
Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation


Abstract
:
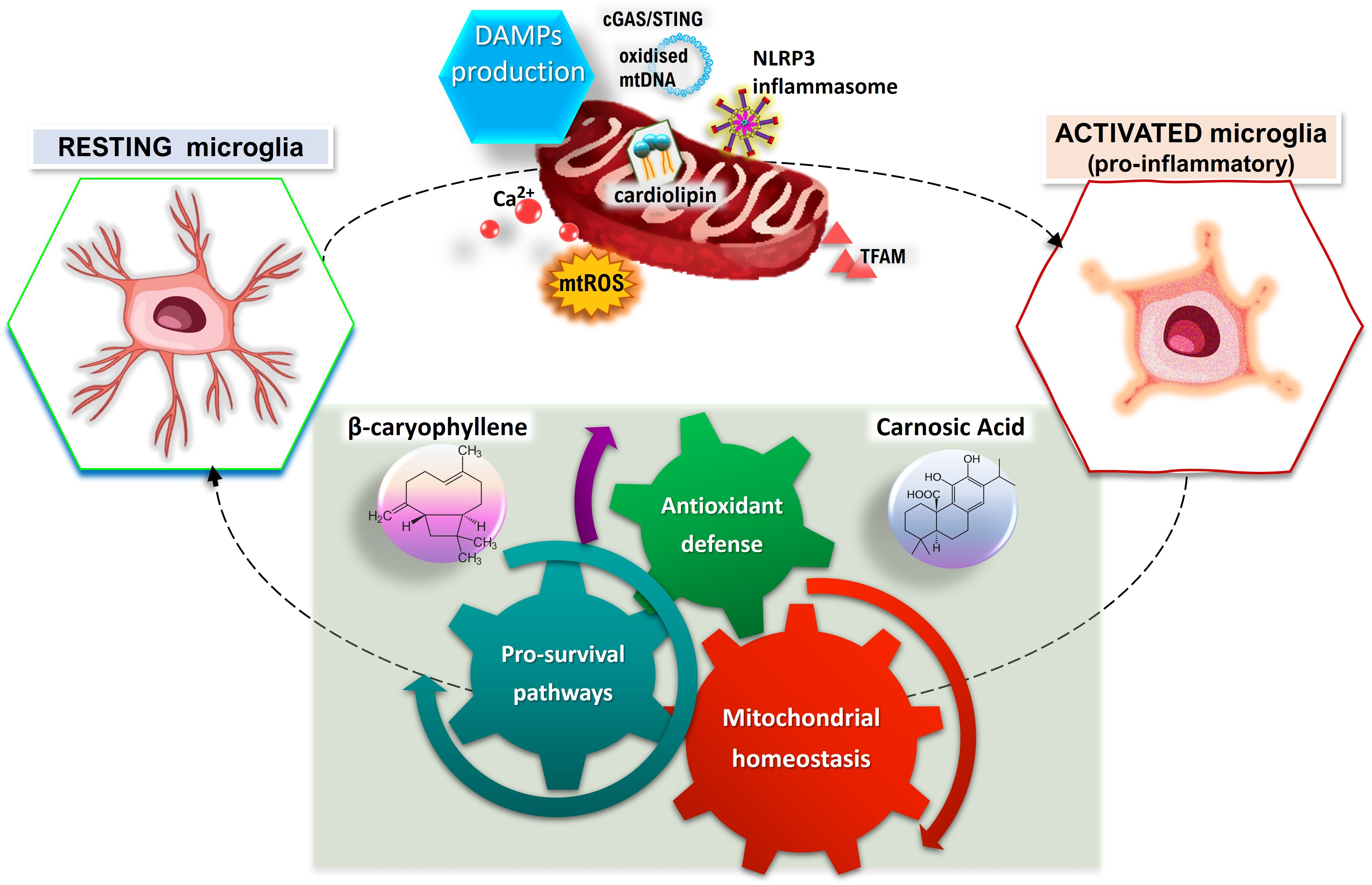{kind=link}
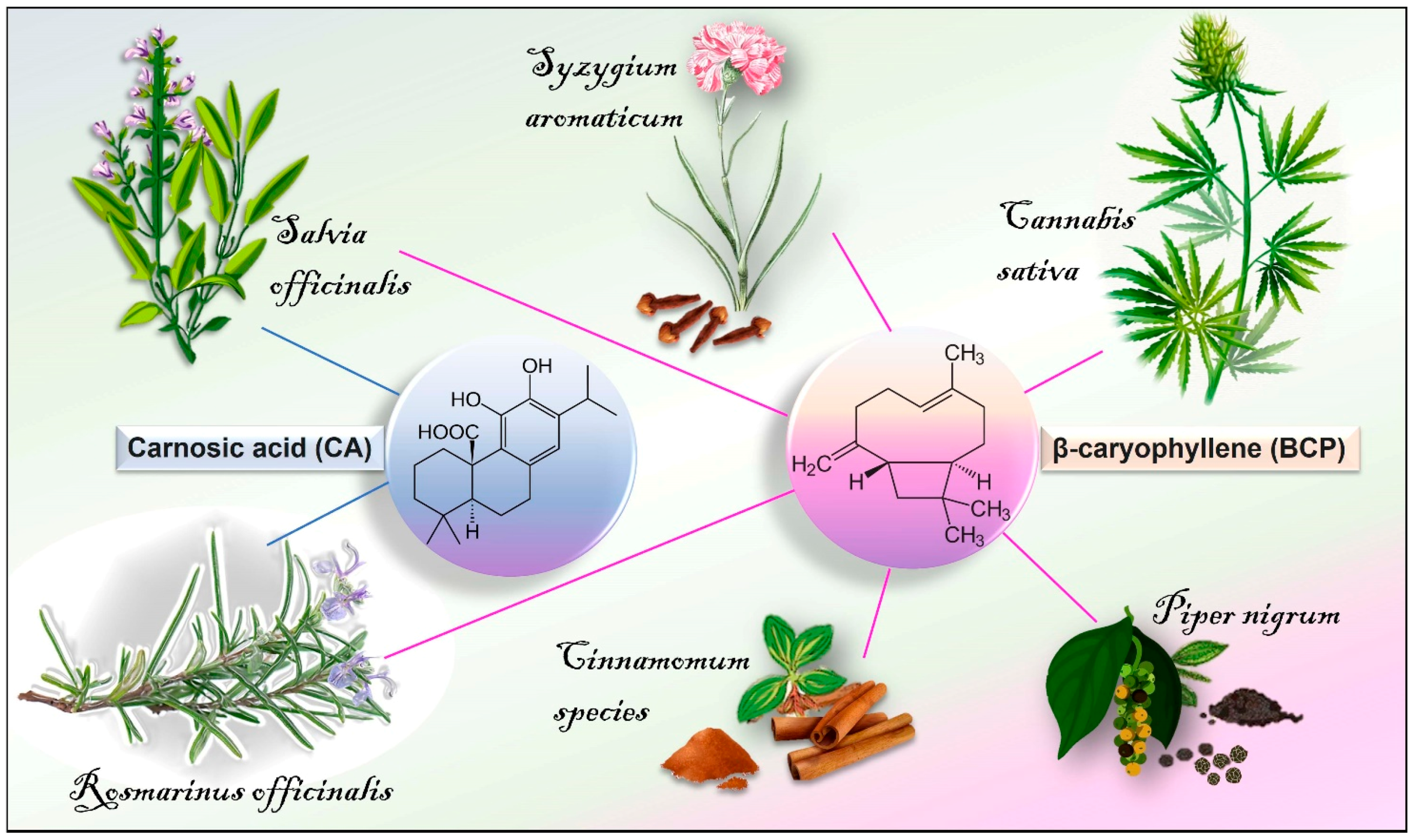{kind=link}
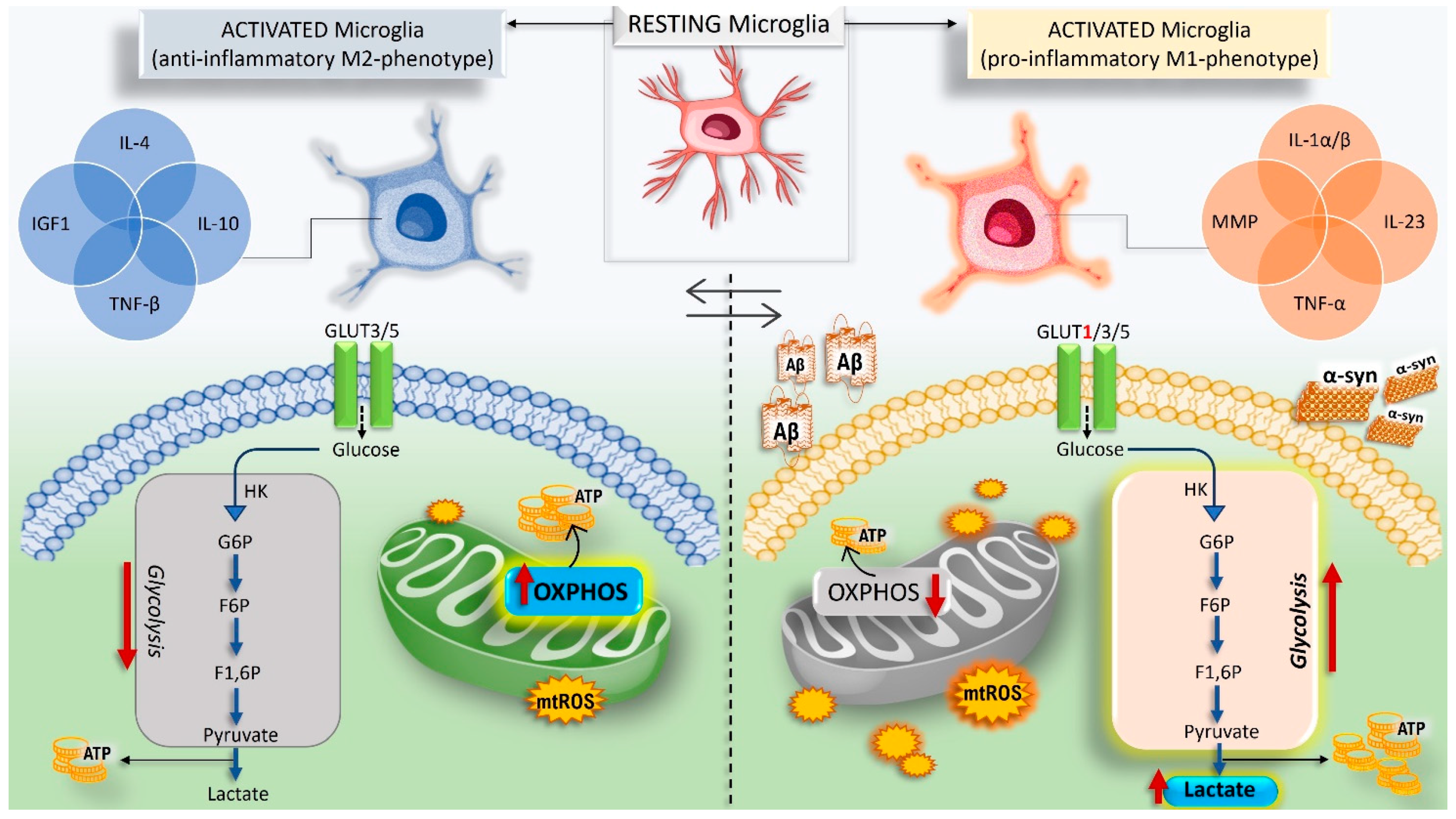{kind=link}
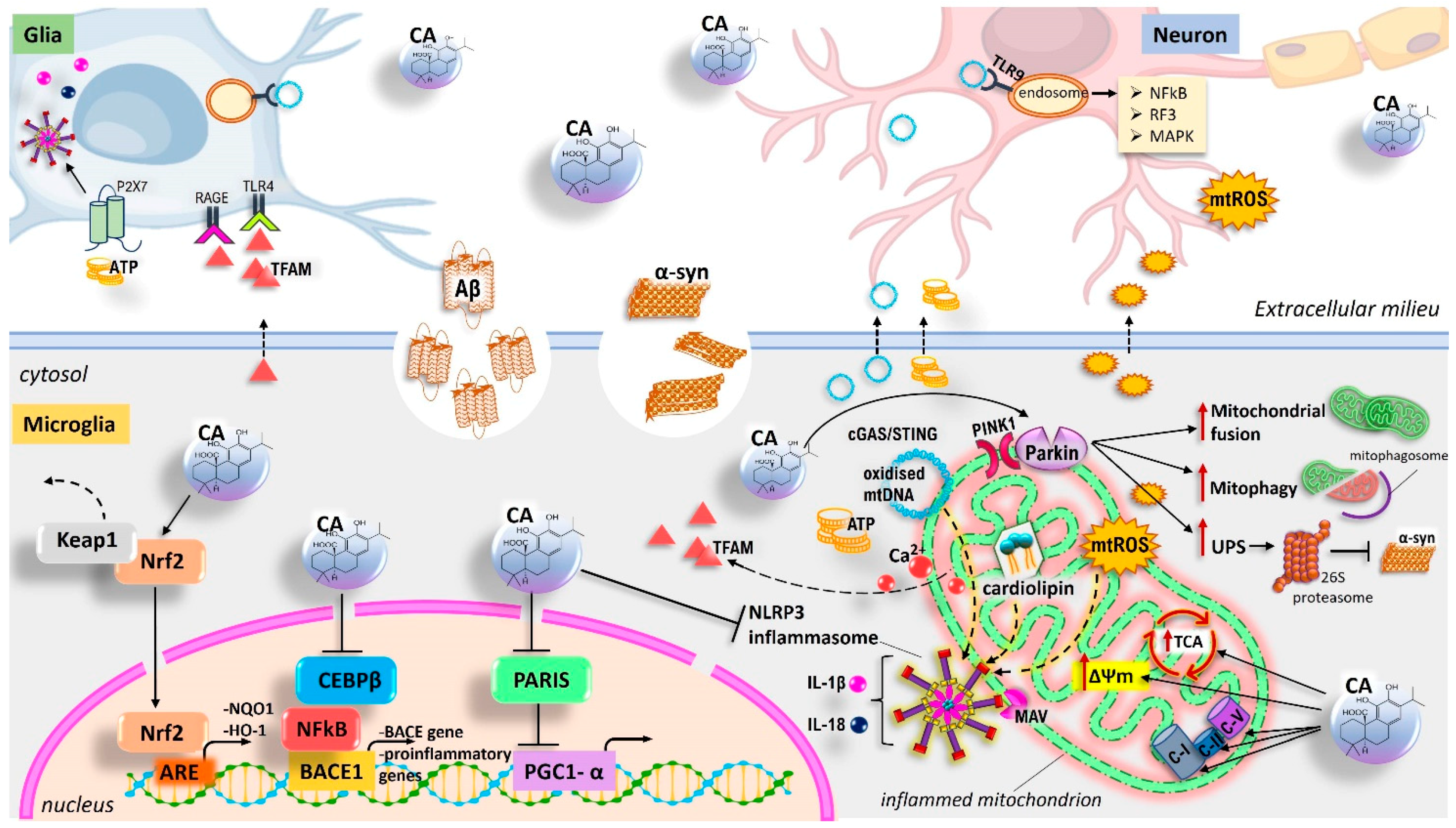{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Oxidative Stress and Inflammation as Common Aspects in the Pathogenesis of Chronic Diseases: A General Overview
3. β-Caryophyllene
3.1. Chemistry, Vegetable Sources, and Pharmacokinetics
3.2. Therapeutic Potential and Polypharmacological Activities of β-Caryophyllene
4. Carnosic Acid
4.1. Chemistry, Vegetable Sources, and Pharmacokinetics
4.2. Therapeutic Potential, Biological Activities, and Pharmacological Effects of Carnosic Acid
5. Neuroprotective Potential of β-Caryophyllene and Carnosic Acid in CNS and Visual System
5.1. Mitochondria as a “Trait-D’Union” in the Complex Interplay between Neuroinflammation and Neurodegeneration
5.2. CA and BCP as a Promising Immunomodulatory Strategy to Maintain Glial and Mitochondrial Homeostasis
5.3. CA Drives the Shift of Microglia Polarization in Neuroinflammation and Ameliorates Neurodegeneration in Models of AD and PD
