
The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment


Abstract
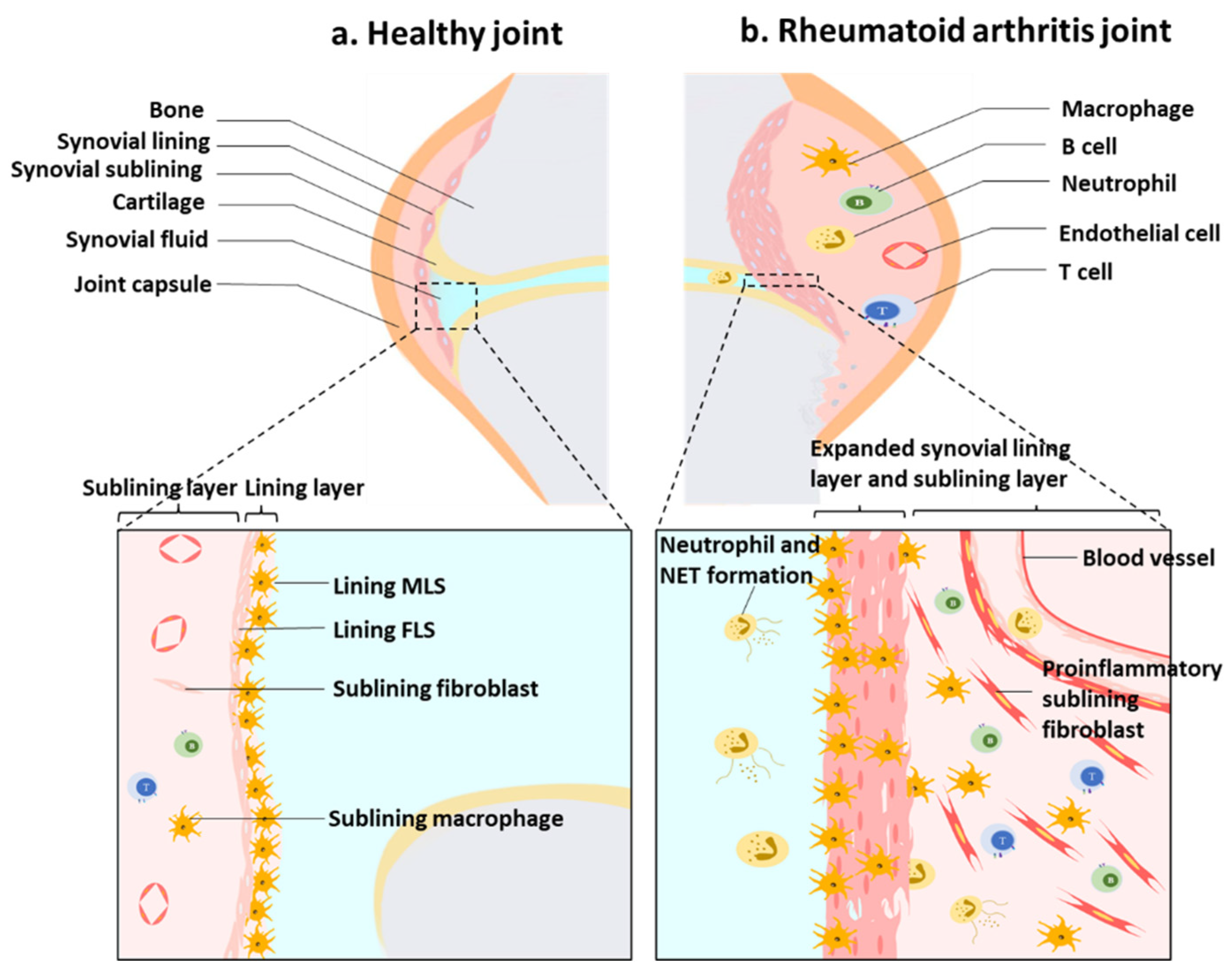
:1. Introduction
2. The Production of ROS in Synovitis
3. ROS and Synovium Stromal Cells
3.1. The Role of ROS in FLSs
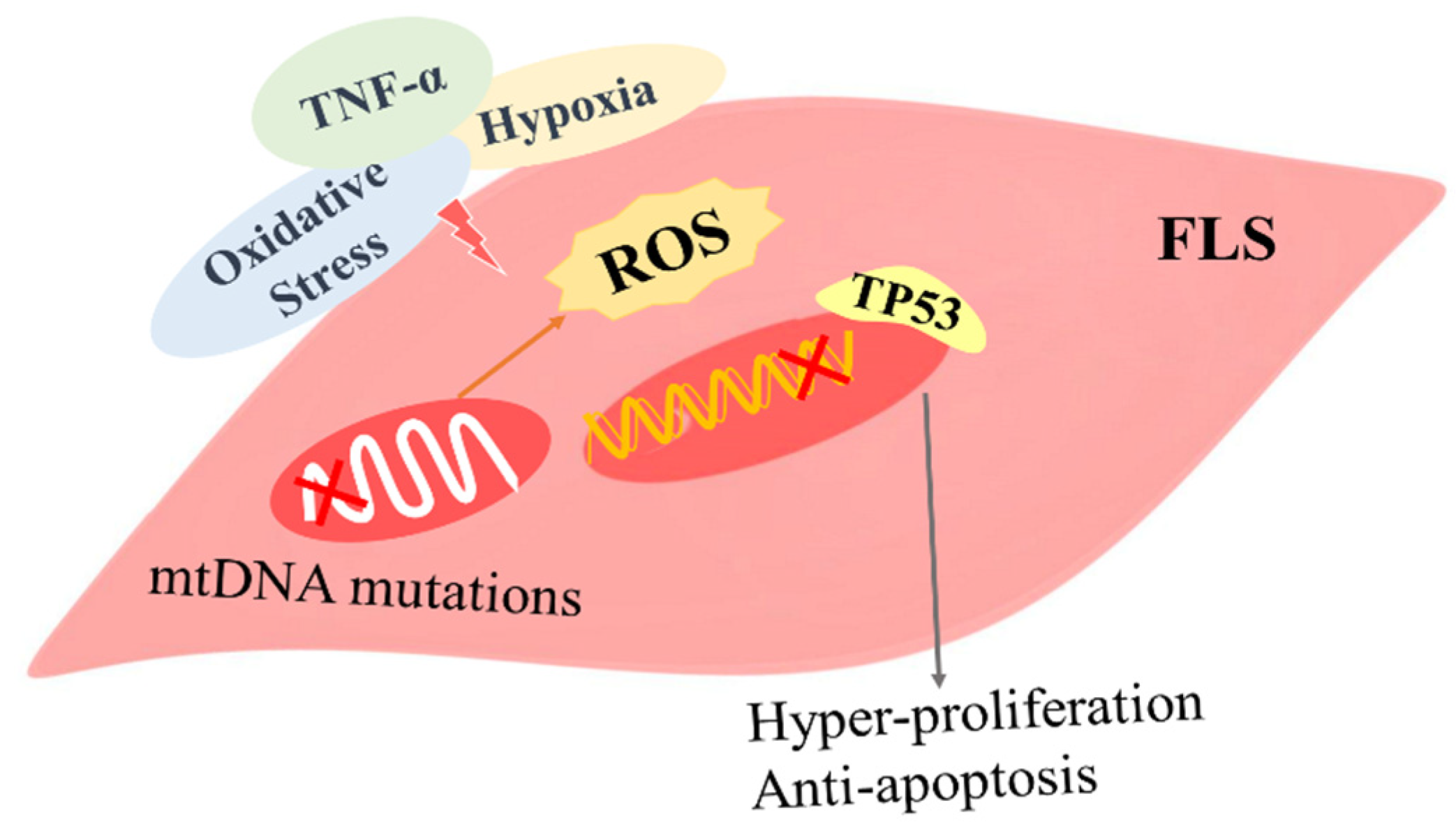
3.1.1. The Role of ROS-Induced Gene Mutations in FLSs
3.1.2. The Role of ROS-Related Autophagy and Apoptosis in FLSs
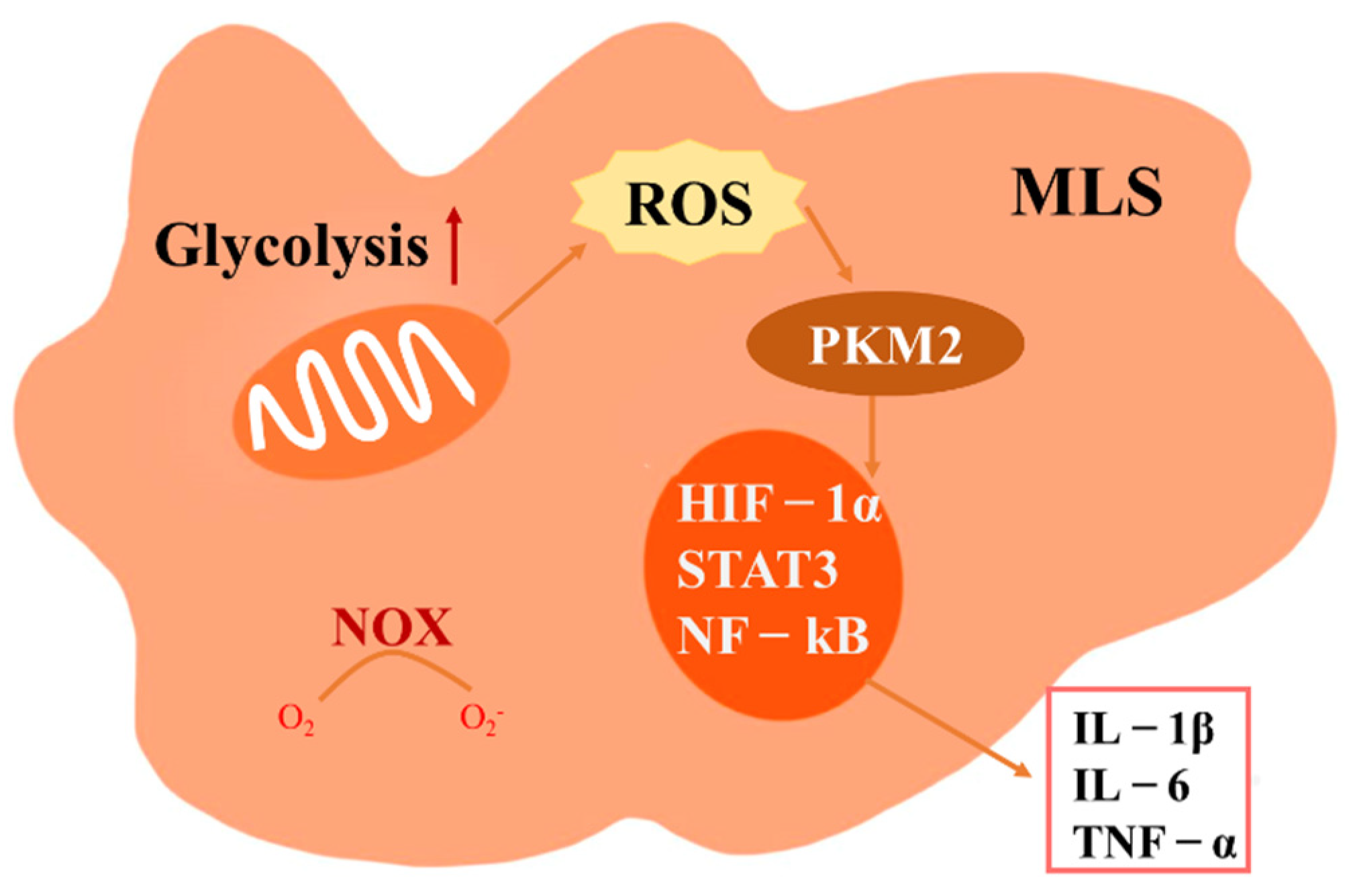
3.2. The Role of ROS in MLS
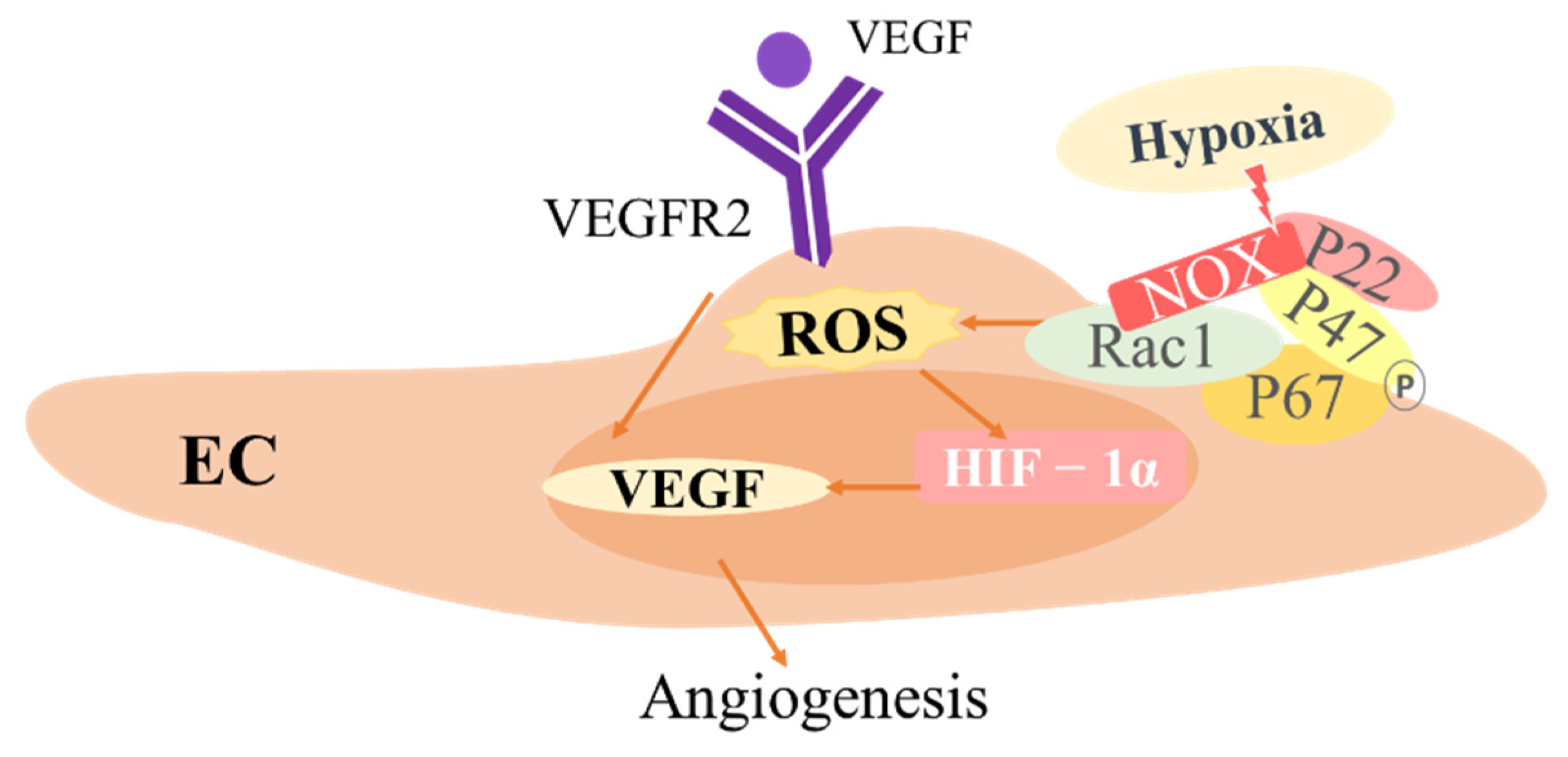
3.3. The Role of ROS in ECs
4. ROS and Synovial Immune Cells
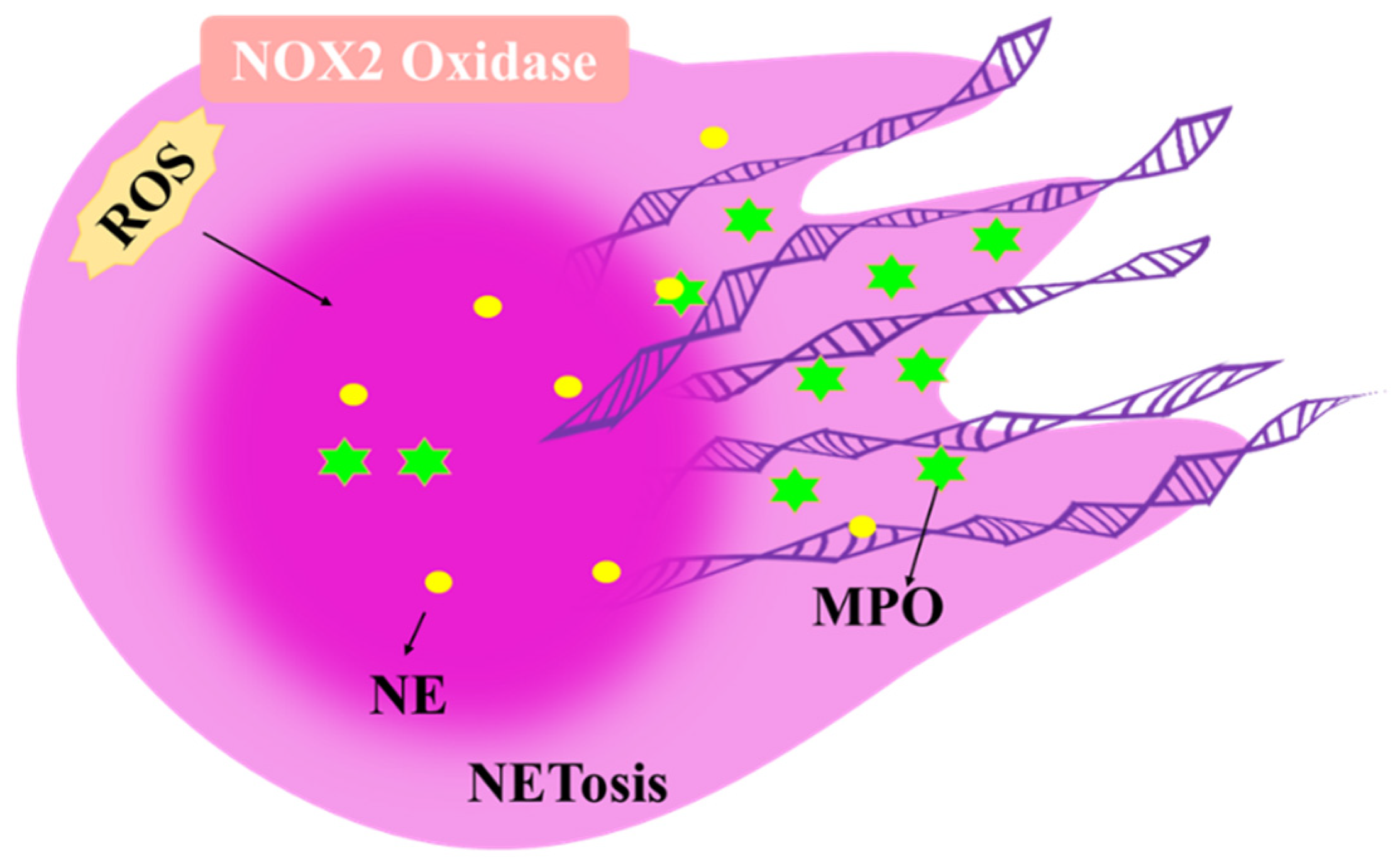
4.1. The Role of ROS in Neutrophils
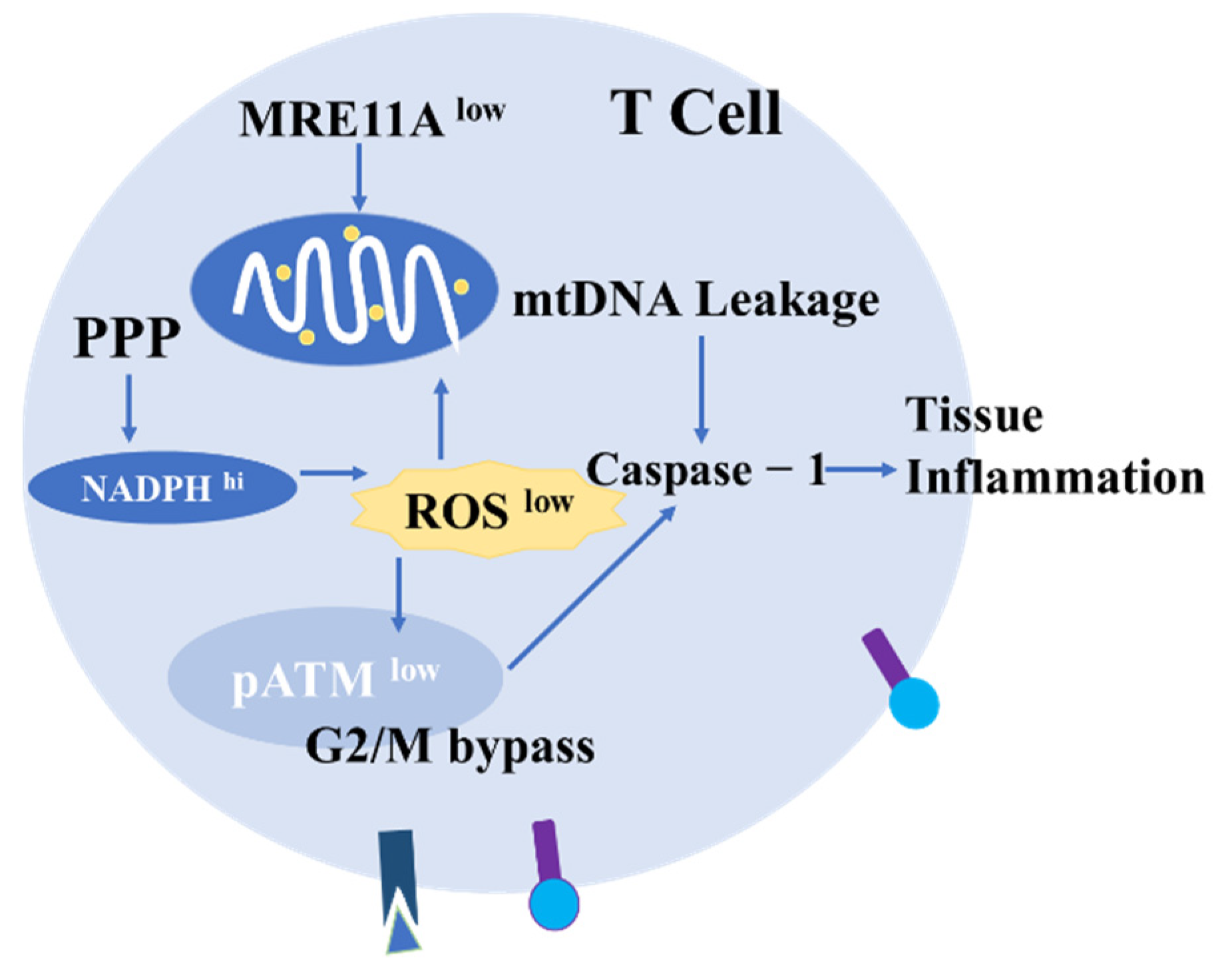
4.2. The Role of ROS in T Cells
4.3. The Role of ROS in B Cells
5. Targeted ROS Therapy for RA
5.1. Antioxidant Therapy
5.2. Oxidant-Promoting Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid arthritis and the mucosal origins hypothesis: Protection turns to destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Ospelt, C.; Gay, S.; Midwood, K.S. Location, location, location: How the tissue microenvironment affects inflammation in RA. Nat. Rev. Rheumatol. 2021, 17, 195–212. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 2019, 25, 215–227. [Google Scholar] [CrossRef]
- Hoffmann, M.H.; Griffiths, H.R. The dual role of Reactive Oxygen Species in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic. Biol. Med. 2018, 125, 62–71. [Google Scholar] [CrossRef]
