roca, P. Age and Sex-Related Changes in Rat Brain Mitochondrial Oxidative Status. Exp. Gerontol. 2011, 46, 923–928. [Google Scholar] [CrossRef]
- Carrascal, L.; Gorton, E.; Pardillo-Díaz, R.; Perez-García, P.; Gómez-Oliva, R.; Castro, C.; Nunez-Abades, P. Age-Dependent Vulnerability to Oxidative Stress of Postnatal Rat Pyramidal Motor Cortex Neurons. Antioxidants 2020, 9, 1307. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial Signatures and Their Role in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Tricoire, L.; Tania, V. Neuronal Nitric Oxide Synthase Expressing Neurons: A Journey from Birth to Neuronal Circuits. Front. Neural Circuits 2012, 6, 82. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Neuendorf, R.; Harding, A.; Stello, N.; Hanes, D.; Wahbeh, H. Depression and Anxiety in Patients with Inflammatory Bowel Disease: A Systematic Review. J. Psychosom. Res. 2016, 87, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.J.; Espinosa-Oliva, A.M.; Oliva-Martin, M.J.; Carrillo-Jimenez, A.; Venero, J.L.; de Pablos, R.M. Collateral Damage: Contribution of Peripheral Inflammation to Neurodegenerative Diseases. Curr. Top. Med. Chem. 2015, 15, 2193–2210. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J. Concepts of Neural Nitric Oxide-Mediated Transmission. Eur. J. Neurosci. 2008, 27, 2783–2802. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Mechanisms of Inflammatory Neurodegeneration: INOS and NADPH Oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef]
- Carrascal, L.; Vázquez-Carretero, M.D.; García-Miranda, P.; Fontán-Lozano, Á.; Calonge, M.L.; Ilundáin, A.A.; Castro, C.; Nunez-Abades, P.; Peral, M.J. Acute Colon Inflammation Triggers Primary Motor Cortex Glial Activation, Neuroinflammation, Neuronal Hyperexcitability, and Motor Coordination Deficits. Int. J. Mol. Sci. 2022, 23, 5347. [Google Scholar] [CrossRef]
- Griego, E.; Santiago-Jiménez, G.; Galván, E.J. Systemic Administration of Lipopolysaccharide Induces Hyperexcitability of Prelimbic Neurons via Modulation of Sodium and Potassium Currents. Neurotoxicology 2022, 91, 128–139. [Google Scholar] [CrossRef]
- Czapski, G.A.; Cakala, M.; Chalimoniuk, M.; Gajkowska, B.; Strosznajder, J.B. Role of Nitric Oxide in the Brain during Lipopolysaccharide-evoked Systemic Inflammation. J. Neurosci. Res. 2007, 85, 1694–1703. [Google Scholar] [CrossRef]
- Beck, H.; Yaari, Y. Plasticity of Intrinsic Neuronal Properties in CNS Disorders. Nat. Rev. Neurosci. 2008, 9, 357–369. [Google Scholar] [CrossRef]
- Griego, E.; Hernández-Frausto, M.; Márquez, L.A.; Lara-Valderrabano, L.; López Rubalcava, C.; Galván, E.J. Activation of D1/D5 Receptors Ameliorates Decreased Intrinsic Excitability of Hippocampal Neurons Induced by Neonatal Blockade of N-Methyl-d-Aspartate Receptors. Br. J. Pharmacol. 2021, 179, 1695–1715. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardillo-Díaz, R.; Pérez-García, P.; Castro, C.; Nunez-Abades, P.; Carrascal, L. Oxidative Stress as a Potential Mechanism Underlying Membrane Hyperexcitability in Neurodegenerative Diseases. Antioxidants 2022, 11, 1511. https://doi.org/10.3390/antiox11081511
Pardillo-Díaz R, Pérez-García P, Castro C, Nunez-Abades P, Carrascal L. Oxidative Stress as a Potential Mechanism Underlying Membrane Hyperexcitability in Neurodegenerative Diseases. Antioxidants. 2022; 11(8):1511. https://doi.org/10.3390/antiox11081511
Chicago/Turabian StylePardillo-Díaz, Ricardo, Patricia Pérez-García, Carmen Castro, Pedro Nunez-Abades, and Livia Carrascal. 2022. "Oxidative Stress as a Potential Mechanism Underlying Membrane Hyperexcitability in Neurodegenerative Diseases" Antioxidants 11, no. 8: 1511. https://doi.org/10.3390/antiox11081511
APA StylePardillo-Díaz, R., Pérez-García, P., Castro, C., Nunez-Abades, P., & Carrascal, L. (2022). Oxidative Stress as a Potential Mechanism Underlying Membrane Hyperexcitability in Neurodegenerative Diseases. Antioxidants, 11(8), 1511. https://doi.org/10.3390/antiox11081511