5.4. Mechanisms of CA-Activated Neuroprotection by Modulating Mitochondrial Homeostasis
5.5. In Vitro Experimental Models
5.6. In Vivo and Ex Vivo Experimental Models
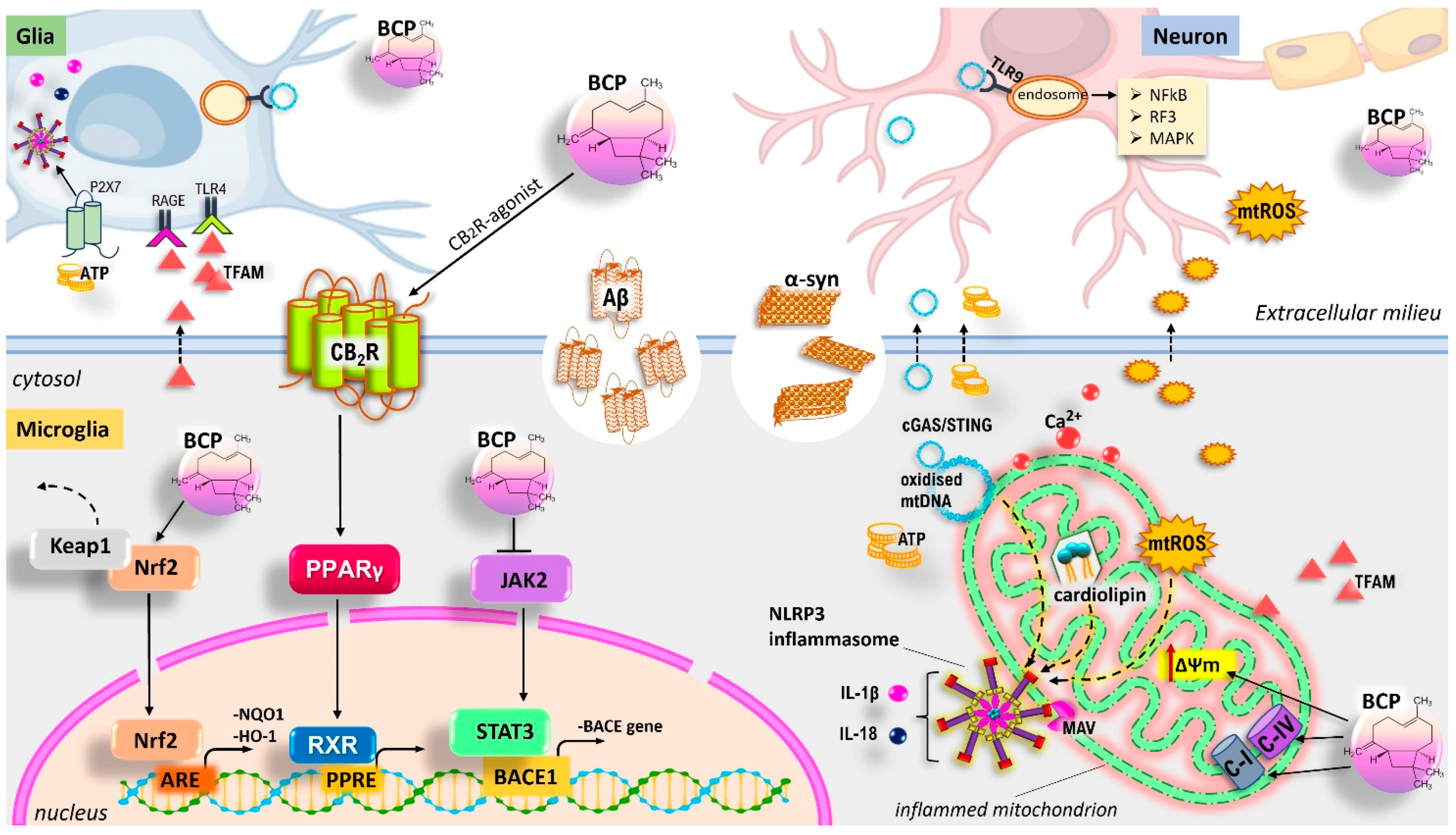
5.7. BCP and Its Neuroprotective Functions: CB2R-Mediated Regulation of the Glia-Mediated Neuroinflammation and Improvement of Neurodegeneration in Model of AD and PD
5.8. Neuroprotective Role of BCP Via Mitochondrial Homeostasis Management
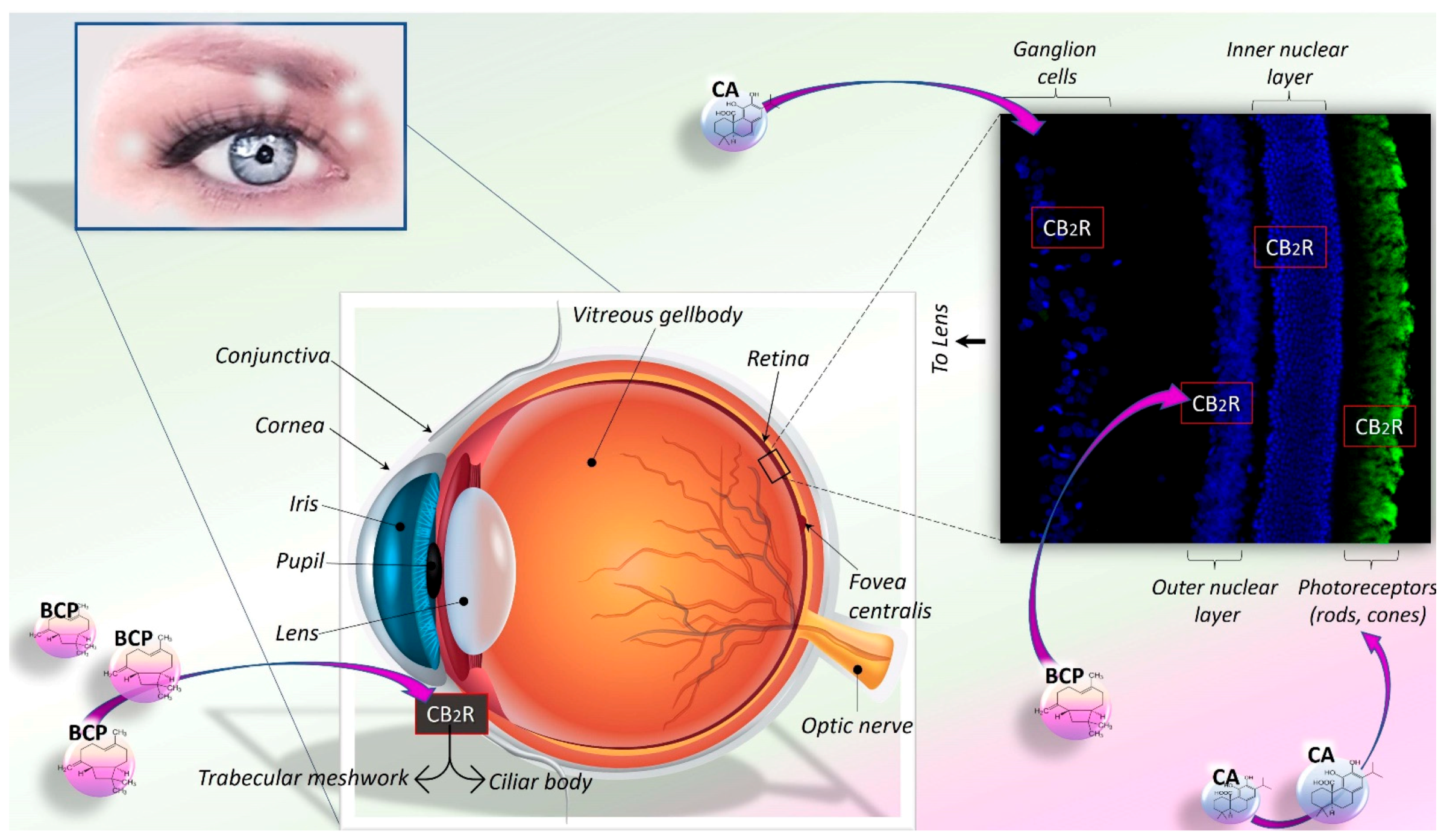
6. CA and BCP: Therapeutic Potential in Eye-Related Diseases
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statements
Acknowledgments
Conflicts of Interest
References
- World Health Organization Noncommunicable Diseases (NCD). 2019. Available online: https://www.who.int/gho/ncd/mortality_morbidity/en/ (accessed on 3 January 2020).
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; AlMazroa, M.A.; Amann, M.; Anderson, H.R.; Andrews, K.G.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef] [Green Version]
- WHO. Global Action Plan for the Prevention and Control of Noncommunicable Diseases 2013–2020; World Health Organization: Geneva, Switzerland, 2013; Available online: http://apps.who.int/iris/bitstream/10665/94384/1/9789241506236_eng.pdf (accessed on 11 February 2016).