- Jakubczyk, K.; Dec, K.; Kałduńska, J.; Kawczuga, D.; Kochman, J.; Janda, K. Reactive oxygen species—Sources, functions, oxidative damage. Pol. Merkur. Lek. 2020, 48, 124–127. [Google Scholar]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- da Fonseca, L.J.S.; Nunes-Souza, V.; Goulart, M.O.F.; Rabelo, L.A. Oxidative Stress in Rheumatoid Arthritis: What the Future Might Hold regarding Novel Biomarkers and Add-On Therapies. Oxid. Med. Cell. Longev. 2019, 2019, 7536805. [Google Scholar] [CrossRef] [Green Version]
- Phull, A.-R.; Nasir, B.; Haq, I.U.; Kim, S.J. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem. Biol. Interact. 2018, 281, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Kundu, S.; Ghosh, P.; De, S.; Ghosh, A.; Chatterjee, M. Correlation of oxidant status with oxidative tissue damage in patients with rheumatoid arthritis. Clin. Rheumatol. 2014, 33, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.; Hong, W.; Zhang, P.; Wang, X.; Körner, H.; Wei, W. Ontology and Function of Fibroblast-Like and Macrophage-Like Synoviocytes: How Do They Talk to Each Other and Can They Be Targeted for Rheumatoid Arthritis Therapy? Front. Immunol. 2018, 9, 1467. [Google Scholar] [CrossRef] [PubMed]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.Á.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef]