- Beard, J.R.; Officer, A.; de Carvalho, I.A.; Sadana, R.; Pot, A.M.; Michel, J.-P.; Lloyd-Sherlock, P.; Epping-Jordan, J.E.; Peeters, G.M.E.E.G.; Mahanani, W.R.; et al. The World report on ageing and health: A policy framework for healthy ageing. Lancet 2016, 387, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef]
- Devasagayam, T.P.A.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Phys. India 2004, 52, 794–804. [Google Scholar]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic Inflammation and Oxidative Stress as a Major Cause of Age—Related Diseases and Cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef]
- Camps, J.; García-Heredia, A. Introduction: Oxidation and Inflammation, A Molecular Link between Non-communicable Diseases. Adv. Exp. Med. Biol. 2014, 824, 1–4. [Google Scholar]
- Peña-Oyarzun, D.; Bravo-Sagua, R.; Vegas, A.D.; Aleman, L.; Chiong, M.; Garcia, L.; Bambs, C.; Troncoso, R.; Cifuentes, M.; Morselli, E.; et al. Autophagy and oxidative stress in non-communicable diseases: A matter of the inflammatory state? Free Radic. Biol. Med. 2018, 124, 61–78. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The Role of Oxidative Stress and Inflammation in Cardiovascular Aging. BioMed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef]
- Hulsmans, M.; Holvoet, P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell. Mol. Med. 2009, 14, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Closa, D.; Puy, E.F. Oxygen Free Radicals and the Systemic Inflammatory Response. IUBMB Life 2004, 56, 185–191. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Di Gioacchino, M.; Tonacci, A.; Musolino, C.; Gangemi, S. Immunopathology of SARS-CoV-2 Infection: Immune Cells and Mediators, Prognostic Factors, and Immune-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 4782. [Google Scholar] [CrossRef]
- Iorio, R.; Castellucci, A.; Rossi, G.; Cinque, B.; Cifone, M.G.; Macchiarelli, G.; Cecconi, S. Mancozeb affects mitochondrial activity, redox status and ATP production in mouse granulosa cells. Toxicol. In Vitro 2015, 30, 438–445. [Google Scholar] [CrossRef]
- Petricca, S.; Flati, V.; Celenza, G.; Di Gregorio, J.; Lizzi, A.R.; Luzi, C.; Cristiano, L.; Cinque, B.; Rossi, G.; Festuccia, C.; et al. Tebuconazole and Econazole Act Synergistically in Mediating Mitochondrial Stress, Energy Imbalance, and Sequential Activation of Autophagy and Apoptosis in Mouse Sertoli TM4 Cells: Possible Role of AMPK/ULK1 Axis. Toxicol. Sci. 2019, 169, 209–223. [Google Scholar] [CrossRef]
- Petricca, S.; Celenza, G.; Luzi, C.; Cinque, B.; Lizzi, A.R.; Franceschini, N.; Festuccia, C.; Iorio, R. Synergistic Activity of Ketoconazole and Miconazole with Prochloraz in Inducing Oxidative Stress, GSH Depletion, Mitochondrial Dysfunction, and Apoptosis in Mouse Sertoli TM4 Cells. Int. J. Mol. Sci. 2022, 23, 5429. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The Multifaceted Role of Nrf2 in Mitochondrial Function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radic. Biol. Med. 2021, 179, 133–143. [Google Scholar] [CrossRef]
- Sabouny, R.; Fraunberger, E.; Geoffrion, M.; Ng, A.C.-H.; Baird, S.D.; Screaton, R.A.; Milne, R.; McBride, H.; Shutt, T. The Keap1–Nrf2 Stress Response Pathway Promotes Mitochondrial Hyperfusion Through Degradation of the Mitochondrial Fission Protein Drp1. Antioxid. Redox Signal. 2017, 27, 1447–1459. [Google Scholar] [CrossRef]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Ferrer, I.J.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.-Y.; Liu, Q.-R.; Shen, H.; Xi, Z.-X.; et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 2017, 95, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2019, 16, 9–29. [Google Scholar] [CrossRef]
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56 (Suppl. S1), 244–253. [Google Scholar] [CrossRef] [Green Version]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Muccioli, G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; Lopez-Rodriguez, M.L.; Bajjalieh, S.; Stella, N. Identification of a Novel Endocannabinoid-Hydrolyzing Enzyme Expressed by Microglial Cells. J. Neurosci. 2007, 27, 2883–2889. [Google Scholar] [CrossRef] [Green Version]
- Young, A.P.; Denovan-Wright, E.M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 806417. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, S.; Jones, J.; Lee, G.J. Plant-Based Dietary Patterns, Plant Foods, and Age-Related Cognitive Decline. Adv. Nutr. 2019, 10, S422–S436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anyene, I.C.; Ergas, I.J.; Kwan, M.L.; Roh, J.M.; Ambrosone, C.B.; Kushi, L.H.; Feliciano, E.M.C. Plant-Based Dietary Patterns and Breast Cancer Recurrence and Survival in the Pathways Study. Nutrients 2021, 13, 3374. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.H.; Cheong, H.C.; Tu, Y.-K.; Kuo, P.-H. Association between Plant-Based Dietary Patterns and Risk of Cardiovascular Disease: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. Nutrients 2021, 13, 3952. [Google Scholar] [CrossRef]
- Zahedipour, F.; Hosseini, S.A.; Henney, N.; Barreto, G.; Sahebkar, A. Phytochemicals as inhibitors of tumor necrosis factor alpha and neuroinflammatory responses in neurodegenerative diseases. Neural Regen. Res. 2022, 17, 1675–1684. [Google Scholar] [CrossRef]
- Zeng, Y.; Xiong, Y.; Yang, T.; Wang, Y.; Zeng, J.; Zhou, S.; Luo, Y.; Li, L. Icariin and its metabolites as potential protective phytochemicals against cardiovascular disease: From effects to molecular mechanisms. Biomed. Pharmacother. 2022, 147, 112642. [Google Scholar] [CrossRef]
- Bag, S.; Mondal, A.; Majumder, A.; Banik, A. Tea and its phytochemicals: Hidden health benefits & modulation of signaling cascade by phytochemicals. Food Chem. 2021, 371, 131098. [Google Scholar] [CrossRef]
- Cote, B.; Elbarbry, F.; Bui, F.; Su, J.W.; Seo, K.; Nguyen, A.; Lee, M.; Rao, D.A. Mechanistic Basis for the Role of Phytochemicals in Inflammation-Associated Chronic Diseases. Molecules 2022, 27, 781. [Google Scholar] [CrossRef]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.-Z.; Xie, X.-Q.; Altmann, K.-H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [Green Version]
- Gertsch, J. Antiinflammatory cannabinoids in diet—Towards a better understanding of CB2 receptor action? Commun. Integr. Biol. 2008, 1, 26–28. [Google Scholar] [CrossRef]
- Sain, S.; Naoghare, P.K.; Saravana Devi, S.; Daiwile, A.; Krishnamurthi, K.; Arrigo, P.; Chakrabarti, T. Beta Caryophyllene and Caryophyllene Oxide, Isolated from Aegle Marmelos, as the Potent Anti-inflammatory Agents against Lymphoma and Neuroblastoma Cells. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2014, 13, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Rufino, A.T.; Ribeiro, M.; Sousa, C.; Judas, F.; Salgueiro, L.; Cavaleiro, C.; Mendes, A.F. Evaluation of the anti-inflammatory, anti-catabolic and pro-anabolic effects of E-caryophyllene, myrcene and limonene in a cell model of osteoarthritis. Eur. J. Pharmacol. 2015, 750, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, C.; Dai, X.; Ao, Y.; Li, Y. Inhibitory effect of trans-caryophyllene (TC) on leukocyte-endothelial attachment. Toxicol. Appl. Pharmacol. 2017, 329, 326–333. [Google Scholar] [CrossRef]
- Youssef, D.A.; El-Fayoumi, H.M.; Mahmoud, M.F. Beta-caryophyllene protects against diet-induced dyslipidemia and vascular inflammation in rats: Involvement of CB2 and PPAR-γ receptors. Chem.-Biol. Interact. 2018, 297, 16–24. [Google Scholar] [CrossRef]
- Youssef, D.A.; El-Fayoumi, H.M.; Mahmoud, M.F. Beta-caryophyllene alleviates diet-induced neurobehavioral changes in rats: The role of CB2 and PPAR-γ receptors. Biomed. Pharmacother. 2018, 110, 145–154. [Google Scholar] [CrossRef]
- Birtić, S.; Dussort, P.; Pierre, F.-X.; Bily, A.C.; Roller, M. Carnosic acid. Phytochemistry 2015, 115, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Keam, S.; Megawati, D.; Patel, S.K.; Tiwari, R.; Dhama, K.; Harapan, H. Immunopathology and immunotherapeutic strategies in severe acute respiratory syndrome coronavirus 2 infection. Rev. Med. Virol. 2020, 30, e2123. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrona, D. Neural–immune interactions: An integrative view of the bidirectional relationship between the brain and immune systems. J. Neuroimmunol. 2006, 172, 38–58. [Google Scholar] [CrossRef]
- Han, R. Plasma lipoproteins are important components of the immune system. Microbiol. Immunol. 2010, 54, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Spilianakis, C.G.; Flavell, R.A. How are TH1 and TH2 effector cells made? Curr. Opin. Immunol. 2009, 21, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2015, 13, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2015, 16, 7–21. [Google Scholar] [CrossRef]
- Ojcius, D.; Saïd-Sadier, N. Alarmins, inflammasomes and immunity. Biomed. J. 2012, 35, 437–449. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Crockett-Torabi, E.; Ward, P.A. The role of leukocytes in tissue injury. Eur. J. Anaesthesiol. 1996, 13, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, J.L.; Tate, M.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.; Mansell, A. Activation of the NLRP3 Inflammasome by IAV Virulence Protein PB1-F2 Contributes to Severe Pathophysiology and Disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, S.; Lan, X.; Wen, H.; Lederman, R.; Chawla, A.; Attia, M.; Bongu, R.P.; Husain, M.; Mikulak, J.; Saleem, M.A.; et al. HIV Promotes NLRP3 Inflammasome Complex Activation in Murine HIV-Associated Nephropathy. Am. J. Pathol. 2015, 186, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, R.R.; Wieser, M.E.; Ganga, R.R.; Barathi, V.A.; Lakshminarayanan, R.; Mohan, R.R.; Hainsworth, D.P.; Chaurasia, S.S. NOD-like Receptors in the Eye: Uncovering Its Role in Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 899. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, A.; Lv, J.; Zhang, Q.; Ran, Y.; Wei, C.; Wu, J. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur. J. Med. Chem. 2019, 185, 111822. [Google Scholar] [CrossRef]
- Salzano, S.; Checconi, P.; Hanschmann, E.-M.; Lillig, C.H.; Bowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef] [Green Version]
- Warnatsch, A.; Tsourouktsoglou, T.-D.; Branzk, N.; Wang, Q.; Reincke, S.; Herbst, S.; Gutierrez, M.; Papayannopoulos, V. Reactive Oxygen Species Localization Programs Inflammation to Clear Microbes of Different Size. Immunity 2017, 46, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-κB and AP-1 Connection: Mechanism of NF-κB-Dependent Regulation of AP-1 Activity. Mol. Cell. Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesarwani, P.; Murali, A.K.; Al-Khami, A.A.; Mehrotra, S. Redox Regulation of T-Cell Function: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2013, 18, 1497–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manicone, A.M.; McGuire, J.K. Matrix Metalloproteinases as Modulators of Inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirantes, C.; Passegue, E.; Pietras, E.M. Pro-inflammatory cytokines: Emerging players regulating HSC function in normal and diseased hematopoiesis. Exp. Cell Res. 2014, 329, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaptoge, S.; Seshasai, S.R.K.; Jørgensen, T.; Danesh, J.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.A. Role of interleukins in obesity: Implications for metabolic disease. Trends Endocrinol. Metab. 2014, 25, 312–319. [Google Scholar] [CrossRef]
- EssOilDB. Available online: https://nipgr.ac.in/Essoildb/ (accessed on 10 March 2022).