- Singh, J.A.; Arayssi, T.; Duray, P.; Schumacher, H.R. Immunohistochemistry of normal human knee synovium: A quantitative study. Ann. Rheum. Dis. 2004, 63, 785–790. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.T.; Biniecka, M.; Kennedy, A.; McCormick, J.; Fitzgerald, O.; Bresnihan, B.; Buggy, D.; Taylor, C.T.; O’Sullivan, J.; Fearon, U.; et al. Synovial tissue hypoxia and inflammation in vivo. Ann. Rheum. Dis. 2010, 69, 1389–1395. [Google Scholar] [CrossRef]
- Biniecka, M.; Canavan, M.; McGarry, T.; Gao, W.; McCormick, J.; Cregan, S.; Gallagher, L.; Smith, T.; Phelan, J.J.; Ryan, J.; et al. Dysregulated bioenergetics: A key regulator of joint inflammation. Ann. Rheum. Dis. 2016, 75, 2192–2200. [Google Scholar] [CrossRef]
- Kuksal, N.; Chalker, J.; Mailloux, R.J. Progress in understanding the molecular oxygen paradox—Function of mitochondrial reactive oxygen species in cell signaling. Biol. Chem. 2017, 398, 1209–1227. [Google Scholar] [CrossRef]
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum. 2011, 63, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Harty, L.C.; Biniecka, M.; O’Sullivan, J.; Fox, E.; Mulhall, K.; Veale, D.J.; Fearon, U. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann. Rheum. Dis. 2012, 71, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yu, S.; Wang, D.; Chen, S.; Chen, S.; Zheng, Y.; Wang, N.; Chen, S.; Li, J.; Shen, B. Germline and somatic mtDNA mutation spectrum of rheumatoid arthritis patients in the Taizhou area, China. Rheumatology 2020, 59, 2982–2991. [Google Scholar] [CrossRef] [PubMed]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Khojah, H.M.; Ahmed, S.; Abdel-Rahman, M.S.; Hamza, A.-B. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic. Biol. Med. 2016, 97, 285–291. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Phillips, D.C.; Dias, H.K.I.; Kitas, G.D.; Griffiths, H.R. Aberrant reactive oxygen and nitrogen species generation in rheumatoid arthritis (RA): Causes and consequences for immune function, cell survival, and therapeutic intervention. Antioxid. Redox Signal. 2010, 12, 743–785. [Google Scholar] [CrossRef]
- Kardeş, S.; Karagülle, M.; Durak, İ.; Avcı, A.; Karagülle, M.Z. Association of oxidative stress with clinical characteristics in patients with rheumatoid arthritis. Eur. J. Clin. Investig. 2018, 48, e12858. [Google Scholar] [CrossRef]
- Biniecka, M.; Kennedy, A.; Fearon, U.; Ng, C.T.; Veale, D.J.; O’Sullivan, J.N. Oxidative damage in synovial tissue is associated with in vivo hypoxic status in the arthritic joint. Ann. Rheum. Dis. 2010, 69, 1172–1178. [Google Scholar] [CrossRef]
- Balogh, E.; Veale, D.J.; McGarry, T.; Orr, C.; Szekanecz, Z.; Ng, C.-T.; Fearon, U.; Biniecka, M. Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Šteňová, E.; Bakošová, M.; Lauková, L.; Celec, P.; Vlková, B. Biological Anti-TNF- Therapy and Markers of Oxidative and Carbonyl Stress in Patients with Rheumatoid Arthritis. Oxid. Med. Cell. Longev. 2021, 2021, 5575479. [Google Scholar] [CrossRef]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, R234–R240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, H.R. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun. Rev. 2008, 7, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, U.; Anwar, A.; Savage, R.S.; Thornalley, P.J.; Rabbani, N. Protein oxidation, nitration and glycation biomarkers for early-stage diagnosis of osteoarthritis of the knee and typing and progression of arthritic disease. Arthritis Res. Ther. 2016, 18, 250. [Google Scholar] [CrossRef] [Green Version]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein oxidation—Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Dabbagh, A.J.; Trenam, C.W.; Morris, C.J.; Blake, D.R. Iron in joint inflammation. Ann. Rheum. Dis. 1993, 52, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Hou, H.; Luo, D.; An, R.; Zhao, Y.; Qiu, C. ROS-Dependent Lipid Peroxidation and Reliant Antioxidant Ferroptosis-Suppressor-Protein 1 in Rheumatoid Arthritis: A Covert Clue for Potential Therapy. Inflammation 2021, 44, 35–47. [Google Scholar] [CrossRef]
- Zhao, T.; Yang, Q.; Xi, Y.; Xie, Z.; Shen, J.; Li, Z.; Li, Z.; Qin, D. Ferroptosis in Rheumatoid Arthritis: A Potential Therapeutic Strategy. Front. Immunol. 2022, 13, 779585. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Newkirk, M.M.; Goldbach-Mansky, R.; Lee, J.; Hoxworth, J.; McCoy, A.; Yarboro, C.; Klippel, J.; El-Gabalawy, H.S. Advanced glycation end-product (AGE)-damaged IgG and IgM autoantibodies to IgG-AGE in patients with early synovitis. Arthritis Res. Ther. 2003, 5, R82–R90. [Google Scholar] [CrossRef] [PubMed]