- Maffei, M.E. Plant Natural Sources of the Endocannabinoid (E)-β-Caryophyllene: A Systematic Quantitative Analysis of Published Literature. Int. J. Mol. Sci. 2020, 21, 6540. [Google Scholar] [CrossRef]
- Schmitt, D.; Levy, R.; Carroll, B. Toxicological Evaluation of β-Caryophyllene Oil: Subchronic Toxicity in Rats. Int. J. Toxicol. 2016, 35, 558–567. [Google Scholar] [CrossRef]
- da Silva Oliveira, G.L.; Machado, K.C.; Machado, K.C.; Feitosa, C.M.; de Castro Almeida, F.R. Non-clinical toxicity of β -caryophyllene, a dietary cannabinoid: Absence of adverse effects in female Swiss mice. Regul. Toxicol. Pharmacol. 2018, 92, 338–346. [Google Scholar] [CrossRef]
- Hashiesh, H.M.; Meeran, M.F.N.; Sharma, C.; Sadek, B.; Al Kaabi, J.; Ojha, S.K. Therapeutic Potential of β-Caryophyllene: A Dietary Cannabinoid in Diabetes and Associated Complications. Nutrients 2020, 12, 2963. [Google Scholar] [CrossRef]
- Liu, H.; Yang, G.; Tang, Y.; Cao, D.; Qi, T.; Qi, Y.; Fan, G. Physicochemical characterization and pharmacokinetics evaluation of β-caryophyllene/β-cyclodextrin inclusion complex. Int. J. Pharm. 2013, 450, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Baldissera, M.D.; Souza, C.F.; Grando, T.H.; Doleski, P.H.; Boligon, A.A.; Stefani, L.M.; Monteiro, S.G. Hypolipidemic effect of β-caryophyllene to treat hyperlipidemic rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y.; Kishi, C.; Sugiura, Y.; Yoshioka, Y.; Matsumura, S.; Moriyama, T.; Zaima, N. Distribution of inhaled volatile β-caryophyllene and dynamic changes of liver metabolites in mice. Sci. Rep. 2021, 11, 1728. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Peng, J.; Zhong, J.; Yang, M.; Pang, J.; Lou, J.; Li, M.; An, R.; Zhang, Q.; Xu, L.; et al. β-Caryophyllene protectsin vitroneurovascular unit against oxygen-glucose deprivation and re-oxygenation-induced injury. J. Neurochem. 2016, 139, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, M. Characterization of Allosteric Modulators of CB2 Receptors as Novel Therapeutics for Inflammatory Diseases. Ph.D. Thesis, University of Arkansas for Medical Sciences, Little Rock, AR, USA, 2011; p. 198. [Google Scholar]
- Christopoulos, A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef]
- Melancon, B.J.; Hopkins, C.R.; Wood, M.R.; Emmitte, K.A.; Niswender, C.M.; Christopoulos, A.; Conn, P.J.; Lindsley, C.W. Allosteric Modulation of Seven Transmembrane Spanning Receptors: Theory, Practice, and Opportunities for Central Nervous System Drug Discovery. J. Med. Chem. 2011, 55, 1445–1464. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of G Protein-Coupled Receptors: An Emerging Approach of Drug Discovery. Austin J. Pharmacol. Ther. 2014, 2, 1101. [Google Scholar]
- Gonçalves, E.C.D.; Baldasso, G.M.; Bicca, M.A.; Paes, R.S.; Capasso, R.; Dutra, R.C. Terpenoids, Cannabimimetic Ligands, beyond the Cannabis Plant. Molecules 2020, 25, 1567. [Google Scholar] [CrossRef] [Green Version]
- Howlett, A.C.; Abood, M.E. CB 1 and CB 2 Receptor Pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar]
- Rieder, S.A.; Chauhan, A.; Singh, U.; Nagarkatti, M.; Nagarkatti, P. Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology 2010, 215, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Karmaus, P.W.F.; Chen, W.; Kaplan, B.; Kaminski, N.E. Δ9-Tetrahydrocannabinol Suppresses Cytotoxic T Lymphocyte Function Independent of CB1 and CB2, Disrupting Early Activation Events. J. Neuroimmune Pharmacol. 2011, 7, 843–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Zeng, Z.; Wang, B.; Guo, S. Trans-caryophyllene inhibits amyloid β (Aβ) oligomer-induced neuroinflammation in BV-2 microglial cells. Int. Immunopharmacol. 2017, 51, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ma, W.; Du, J. β-Caryophyllene (BCP) ameliorates MPP+ induced cytotoxicity. Biomed. Pharmacother. 2018, 103, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Shafiee-Nick, R. Promising neuroprotective effects of β-caryophyllene against LPS-induced oligodendrocyte toxicity: A mechanistic study. Biochem. Pharmacol. 2018, 159, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Shafiee-Nick, R. The protective effects of β-caryophyllene on LPS-induced primary microglia M1/M2 imbalance: A mechanistic evaluation. Life Sci. 2019, 219, 40–73. [Google Scholar] [CrossRef]
- Assis, L.; Straliotto, M.; Engel, D.; Hort, M.; Dutra, R.; de Bem, A. β-Caryophyllene protects the C6 glioma cells against glutamate-induced excitotoxicity through the Nrf2 pathway. Neuroscience 2014, 279, 220–231. [Google Scholar] [CrossRef]
- Borgonetti, V.; Benatti, C.; Governa, P.; Isoldi, G.; Pellati, F.; Alboni, S.; Tascedda, F.; Montopoli, M.; Galeotti, N.; Manetti, F.; et al. Non-psychotropic Cannabis sativa L. phytocomplex modulates microglial inflammatory response through CB2 receptors-, endocannabinoids-, and NF-κB-mediated signaling. Phytother. Res. 2022, 36, 2246–2263. [Google Scholar] [CrossRef]
- Askari, V.R.; Baradaran Rahimi, V.; Tabatabaee, S.A.; Shafiee-Nick, R. Combination of Imipramine, a sphingomyelinase inhibitor, and β-caryophyllene improve their therapeutic effects on experimental autoimmune encephalomyelitis (EAE). Int. Immunopharmacol. 2019, 77, 105923. [Google Scholar] [CrossRef]
- Ku, C.-M.; Lin, J.-Y. Anti-inflammatory effects of 27 selected terpenoid compounds tested through modulating Th1/Th2 cytokine secretion profiles using murine primary splenocytes. Food Chem. 2013, 141, 1104–1113. [Google Scholar] [CrossRef]
- Jha, N.K.; Sharma, C.; Hashiesh, H.M.; Arunachalam, S.; Meeran, M.N.; Javed, H.; Patil, C.R.; Goyal, S.N.; Ojha, S. β-Caryophyllene, A Natural Dietary CB2 Receptor Selective Cannabinoid can be a Candidate to Target the Trinity of Infection, Immunity, and Inflammation in COVID-19. Front. Pharmacol. 2021, 12, 590201. [Google Scholar] [CrossRef]
- Oboh, G.; Olasehinde, T.A.; Ademosun, A.O. Essential Oil from Lemon Peels Inhibit Key Enzymes Linked to Neurodegenerative Conditions and Pro-oxidant Induced Lipid Peroxidation. J. Oleo Sci. 2014, 63, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, A.; Saikia, S.K.; Shukla, V.; Asthana, J.; Akhoon, B.A.; Pandey, R. Beta-caryophyllene modulates expression of stress response genes and mediates longevity in Caenorhabditis elegans. Exp. Gerontol. 2014, 57, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Alberti, T.B.; Barbosa, W.L.; Vieira, J.L.; Raposo, N.R.; Dutra, R.C. (−)-β-Caryophyllene, a CB2 Receptor-Selective Phytocannabinoid, Suppresses Motor Paralysis and Neuroinflammation in a Murine Model of Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 691. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, L.P.; Daphney, C.M.; Oppong-Damoah, A.; Uchakin, P.N.; Abney, S.E.; Uchakina, O.N.; Khusial, R.D.; Akil, A.; Murnane, K.S. The cannabinoid receptor 2 agonist, β-caryophyllene, improves working memory and reduces circulating levels of specific proinflammatory cytokines in aged male mice. Behav. Brain Res. 2019, 372, 112012. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Hurtado, P.; González-Castañeda, R.E.; Beas-Zarate, C.; Flores-Soto, M.E.; Viveros-Paredes, J.M. β-Caryophyllene Reduces DNA Oxidation and the Overexpression of Glial Fibrillary Acidic Protein in the Prefrontal Cortex and Hippocampus of d-Galactose-Induced Aged BALB/c Mice. J. Med. Food 2020, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Segat, G.C.; Manjavachi, M.N.; Matias, D.O.; Passos, G.F.; Freitas, C.S.; da Costa, R.; Calixto, J.B. Antiallodynic effect of β-caryophyllene on paclitaxel-induced peripheral neuropathy in mice. Neuropharmacology 2017, 125, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Meeran, M.N.; Al Taee, H.; Azimullah, S.; Tariq, S.; Adeghate, E.; Ojha, S. β-Caryophyllene, a natural bicyclic sesquiterpene attenuates doxorubicin-induced chronic cardiotoxicity via activation of myocardial cannabinoid type-2 (CB2) receptors in rats. Chem. Biophys. Interact. 2019, 304, 158–167. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [Green Version]
- Poddighe, L.; Carta, G.; Serra, M.P.; Melis, T.; Boi, M.; Lisai, S.; Murru, E.; Muredda, L.; Collu, M.; Banni, S.; et al. Acute administration of beta-caryophyllene prevents endocannabinoid system activation during transient common carotid artery occlusion and reperfusion. Lipids Health Dis. 2018, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Silva, M.; Correa, L.B.; Candéa, A.L.P.; Cavalher-Machado, S.C.; Barbosa, H.S.; Rosas, E.C.; Henriques, M.G. The cannabinoid 2 receptor agonist β-caryophyllene modulates the inflammatory reaction induced by Mycobacterium bovis BCG by inhibiting neutrophil migration. Inflamm. Res. 2016, 65, 869–879. [Google Scholar] [CrossRef]
- Bento, A.F.; Marcon, R.; Dutra, R.C.; Claudino, R.F.; Cola, M.; Leite, D.F.P.; Calixto, J.B. β-Caryophyllene Inhibits Dextran Sulfate Sodium-Induced Colitis in Mice through CB2 Receptor Activation and PPARγ Pathway. Am. J. Pathol. 2011, 178, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.S.; Mohamed, M.E. β-Caryophyllene as a Potential Protective Agent Against Myocardial Injury: The Role of Toll-Like Receptors. Molecules 2019, 24, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, B.; Mukhopadhyay, P.; Kechrid, M.; Patel, V.; Tanchian, G.; Wink, D.A.; Gertsch, J.; Pacher, P. β-Caryophyllene ameliorates cisplatin-induced nephrotoxicity in a cannabinoid 2 receptor-dependent manner. Free Radic. Biol. Med. 2012, 52, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, D.; Chen, Y.; Yang, M. β-Caryophyllene inhibits high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation in mesangial cells. Int. Immunopharmacol. 2020, 84, 106556. [Google Scholar] [CrossRef]
- Refaat, B.; El-Boshy, M. Protective antioxidative and anti-inflammatory actions of β-caryophyllene against sulfasalazine-induced nephrotoxicity in rat. Exp. Biol. Med. 2022, 247, 691–699. [Google Scholar] [CrossRef]
- Calleja, M.A.; Vieites, J.M.; Montero-Melendez, T.; Torres, M.I.; Faus, M.J.; Gil, A.; Suárez, A. The antioxidant effect of β-caryophyllene protects rat liver from carbon tetrachloride-induced fibrosis by inhibiting hepatic stellate cell activation. Br. J. Nutr. 2012, 109, 394–401. [Google Scholar] [CrossRef]
- Klauke, A.-L.; Racz, I.; Pradier, B.; Markert, A.; Zimmer, A.M.; Gertsch, J.; Zimmer, A. The cannabinoid CB2 receptor-selective phytocannabinoid beta-caryophyllene exerts analgesic effects in mouse models of inflammatory and neuropathic pain. Eur. Neuropsychopharmacol. 2014, 24, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Dong, Z.; Liu, S. β-Caryophyllene Ameliorates the Alzheimer-Like Phenotype in APP/PS1 Mice through CB2 Receptor Activation and the PPARγ Pathway. Pharmacology 2014, 94, 1–12. [Google Scholar] [CrossRef]
- Tian, X.; Liu, H.; Xiang, F.; Xu, L.; Dong, Z. β-Caryophyllene protects against ischemic stroke by promoting polarization of microglia toward M2 phenotype via the TLR4 pathway. Life Sci. 2019, 237, 116915. [Google Scholar] [CrossRef]
- Aly, E.; Khajah, M.A.; Masocha, W. β-Caryophyllene, a CB2-Receptor-Selective Phytocannabinoid, Suppresses Mechanical Allodynia in a Mouse Model of Antiretroviral-Induced Neuropathic Pain. Molecules 2020, 25, 106. [Google Scholar] [CrossRef] [Green Version]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Muthuramalingam, P.; Jeyasri, R.; Valliammai, A.; Selvaraj, A.; Karthika, C.; Gowrishankar, S.; Pandian, S.K.; Ramesh, M.; Chen, J.-T. Global multi-omics and systems pharmacological strategy unravel the multi-targeted therapeutic potential of natural bioactive molecules against COVID-19: An in silico approach. Genomics 2020, 112, 4486–4504. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Sun, T.; Wang, X. Activation of type 2 cannabinoid receptors (CB2R) promotes fatty acid oxidation through the SIRT1/PGC-1α pathway. Biochem. Biophys. Res. Commun. 2013, 436, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Kamikubo, R.; Kai, K.; Tsuji-Naito, K.; Akagawa, M. β-Caryophyllene attenuates palmitate-induced lipid accumulation through AMPK signaling by activating CB2 receptor in human HepG2 hepatocytes. Mol. Nutr. Food Res. 2016, 60, 2228–2242. [Google Scholar] [CrossRef]
- Xu, G.; Huang, K.; Zhou, J. Hepatic AMP Kinase as a Potential Target for Treating Nonalcoholic Fatty Liver Disease: Evidence from Studies of Natural Products. Curr. Med. Chem. 2018, 25, 889–907. [Google Scholar] [CrossRef]
- O’Sullivan, S.; Kendall, D. Cannabinoid activation of peroxisome proliferator-activated receptors: Potential for modulation of inflammatory disease. Immunobiology 2010, 215, 611–616. [Google Scholar] [CrossRef]
- Decara, J.; Rivera, P.; López-Gambero, A.J.; Serrano, A.; Pavón, F.J.; Baixeras, E.; De Fonseca, F.R.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef]
- Iannotti, F.; Vitale, R. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef]
- Irrera, N.; D’Ascola, A.; Pallio, G.; Bitto, A.; Mazzon, E.; Mannino, F.; Squadrito, V.; Arcoraci, V.; Minutoli, L.; Campo, G.M.; et al. β-Caryophyllene Mitigates Collagen Antibody Induced Arthritis (CAIA) in Mice Through a Cross-Talk between CB2 and PPAR-γ Receptors. Biomolecules 2019, 9, 326. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Jia, Y.; Lee, J.H.; Jun, H.-J.; Lee, H.-S.; Hwang, K.-Y.; Lee, S.-J. Trans-Caryophyllene is a natural agonistic ligand for peroxisome proliferator-activated receptor-α. Bioorg. Med. Chem. Lett. 2014, 24, 3168–3174. [Google Scholar] [CrossRef]
- Cho, H.-I.; Hong, J.-M.; Choi, J.-W.; Choi, H.-S.; Kwak, J.H.; Lee, D.-U.; Lee, S.K.; Lee, S.-M. β-Caryophyllene alleviates d-galactosamine and lipopolysaccharide-induced hepatic injury through suppression of the TLR4 and RAGE signaling pathways. Eur. J. Pharmacol. 2015, 764, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Y.; Yang, F.; Chen, J.-H.; Ren, G.-F. β-Caryophyllene Ameliorates MSU-Induced Gouty Arthritis and Inflammation Through Inhibiting NLRP3 and NF-κB Signal Pathway: In Silico and In Vivo. Front. Pharmacol. 2021, 12, 651305. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Purk, A.; Kaur, M.; Soini, H.A.; Novotny, M.V.; Davis, K.; Kao, C.C.; Matsunami, H.; Mescher, A. Beta-caryophyllene enhances wound healing through multiple routes. PLoS ONE 2019, 14, e0216104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paula-Freire, L.; Andersen, M.; Gama, V.; Molska, G.; Carlini, E. The oral administration of trans-caryophyllene attenuates acute and chronic pain in mice. Phytomedicine 2014, 21, 356–362. [Google Scholar] [CrossRef] [PubMed]
- de Morais Oliveira-Tintino, C.D.; Pessoa, R.T.; Fernandes, M.N.M.; Alcântara, I.S.; da Silva BA, F.; de Oliveira, M.R.C.; Martins, A.O.B.P.B.; do Socorro da Silva, M.; Tintino, S.R.; Rodrigues, F.F.G.; et al. Anti-inflammatory and anti-edematogenic action of the Croton campestris A. St.-Hil (Euphorbiaceae) essential oil and the compound β-caryophyllene in in vivo models. Phytomedicine 2018, 41, 82–95. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Mysliveckova, Z.; Szotakova, B.; Spicakova, A.; Lnenickova, K.; Ambroz, M.; Kubicek, V.; Krasulova, K.; Anzenbacher, P.; Skalova, L. The inhibitory effects of β-caryophyllene, β-caryophyllene oxide and α-humulene on the activities of the main drug-metabolizing enzymes in rat and human liver in vitro. Chem. Biol. Interact. 2017, 278, 123–128. [Google Scholar] [CrossRef]
- Hill, R.A.; Connolly, J.D. Triterpenoids. Nat. Prod. Rep. 2013, 30, 1028–1065. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.B.; Rai, D.K.; Brunton, N.P.; Martin-Diana, A.B.; Barry-Ryan, A.C. Characterization of Phenolic Composition in Lamiaceae Spices by LC-ESI-MS/MS. J. Agric. Food Chem. 2010, 58, 10576–10581. [Google Scholar] [CrossRef]
- del Baño, M.J.; Lorente, J.; Castillo, J.; Benavente-García, O.; del Río, J.A.; Ortuño, A.; Quirin, A.K.-W.; Gerard, D. Phenolic Diterpenes, Flavones, and Rosmarinic Acid Distribution during the Development of Leaves, Flowers, Stems, and Roots of Rosmarinus officinalis. Antioxidant Activity. J. Agric. Food Chem. 2003, 51, 4247–4253. [Google Scholar] [CrossRef]
- Luis, J.; Johnson, C. Seasonal variations of rosmarinic and carnosic acids in rosemary extracts. Analysis of their in vitro antiradical activity. Span. J. Agric. Res. 2005, 3, 106. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Kosaka, K.; Itoh, K.; Kobayashi, A.; Yamamoto, M.; Shimojo, Y.; Kitajima, C.; Cui, J.; Kamins, J.; Okamoto, S.-I.; et al. Carnosic acid, acatechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J. Neurochem. 2008, 104, 1116–1131. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, M.R.; Villalba, R.G.; Larrosa, M.; Yáñez-Gascón, M.J.; Fromentin, E.; Flanagan, J.; Roller, M.; Tomas-Barberan, F.; Espín, J.C.; García-Conesa, M.-T. Bioavailability of the major bioactive diterpenoids in a rosemary extract: Metabolic profile in the intestine, liver, plasma, and brain of Zucker rats. Mol. Nutr. Food Res. 2013, 57, 1834–1846. [Google Scholar] [CrossRef] [PubMed]
- Doolaege, E.H.A.; Raes, K.; De Vos, F.; Verhé, R.; De Smet, S. Absorption, Distribution and Elimination of Carnosic Acid, A Natural Antioxidant from Rosmarinus officinalis, in Rats. Plant Foods Hum. Nutr. 2011, 66, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Erkan, N.; Ayranci, G.; Ayranci, E. Antioxidant activities of rosemary (Rosmarinus officinalis L.) extract, blackseed (Nigella sativa L.) essential oil, carnosic acid, rosmarinic acid and sesamol. Food Chem. 2008, 110, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Fons, L.; Garzón, M.T.; Micol, V. Relationship between the Antioxidant Capacity and Effect of Rosemary (Rosmarinus officinalis L.) Polyphenols on Membrane Phospholipid Order. J. Agric. Food Chem. 2009, 58, 161–171. [Google Scholar] [CrossRef]
- Loussouarn, M.; Krieger-Liszkay, A.; Svilar, L.; Bily, A.; Birtić, S.; Havaux, M. Carnosic Acid and Carnosol, Two Major Antioxidants of Rosemary, Act through Different Mechanisms. Plant Physiol. 2017, 175, 1381–1394. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: Development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Research 2017, 6, 2138. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, M.R.; Ferreira, G.C.; Schuck, P.F. Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in SH-SY5Y cells: Role for PI3K/Akt/Nrf2 pathway. Toxicol. In Vitro 2016, 32, 41–54. [Google Scholar] [CrossRef]
- Satoh, T.; Izumi, M.; Inukai, Y.; Tsutsumi, Y.; Nakayama, N.; Kosaka, K.; Shimojo, Y.; Kitajima, C.; Itoh, K.; Yokoi, T.; et al. Carnosic acid protects neuronal HT22 Cells through activation of the antioxidant-responsive element in free carboxylic acid- and catechol hydroxyl moieties-dependent manners. Neurosci. Lett. 2008, 434, 260–265. [Google Scholar] [CrossRef]
- Satoh, T.; Saitoh, S.; Hosaka, M.; Kosaka, K. Simple ortho- and para-hydroquinones as compounds neuroprotective against oxidative stress in a manner associated with specific transcriptional activation. Biochem. Biophys. Res. Commun. 2009, 379, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Tabuchi, T.; Takahashi, T.; Kosaka, K.; Satoh, T. Activated Glutathione Metabolism Participates in Protective Effects of Carnosic Acid against Oxidative Stress in Neuronal HT22 Cells. Planta Med. 2009, 76, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Tena, M.T.; Valcárcel, M.; Hidalgo, P.J.; Ubera, J.L. Supercritical Fluid Extraction of Natural Antioxidants from Rosemary: Comparison with Liquid Solvent Sonication. Anal. Chem. 1997, 69, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.M.; Faustino, C.; Garcia, C.; Ladeiras, D.; Reis, C.P.; Rijo, P. Rosmarinus officinalis L.: An update review of its phytochemistry and biological activity. Future Sci. OA 2018, 4, FSO283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagitai, M.; Itoh, S.; Kitagawa, T.; Takenouchi, T.; Kitani, H.; Satoh, T. Carnosic acid, a pro-electrophilic compound, inhibits LPS-induced activation of microglia. Biochem. Biophys. Res. Commun. 2012, 418, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Hadad, N.; Levy, R. Combination of EPA with Carotenoids and Polyphenol Synergistically Attenuated the Transformation of Microglia to M1 Phenotype Via Inhibition of NF-κB. NeuroMol. Med. 2017, 19, 436–451. [Google Scholar] [CrossRef]