- Strollo, R.; Ponchel, F.; Malmström, V.; Rizzo, P.; Bombardieri, M.; Wenham, C.Y.; Landy, R.; Perret, D.; Watt, F.; Corrigall, V.M.; et al. Autoantibodies to posttranslationally modified type II collagen as potential biomarkers for rheumatoid arthritis. Arthritis Rheum. 2013, 65, 1702–1712. [Google Scholar] [CrossRef]
- Monu; Agnihotri, P.; Biswas, S. AGE/Non-AGE Glycation: An Important Event in Rheumatoid Arthritis Pathophysiology. Inflammation 2022, 45, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Hui, Z.; Wei, W.; Yang, J.; Chen, Z.; Guo, B.; Xing, F.; Zhang, X.; Pan, L.; Xu, J. Hypotonic stress promotes ATP release, reactive oxygen species production and cell proliferation via TRPV4 activation in rheumatoid arthritis rat synovial fibroblasts. Biochem. Biophys. Res. Commun. 2017, 486, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Michalick, L.; Kuebler, W.M. TRPV4-A Missing Link Between Mechanosensation and Immunity. Front. Immunol. 2020, 11, 413. [Google Scholar] [CrossRef] [Green Version]
- Valencia, X.; Higgins, J.M.G.; Kiener, H.P.; Lee, D.M.; Podrebarac, T.A.; Dascher, C.C.; Watts, G.F.M.; Mizoguchi, E.; Simmons, B.; Patel, D.D.; et al. Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J. Exp. Med. 2004, 200, 1673–1679. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Koh, J.H.; Leng, L.; Kim, W.-U.; Bucala, R. The Tumor-Like Phenotype of Rheumatoid Synovium: Molecular Profiling and Prospects for Precision Medicine. Arthritis Rheumatol. 2018, 70, 637–652. [Google Scholar] [CrossRef]