- Chae, I.G.; Yu, M.H.; Im, N.-K.; Jung, Y.T.; Lee, J.; Chun, K.-S.; Lee, I.-S. Effect of Rosemarinus officinalis L. on MMP-9, MCP-1 Levels, and Cell Migration in RAW 264.7 and Smooth Muscle Cells. J. Med. Food 2012, 15, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-F.; Su, J.-D.; Chiu, C.-H.; Peng, C.-C.; Chang, C.-H.; Sung, T.-Y.; Huang, S.-H.; Lee, W.-C.; Chyau, C.-C. Anti-Inflammatory Effects of Supercritical Carbon Dioxide Extract and Its Isolated Carnosic Acid from Rosmarinus officinalis Leaves. J. Agric. Food Chem. 2011, 59, 3674–3685. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Ferreira, G.C.; Schuck, P.F.; dal Bosco, S.M. Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem. Biol. Interact. 2015, 242, 396–406. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Peres, A.; Ferreira, G.C.; Schuck, P.F.; Bosco, S.M.D. Carnosic Acid Affords Mitochondrial Protection in Chlorpyrifos-Treated Sh-Sy5y Cells. Neurotox. Res. 2016, 30, 367–379. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Ferreira, G.D.C.; Peres, A.; Bosco, S.M.D. Carnosic Acid Suppresses the H2O2-Induced Mitochondria-Related Bioenergetics Disturbances and Redox Impairment in SH-SY5Y Cells: Role for Nrf2. Mol. Neurobiol. 2017, 55, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Taram, F.; Ignowski, E.; Duval, N.; Linseman, D.A. Neuroprotection Comparison of Rosmarinic Acid and Carnosic Acid in Primary Cultures of Cerebellar Granule Neurons. Molecules 2018, 23, 2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwager, J.; Richard, N.; Fowler, A.; Seifert, N.; Raederstorff, D. Carnosol and Related Substances Modulate Chemokine and Cytokine Production in Macrophages and Chondrocytes. Molecules 2016, 21, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.; Ishitobi, H.; Miyaki, S.; Kawaoka, T.; Kayashima, T.; Matsubara, K. Carnosic acid protects starvation-induced SH-SY5Y cell death through Erk1/2 and Akt pathways, autophagy, and FoxO3a. Int. J. Food Sci. Nutr. 2016, 67, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Thummuri, D.; Naidu, V.G.M.; Chaudhari, P. Carnosic acid attenuates RANKL-induced oxidative stress and osteoclastogenesis via induction of Nrf2 and suppression of NF-κB and MAPK signalling. J. Mol. Med. 2017, 95, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Wang, X.; Sun, H.; Qin, Y.; Fu, M. Carnosic acid (CA) attenuates collagen-induced arthritis in db/db mice via inflammation suppression by regulating ROS-dependent p38 pathway. Free Radic. Biol. Med. 2017, 108, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Xia, Z.; Shao, N.; Li, B.; Xue, L.; Peng, Y.; Zhi, F.; Yang, Y. Carnosic acid prevents dextran sulfate sodium-induced acute colitis associated with the regulation of the Keap1/Nrf2 pathway. Sci. Rep. 2017, 7, 11036. [Google Scholar] [CrossRef]
- Wang, L.-C.; Wei, W.-H.; Zhang, X.-W.; Liu, D.; Zeng, K.-W.; Tu, P.-F. An Integrated Proteomics and Bioinformatics Approach Reveals the Anti-inflammatory Mechanism of Carnosic Acid. Front. Pharmacol. 2018, 9, 370. [Google Scholar] [CrossRef] [Green Version]
- Song, H.-M.; Li, X.; Liu, Y.-Y.; Lu, W.-P.; Cui, Z.-H.; Zhou, L.; Yao, D.; Zhang, H.-M. Carnosic acid protects mice from high-fat diet-induced NAFLD by regulating MARCKS. Int. J. Mol. Med. 2018, 42, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Maione, F.; Cantone, V.; Pace, S.; Chini, M.G.; Bisio, A.; Romussi, G.; Pieretti, S.; Werz, O.; Koeberle, A.; Mascolo, N.; et al. Anti-inflammatory and analgesic activity of carnosol and carnosic acid in vivo and in vitro and in silico analysis of their target interactions. Br. J. Pharmacol. 2017, 174, 1497–1508. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Q.; Liu, Z.; Wang, Y.; Xiao, H.; Wu, W.; Xiao, C.; Liu, X. Carnosic acid attenuates lipopolysaccharide-induced liver injury in rats via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 2013, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mengoni, E.S.; Vichera, G.; Rigano, L.A.; Rodriguez-Puebla, M.L.; Galliano, S.R.; Cafferata, E.E.; Pivetta, O.H.; Moreno, S.; Vojnov, A.A. Suppression of COX-2, IL-1β and TNF-α expression and leukocyte infiltration in inflamed skin by bioactive compounds from Rosmarinus officinalis L. Fitoterapia 2011, 82, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, J.-J.; Chen, G.-Y.; Wang, W.-W.; Xie, Z.-T.; Tang, G.-F.; Wei, S.-D. Carnosic acid nanoparticles suppress liver ischemia/reperfusion injury by inhibition of ROS, Caspases and NF-κB signaling pathway in mice. Biomed. Pharmacother. 2016, 82, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, L.; Sun, H.; Cao, K. Carnosic acid protects against lipopolysaccharide-induced acute lung injury in mice. Exp. Ther. Med. 2019, 18, 3707–3714. [Google Scholar] [CrossRef]
- Tang, B.; Tang, F.; Wang, Z.; Qi, G.; Liang, X.; Li, B.; Yuan, S.; Liu, J.; Yu, S.; He, S. Upregulation of Akt/NF-κB-regulated inflammation and Akt/Bad-related apoptosis signaling pathway involved in hepatic carcinoma process: Suppression by carnosic acid nanoparticle. Int. J. Nanomed. 2016, 11, 6401–6420. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Shan, W.; Zeng, W.; Hu, Y.; Wang, G.; Tian, X.; Zhang, N.; Shi, X.; Zhao, Y.; Ding, C.; et al. Carnosic acid alleviates chronic alcoholic liver injury by regulating the SIRT1/ChREBP and SIRT1/p66shc pathways in rats. Mol. Nutr. Food Res. 2016, 60, 1902–1911. [Google Scholar] [CrossRef]
- Zhang, Q.-L.; Yang, J.-J.; Zhang, H.-S. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubicin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomed. Pharmacother. 2018, 109, 71–83. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, X.; Zhou, L.; Liu, Z.; Yuan, J.; Cheng, J.; Zhao, J.; Wu, L.; Li, H.; Qiu, H.; et al. Carnosic acid inhibits inflammation response and joint destruction on osteoclasts, fibroblast-like synoviocytes, and collagen-induced arthritis rats. J. Cell. Physiol. 2018, 233, 6291–6303. [Google Scholar] [CrossRef]
- Park, M.-Y.; Sung, M.-K. Carnosic acid attenuates obesity-induced glucose intolerance and hepatic fat accumulation by modulating genes of lipid metabolism in C57BL/6J-ob/ob mice. J. Sci. Food Agric. 2014, 95, 828–835. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Hu, M.; Li, Y.; Cao, X. Carnosic acid alleviates brain injury through NF-κB-regulated inflammation and Caspase-3-associated apoptosis in high fat-induced mouse models. Mol. Med. Rep. 2019, 20, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.M.; Lee, D.K.M.; Wong, D.P.; Wong, R.N.; Yung, K.K.; Cheng, C.H.-K.; Yue, K.K. Ginsenosides attenuate methylglyoxal-induced impairment of insulin signaling and subsequent apoptosis in primary astrocytes. Neuropharmacology 2014, 85, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.M.T.; Lee, D.K.M.; Wong, D.P.K.; Wong, G.T.C.; Yue, K.K.M. Methylglyoxal-induced neuroinflammatory response in in vitro astrocytic cultures and hippocampus of experimental animals. Metab. Brain Dis. 2016, 31, 1055–1064. [Google Scholar] [CrossRef]
- Kemeny, S.; Dery, D.; Loboda, Y.; Rovner, M.; Lev, T.; Zuri, D.; Finberg, J.P.M.; Larisch, S. Parkin Promotes Degradation of the Mitochondrial Pro-Apoptotic ARTS Protein. PLoS ONE 2012, 7, e38837. [Google Scholar] [CrossRef] [Green Version]
- Pike, A.F.; Szabò, I.; Veerhuis, R.; Bubacco, L. The potential convergence of NLRP3 inflammasome, potassium, and dopamine mechanisms in Parkinson’s disease. npj Parkinson’s Dis. 2022, 8, 32. [Google Scholar] [CrossRef]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein–antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef] [PubMed]
- Iborra, S.F.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; López-Cuenca, I.; Rojas, P.; Triviño, A.; Ramírez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; McManus, R.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2019, 25, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, J.; Petricca, S.; Iorio, R.; Toniato, E.; Flati, V. Mitochondrial and metabolic alterations in cancer cells. Eur. J. Cell Biol. 2022, 101, 151225. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia reprogram metabolic profiles for phenotype and function changes in central nervous system. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2020, 21, 363–381. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Gallardo, G. Mitochondria fragments fuel the fire of neuroinflammation. Sci. Transl. Med. 2019, 11, eaaz3714. [Google Scholar] [CrossRef]
- de Souza Breda, C.N.; Davanzo, G.G.; Basso, P.J.; Câmara, N.O.S.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.; Höllerhage, M.; Höglinger, G.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2015, 16, 849–857. [Google Scholar] [CrossRef]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-M.; Liu, N.; Qin, Z.-H.; Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The Mitochondrial Antiviral Protein MAVS Associates with NLRP3 and Regulates Its Inflammasome Activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.; et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49, 413–426.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Junkins, R.D.; Kurkjian, C.J.; Holley-Guthrie, E.; Pendse, A.A.; El Morabiti, R.; Petrucelli, A.; Barber, G.N.; Benedict, C.A.; Ting, J.P.-Y. A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 2017, 214, 3611–3626. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ning, X.; Gao, P.; Wu, S.; Sha, M.; Lv, M.; Zhou, X.; Gao, J.; Fang, R.; Meng, G.; et al. Inflammasome Activation Triggers Caspase-1-Mediated Cleavage of cGAS to Regulate Responses to DNA Virus Infection. Immunity 2017, 46, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.-P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS 1-caspase-1 axis. EMBO J. 2018, 37, e99347. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e18. [Google Scholar] [CrossRef]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human immunodeficiency virus Type-1 single-stranded RNA activates the NLRP3 inflammasome and impairs autophagic clearance of damaged mitochondria in human microglia. Glia 2018, 67, 802–824. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson’s disease model. Neuropharmacology 2022, 207, 108963. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-Q.; Ai, W.; Zhu, F.-D.; Zhang, Y.; Guo, M.-S.; Law, B.Y.-K.; Wu, J.-M.; Wong, V.K.-W.; Tang, Y.; Yu, L.; et al. Polygala saponins inhibit NLRP3 inflammasome-mediated neuroinflammation via SHP-2-Mediated mitophagy. Free Radic. Biol. Med. 2021, 179, 76–94. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Li, Y.; Chen, S.; Wen, Y.; Zhang, S.; Luo, E.; Li, X.; Zhong, W.; Zeng, H. Fisetin ameliorates cognitive impairment by activating mitophagy and suppressing neuroinflammation in rats with sepsis-associated encephalopathy. CNS Neurosci. Ther. 2021, 28, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Tsai, C.-W. Carnosic acid protects SH-SY5Y cells against 6-hydroxydopamine-induced cell death through upregulation of parkin pathway. Neuropharmacology 2016, 110, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Tsai, C.-W. Carnosic Acid Attenuates 6-Hydroxydopamine-Induced Neurotoxicity in SH-SY5Y Cells by Inducing Autophagy Through an Enhanced Interaction of Parkin and Beclin1. Mol. Neurobiol. 2016, 54, 2813–2822. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Tsai, C.-W. PINK1/parkin-mediated mitophagy pathway is related to neuroprotection by carnosic acid in SH-SY5Y cells. Food Chem. Toxicol. 2019, 125, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Huang, Y.-N.; Fu, R.-H.; Liao, Y.-H.; Kuo, T.-Y.; Tsai, C.-W. Promotion of mitochondrial biogenesis via the regulation of PARIS and PGC-1α by parkin as a mechanism of neuroprotection by carnosic acid. Phytomedicine 2020, 80, 153369. [Google Scholar] [CrossRef]
- Foresti, R.; Bains, S.K.; Pitchumony, T.S.; De Castro Brás, L.E.; Drago, F.; Dubois-Randé, J.-L.; Bucolo, C.; Motterlini, R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol. Res. 2013, 76, 132–148. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Trudler, D.; Oh, C.-K.; Lipton, S.A. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants 2022, 11, 124. [Google Scholar] [CrossRef]
- Straccia, M.; Gresa-Arribas, N.; Dentesano, G.; Ejarque-Ortiz, A.; Tusell, J.M.; Serratosa, J.; Solà, C.; Saura, J. Pro-inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein β. J. Neuroinflamm. 2011, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Morales-Garcia, J.A.; Gine, E.; Hernandez-Encinas, E.; Aguilar-Morante, D.; Sierra-Magro, A.; Sanz-SanCristobal, M.; Alonso-Gil, S.; Sanchez-Lanzas, R.; Castaño, J.G.; Santos, A.; et al. CCAAT/Enhancer binding protein β silencing mitigates glial activation and neurodegeneration in a rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 13526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, B.; Cogswell, P.C.; Baldwin, A.S. Functional and physical associations between NF-kappa B and C/EBP family members: A Rel domain-bZIP interaction. Mol. Cell. Biol. 1993, 13, 3964–3974. [Google Scholar] [CrossRef] [PubMed]
- Yi-Bin, W.; Xiang, L.; Bing, Y.; Qi, Z.; Fei-Tong, J.; Minghong, W.; Xiangxiang, Z.; Le, K.; Yan, L.; Ping, S.; et al. Inhibition of the CEBPβ-NFκB interaction by nanocarrier-packaged Carnosic acid ameliorates glia-mediated neuroinflammation and improves cognitive function in an Alzheimer’s disease model. Cell Death Dis. 2022, 13, 318. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Rasoolijazi, H.; Joghataie, M.T.; Soleimani, S. Neuroprotective Effects of Carnosic Acid in an Experimental Model of Alzheimer’s Disease in Rats. Cell J. 2011, 13, 39–44. [Google Scholar] [PubMed]
- Rasoolijazi, H.; Azad, N.; Joghataei, M.T.; Kerdari, M.; Nikbakht, F.; Soleimani, M. The Protective Role of Carnosic Acid against Beta-Amyloid Toxicity in Rats. Sci. World J. 2013, 2013, 917082. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef]
- Meng, P.; Yoshida, H.; Matsumiya, T.; Imaizumi, T.; Tanji, K.; Xing, F.; Hayakari, R.; Dempoya, J.; Tatsuta, T.; Aizawa-Yashiro, T.; et al. Carnosic acid suppresses the production of amyloid-β 1–42 by inducing the metalloprotease gene TACE/ADAM17 in SH-SY5Y human neuroblastoma cells. Neurosci. Res. 2013, 75, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Meng, P.; Matsumiya, T.; Tanji, K.; Hayakari, R.; Xing, F.; Wang, L.; Tsuruga, K.; Tanaka, H.; Mimura, J.; et al. Carnosic acid suppresses the production of amyloid-β 1-42 and 1-43 by inducing an α-secretase TACE/ADAM17 in U373MG human astrocytoma cells. Neurosci. Res. 2014, 79, 83–93. [Google Scholar] [CrossRef]
- Meng, P.; Yoshida, H.; Tanji, K.; Matsumiya, T.; Xing, F.; Hayakari, R.; Wang, L.; Tsuruga, K.; Tanaka, H.; Mimura, J.; et al. Carnosic acid attenuates apoptosis induced by amyloid-β 1–42 or 1–43 in SH-SY5Y human neuroblastoma cells. Neurosci. Res. 2015, 94, 1–9. [Google Scholar] [CrossRef]
- Haddad, M.; Perrotte, M.; Ben Khedher, M.R.; Demongin, C.; Lepage, A.; Fülöp, T.; Ramassamy, C. Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 4906. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.P.; Vizuete, A.F.K.; Zin, L.E.F.; de Marques, C.O.; Pacheco, R.F.; Leal, M.B.; Gonçalves, C.-A. The Methylglyoxal/RAGE/NOX-2 Pathway is Persistently Activated in the Hippocampus of Rats with STZ-Induced Sporadic Alzheimer’s Disease. Neurotox. Res. 2022, 40, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-G.; Wang, X.; Zhou, T.-T.; Wu, X.-F.; Peng, Y.; Zhang, W.-Q.; Li, S.; Zhao, J. Scorpion Venom Heat-Resistant Peptide Protects Transgenic Caenorhabditis elegans from β-Amyloid Toxicity. Front. Pharmacol. 2016, 7, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Pelt, K.M.; Truttmann, M.C. Caenorhabditis elegans as a model system for studying aging-associated neurodegenerative diseases. Transl. Med. Aging 2020, 4, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zhang, X.; Xiao, J.; Zhong, Q.; Kuang, Y.; Cao, Y.; Chen, Y. Effects on longevity extension and mechanism of action of carnosic acid in Caenorhabditis elegans. Food Funct. 2019, 10, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Qin, Q.; Zhang, Y.; Xie, L.; Xiao, J.; Cao, Y.; Su, Z.; Chen, Y. Carnosic acid ameliorated Aβ-mediated (amyloid-β peptide) toxicity, cholinergic dysfunction and mitochondrial defect in Caenorhabditis elegans of Alzheimer’s Model. Food Funct. 2022, 13, 4624–4640. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Kim, S.; Lee, S.-Y.; Kim, C.-S.; Kim, D.K.; Kim, S.-J.; Chun, H.S. Beneficial effects of carnosic acid on dieldrin-induced dopaminergic neuronal cell death. Neuroreport 2008, 19, 1301–1304. [Google Scholar] [CrossRef]
- Chen, J.-H.; Ou, H.-P.; Lin, C.-Y.; Lin, F.-J.; Wu, C.-R.; Chang, S.-W.; Tsai, C.-W. Carnosic Acid Prevents 6-Hydroxydopamine-Induced Cell Death in SH-SY5Y Cells via Mediation of Glutathione Synthesis. Chem. Res. Toxicol. 2012, 25, 1893–1901. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Chen, J.-H.; Fu, R.-H.; Tsai, C.-W. Induction of Pi Form of Glutathione S-Transferase by Carnosic Acid Is Mediated through PI3K/Akt/NF-κB Pathway and Protects against Neurotoxicity. Chem. Res. Toxicol. 2014, 27, 1958–1966. [Google Scholar] [CrossRef]
- Fu, R.-H.; Lin, C.-Y.; Tsai, C.-W.; Huang, L.-C. Modulation of ARTS and XIAP by Parkin Is Associated with Carnosic Acid Protects SH-SY5Y Cells against 6-Hydroxydopamine-Induced Apoptosis. Mol. Neurobiol. 2017, 55, 1786–1794. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, B.; Nutter, A.; Song, P.; Dolatabadi, N.; Parker, J.; Sanz-Blasco, S.; Newmeyer, T.; Ambasudhan, R.; McKercher, S.R.; et al. Protection from cyanide-induced brain injury by the Nrf2 transcriptional activator carnosic acid. J. Neurochem. 2015, 133, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-R.; Tsai, C.-W.; Chang, S.-W.; Lin, C.-Y.; Huang, L.-C.; Tsai, C.-W. Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: Involvement of antioxidative enzymes induction. Chem.-Biol. Interact. 2015, 225, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Lin, C.-Y.; Wu, C.-R.; Tsai, C.-H.; Tsai, C.-W. Carnosic Acid Alleviates Levodopa-Induced Dyskinesia and Cell Death in 6-Hydroxydopamine-lesioned Rats and in SH-SY5Y Cells. Front. Pharmacol. 2021, 12, 703894. [Google Scholar] [CrossRef] [PubMed]
- Bento-Pereira, C.; Dinkova-Kostova, A.T. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson’s disease. Med. Res. Rev. 2020, 41, 785–802. [Google Scholar] [CrossRef] [PubMed]
- De Souza, I.C.C.; Gobbo, R.C.B.; De Almeida, F.J.S.; Luckachaki, M.D.; De Oliveira, M.R. Carnosic acid depends on glutathione to promote mitochondrial protection in methylglyoxal-exposed SH-SY5Y cells. Metab. Brain Dis. 2021, 36, 471–481. [Google Scholar] [CrossRef]
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef]
- Covill-Cooke, C.; Howden, J.H.; Birsa, N.; Kittler, J.T. Ubiquitination at the mitochondria in neuronal health and disease. Neurochem. Int. 2018, 117, 55–64. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Chen, W.-J.; Fu, R.-H.; Tsai, C.-W. Upregulation of OPA1 by carnosic acid is mediated through induction of IKKγ ubiquitination by parkin and protects against neurotoxicity. Food Chem. Toxicol. 2019, 136, 110942. [Google Scholar] [CrossRef]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Wyse, R.K.; Stott, S.R.; Mursaleen, L.; Matthews, H.; Dawson, V.L.; Dawson, T.M. Waiting for PARIS—A Biological Target in Search of a Drug. J. Parkinson’s Dis. 2022, 12, 95–103. [Google Scholar] [CrossRef]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Administration of the Nrf2–ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic. Biol. Med. 2013, 57, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Nrf2–ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp. Neurol. 2014, 264, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balietti, M.; Giorgetti, B.; Casoli, T.; Solazzi, M.; Tamagnini, F.; Burattini, C.; Aicardi, G.; Fattoretti, P. Early Selective Vulnerability of Synapses and Synaptic Mitochondria in the Hippocampal CA1 Region of the Tg2576 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 34, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Viveros-Paredes, J.M.; González-Castañeda, R.E.; Gertsch, J.; Chaparro-Huerta, V.; López-Roa, R.I.; Vázquez-Valls, E.; Beas-Zarate, C.; Camins-Espuny, A.; Flores-Soto, M.E.; Viveros-Paredes, J.M.; et al. Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP. Pharmaceuticals 2017, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, Y.; Ji, L.; Wu, Y.; Fu, Y.; Lin, L.; Lin, Y.; Zhang, Y. Anti-Alzheimer’s Disease Molecular Mechanism of Acori Tatarinowii Rhizoma Based on Network Pharmacology. Med. Sci. Monit. Basic Res. 2020, 26, e924203. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, H.Y.; Park, H.J.; Shin, H.K.; Hong, K.W.; Kim, C.D. Concurrent Treatment with Taxifolin and Cilostazol on the Lowering of β-Amyloid Accumulation and Neurotoxicity via the Suppression of P-JAK2/P-STAT3/NF-κB/BACE1 Signaling Pathways. PLoS ONE 2016, 11, e0168286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, Q.; Wang, S.; Liao, Z.; Jin, H.; Huang, S.; Hong, X.; Liu, Y.; Pang, J.; Shen, Q.; et al. The Food Additive β-Caryophyllene Exerts Its Neuroprotective Effects Through the JAK2-STAT3-BACE1 Pathway. Front. Aging Neurosci. 2022, 14, 814432. [Google Scholar] [CrossRef]
- Flores-Soto, M.; Corona-Angeles, J.; Tejeda-Martinez, A.; Flores-Guzman, P.; Luna-Mujica, I.; Chaparro-Huerta, V.; Viveros-Paredes, J. β-Caryophyllene exerts protective antioxidant effects through the activation of NQO1 in the MPTP model of Parkinson’s disease. Neurosci. Lett. 2020, 742, 135534. [Google Scholar] [CrossRef]
- Postu, P.A.; Sadiki, F.Z.; El Idrissi, M.; Cioanca, O.; Trifan, A.; Hancianu, M.; Hritcu, L. Pinus halepensis essential oil attenuates the toxic Alzheimer’s amyloid beta (1-42)-induced memory impairment and oxidative stress in the rat hippocampus. Biomed. Pharmacother. 2019, 112, 108673. [Google Scholar] [CrossRef]
- Abuhamdah, S.; Abuhamdah, R.; Howes, M.-J.R.; Al-Olimat, S.; Ennaceur, A.; Chazot, P.L. Pharmacological and neuroprotective profile of an essential oil derived from leaves of A loysia citrodora Palau. J. Pharm. Pharmacol. 2015, 67, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beserra-Filho, J.I.; de Macêdo, A.M.; Leão, A.H.; Bispo, J.M.M.; Santos, J.R.; de Oliveira-Melo, A.J.; Menezes, P.D.P.; Duarte, M.C.; de Souza Araújo, A.A.; Silva, R.H.; et al. Eplingiella fruticosa leaf essential oil complexed with β-cyclodextrin produces a superior neuroprotective and behavioral profile in a mice model of Parkinson’s disease. Food Chem. Toxicol. 2018, 124, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Javed, H.; Azimullah, S.; Haque, M.E. β-Caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol. Cell. Biochem. 2016, 418, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Aggrawal, A.; Pottabathini, R.; Singh, A. Possible neuroprotective mechanisms of clove oil against icv-colchicine induced cognitive dysfunction. Pharmacol. Rep. 2016, 68, 764–772. [Google Scholar] [CrossRef]
- Gupta, N.; Yücel, Y.H. Glaucoma as a neurodegenerative disease. Curr. Opin. Ophthalmol. 2007, 18, 110–114. [Google Scholar] [CrossRef]
- Gauthier, A.C.; Liu, J. Neurodegeneration and Neuroprotection in Glaucoma. Yale J. Biol. Med. 2016, 89, 73–79. [Google Scholar]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Gupta, N.; Ang, L.-C.; De Tilly, L.N.; Bidaisee, L.; Yücel, Y.H. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br. J. Ophthalmol. 2006, 90, 674–678. [Google Scholar] [CrossRef]
- Gupta, N.; Greenberg, G.; De Tilly, L.N.; Gray, B.; Polemidiotis, M.; Yucel, Y.H. Atrophy of the lateral geniculate nucleus in human glaucoma detected by magnetic resonance imaging. Br. J. Ophthalmol. 2008, 93, 56–60. [Google Scholar] [CrossRef]
- Mancino, R.; Martucci, A.; Cesareo, M.; Giannini, C.; Corasaniti, M.T.; Bagetta, G.; Nucci, C. Glaucoma and Alzheimer Disease: One Age-Related Neurodegenerative Disease of the Brain. Curr. Neuropharmacol. 2018, 16, 971–977. [Google Scholar] [CrossRef]
- O’Bryhim, B.E.; Apte, R.S.; Kung, N.; Coble, D.; Van Stavern, G.P. Association of Preclinical Alzheimer Disease with Optical Coherence Tomographic Angiography Findings. JAMA Ophthalmol. 2018, 136, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Satue, M.; Rodrigo, M.J.; Obis, J.; Vilades, E.; Gracia, H.; Otin, S.; Fuertes, M.I.; Alarcia, R.; Crespo, J.A.; Polo, V.; et al. Evaluation of Progressive Visual Dysfunction and Retinal Degeneration in Patients with Parkinson’s Disease. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syc-Mazurek, S.; Libby, R.T. Axon injury signaling and compartmentalized injury response in glaucoma. Prog. Retin. Eye Res. 2019, 73, 100769. [Google Scholar] [CrossRef]
- Tezel, G. A broad perspective on the molecular regulation of retinal ganglion cell degeneration in glaucoma. Prog. Brain Res. 2020, 256, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Zimbrón, L.F.; Zamora-Alvarado, R.; La Paz, L.O.-D.; Velez-Montoya, R.; Zenteno, E.; Gulias-Cañizo, R.; Quiroz-Mercado, H.; Gonzalez-Salinas, R. Age-Related Macular Degeneration: New Paradigms for Treatment and Management of AMD. Oxidat. Med. Cell. Longev. 2018, 2018, 8374647. [Google Scholar] [CrossRef]
- Dhodapkar, R.M.; Martell, D.; Hafler, B.P. Glial-mediated neuroinflammatory mechanisms in age-related macular degeneration. Semin. Immunopathol. 2022, 1–11. [Google Scholar] [CrossRef]
- Rezaie, T.; McKercher, S.R.; Kosaka, K.; Seki, M.; Wheeler, L.; Viswanath, V.; Chun, T.; Joshi, R.; Valencia, M.; Sasaki, S.; et al. Protective Effect of Carnosic Acid, a Pro-Electrophilic Compound, in Models of Oxidative Stress and Light-Induced Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7847–7854. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Tarchick, M.J.; Yu, X.; Beight, C.; Bu, P.; Yu, M. Carnosic acid slows photoreceptor degeneration in the Pde6brd10 mouse model of retinitis pigmentosa. Sci. Rep. 2016, 6, 22632. [Google Scholar] [CrossRef] [Green Version]
- Nagar, S.; Noveral, S.M.; Trudler, D.; Lopez, K.M.; McKercher, S.R.; Han, X.; Yates, J.R.; Piña-Crespo, J.C.; Nakanishi, N.; Satoh, T.; et al. MEF2D haploinsufficiency downregulates the NRF2 pathway and renders photoreceptors susceptible to light-induced oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, E4048–E4056. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.; Markey, M.; Rapp, C.M.; Darrow, R.M.; Ziesel, A.; Organisciak, D. Enhancing the efficacy of AREDS antioxidants in light-induced retinal degeneration. Mol. Vis. 2017, 23, 718–739. [Google Scholar]
- Albalawi, A.; Alhasani, R.H.A.; Biswas, L.; Reilly, J.; Shu, X. Protective effect of carnosic acid against acrylamide-induced toxicity in RPE cells. Food Chem. Toxicol. 2017, 108, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaz-Hayoun, S.; Rinsky, B.; Hagbi-Levi, S.; Grunin, M.; HayaYedid, T.; Chowers, I. Evaluation of antioxidant treatments for the modulation of macrophage function in the context of retinal degeneration. Mol. Vis. 2019, 25, 479–488. [Google Scholar] [PubMed]
- Liang, L.; He, L.; Zhu, M.; Chen, B.; Xiao, C. Protective effects of carnosic acid on retinal ganglion cells in acute ocular hypertension rats. Int. Ophthalmol. 2020, 40, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- AlKahtane, A.A.; Ghanem, E.; Bungau, S.G.; Alarifi, S.; Ali, D.; AlBasher, G.; Alkahtani, S.; Aleya, L.; Abdel-Daim, M.M. Carnosic acid alleviates chlorpyrifos-induced oxidative stress and inflammation in mice cerebral and ocular tissues. Environ. Sci. Pollut. Res. 2020, 27, 11663–11670. [Google Scholar] [CrossRef]
- Ho, K.L.; Yong, P.H.; Wang, C.W.; Kuppusamy, U.R.; Ngo, C.T.; Massawe, F.; Ng, Z.X. Peperomia pellucida (L.) Kunth and eye diseases: A review on phytochemistry, pharmacology and toxicology. J. Integr. Med. 2022, S2095–S4964. [Google Scholar] [CrossRef]
- Iorio, R.; Petricca, S.; Luzi, C.; Bellio, P.; Cristiano, L.; Festuccia, C.; Amicosante, G.; Celenza, G. Lactobacillus sakei Pro-Bio65 Reduces TNF-α Expression and Upregulates GSH Content and Antioxidant Enzymatic Activities in Human Conjunctival Cells. Transl. Vis. Sci. Technol. 2021, 10, 8. [Google Scholar] [CrossRef]

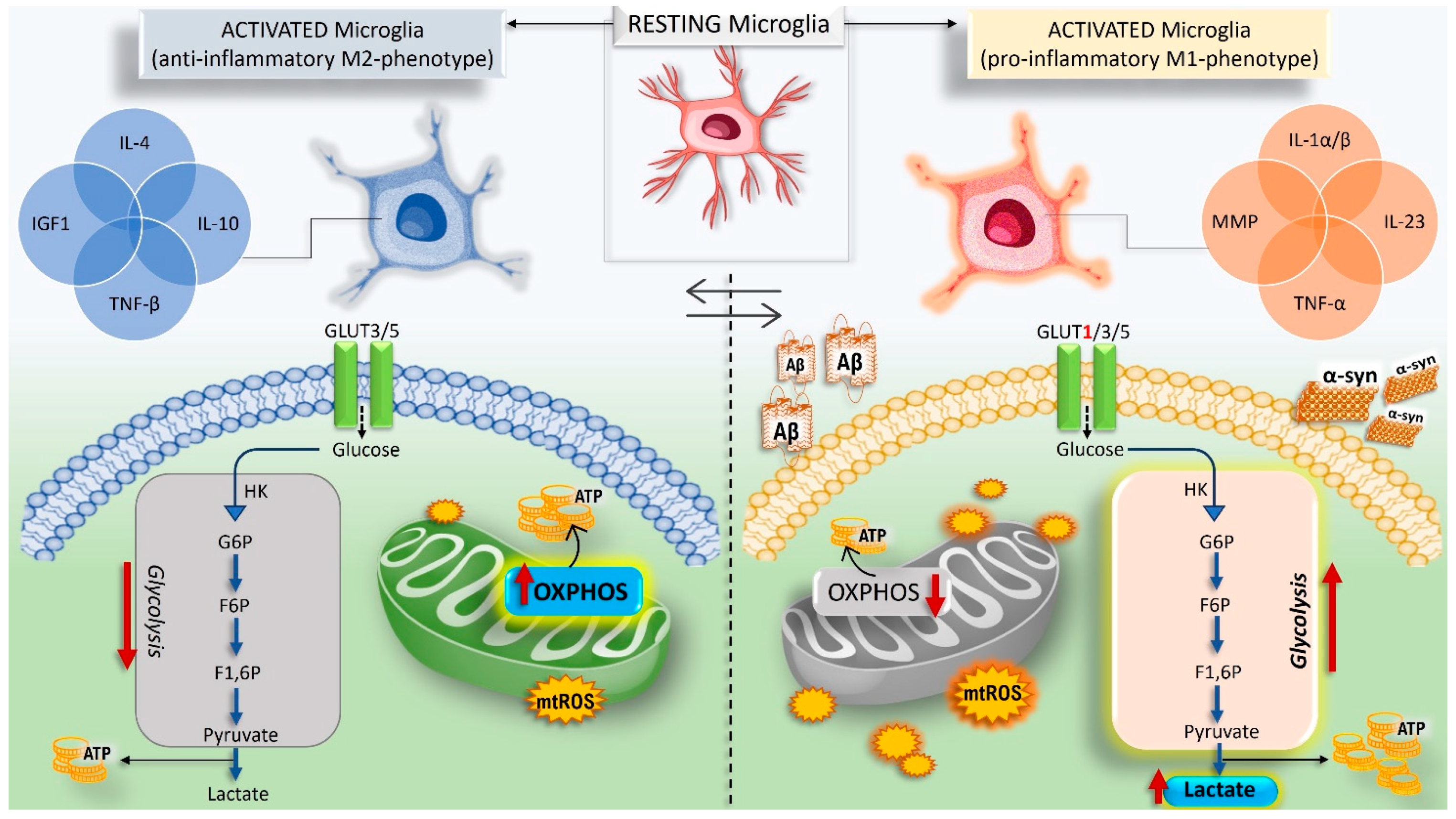
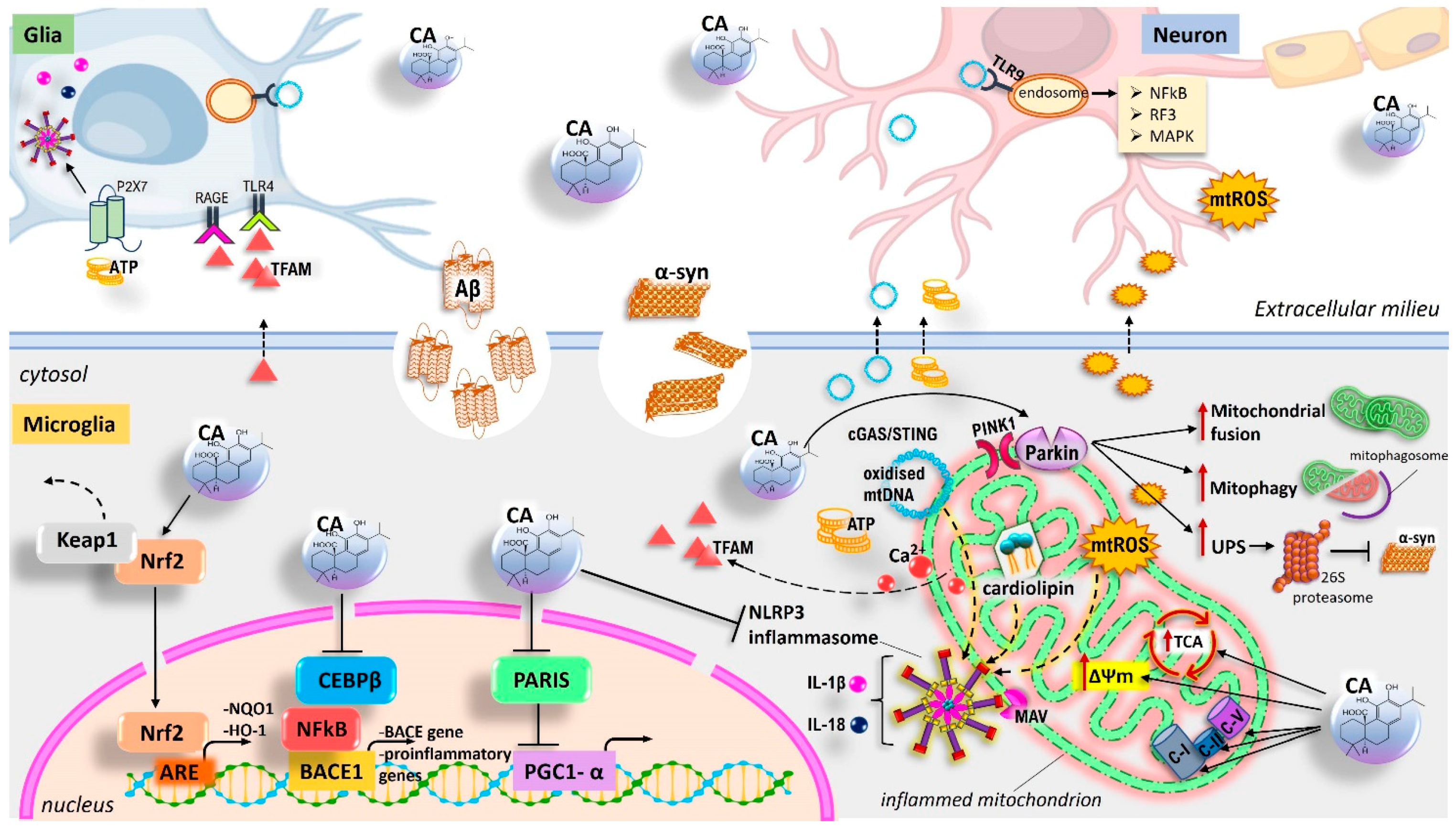
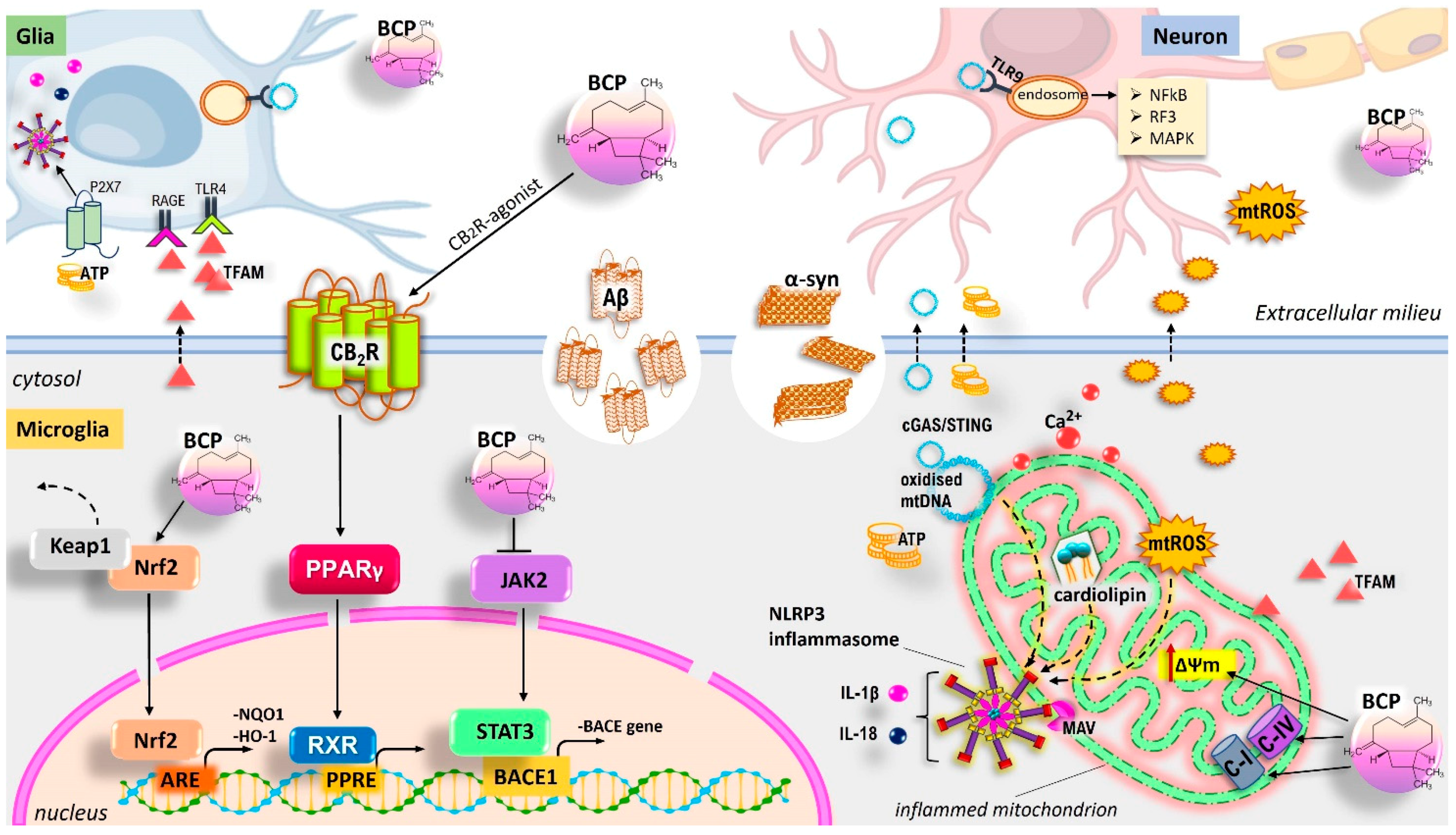
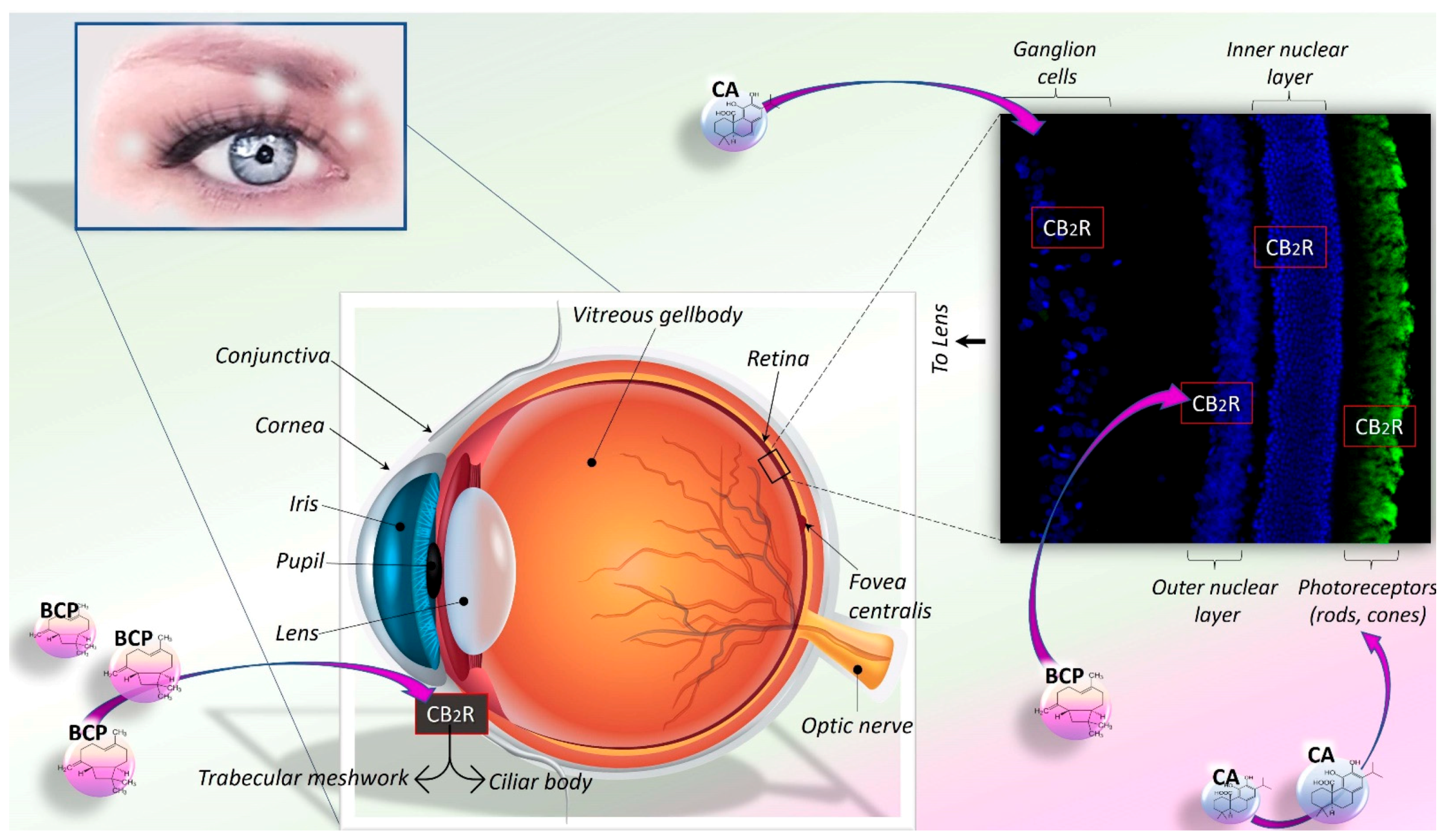
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iorio, R.; Celenza, G.; Petricca, S. Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation. Antioxidants 2022, 11, 1199. https://doi.org/10.3390/antiox11061199
Iorio R, Celenza G, Petricca S. Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation. Antioxidants. 2022; 11(6):1199. https://doi.org/10.3390/antiox11061199
Chicago/Turabian StyleIorio, Roberto, Giuseppe Celenza, and Sabrina Petricca. 2022. "Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation" Antioxidants 11, no. 6: 1199. https://doi.org/10.3390/antiox11061199
APA StyleIorio, R., Celenza, G., & Petricca, S. (2022). Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation. Antioxidants, 11(6), 1199. https://doi.org/10.3390/antiox11061199