- Lee, S.-H.; Chang, D.K.; Goel, A.; Boland, C.R.; Bugbee, W.; Boyle, D.L.; Firestein, G.S. Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J. Immunol. 2003, 170, 2214–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanishi, Y.; Boyle, D.L.; Green, D.R.; Keystone, E.C.; Connor, A.; Zollman, S.; Firestein, G.S. p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R12–R18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Wang, S.; Glorioso, J.C.; Evans, C.H.; Robbins, P.D.; Ghivizzani, S.C.; Oligino, T.J. Gene transfer of p53 to arthritic joints stimulates synovial apoptosis and inhibits inflammation. Mol. Ther. 2001, 3, 901–910. [Google Scholar] [CrossRef]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef] [Green Version]
- Taghadosi, M.; Adib, M.; Jamshidi, A.; Mahmoudi, M.; Farhadi, E. The p53 status in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Immunol. Res. 2021, 69, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Pap, T.; Aupperle, K.R.; Gay, S.; Firestein, G.S.; Gay, R.E. Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum. 2001, 44, 676–681. [Google Scholar] [CrossRef]
- Da Sylva, T.R.; Connor, A.; Mburu, Y.; Keystone, E.; Wu, G.E. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Res. Ther. 2005, 7, R844–R851. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Kwon, J.-E.; Lee, S.-Y.; Lee, E.-J.; Kim, D.S.; Moon, S.-J.; Lee, J.; Kwok, S.-K.; Park, S.-H.; Cho, M.-L. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Z.; Fan, X.; Li, W.; Qu, J.; Dong, C.; Wang, Z.; Ji, Z.; Li, Y. Inhibition of DNM1L and mitochondrial fission attenuates inflammatory response in fibroblast-like synoviocytes of rheumatoid arthritis. J. Cell. Mol. Med. 2020, 24, 1516–1528. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Xia, Y.; Lu, C.; Ye, Q.; Xi, X.; Wang, Q.; Wang, Z.; Wang, C.; Xiao, C. Regulatory Role of the RNA N-Methyladenosine Modification in Immunoregulatory Cells and Immune-Related Bone Homeostasis Associated With Rheumatoid Arthritis. Front. Cell. Dev. Biol. 2020, 8, 627893. [Google Scholar] [CrossRef]
- Shi, W.; Zheng, Y.; Luo, S.; Li, X.; Zhang, Y.; Meng, X.; Huang, C.; Li, J. METTL3 Promotes Activation and Inflammation of FLSs Through the NF-κB Signaling Pathway in Rheumatoid Arthritis. Front. Med. 2021, 8, 607585. [Google Scholar] [CrossRef]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Wei, J.; Cui, X.; Yu, C.; Ni, W.; Bungert, J.; Wu, L.; He, C.; Qian, Z. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 2021, 49, 5779–5797. [Google Scholar] [CrossRef]
- Korb, A.; Pavenstädt, H.; Pap, T. Cell death in rheumatoid arthritis. Apoptosis 2009, 14, 447–454. [Google Scholar] [CrossRef]
- Liu, H.; Pope, R.M. The role of apoptosis in rheumatoid arthritis. Curr. Opin. Pharm. 2003, 3, 317–322. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, H.; Wu, Y.; He, Z.; Qin, Y.; Shen, Q. The Autophagy Level Is Increased in the Synovial Tissues of Patients with Active Rheumatoid Arthritis and Is Correlated with Disease Severity. Mediat. Inflamm. 2017, 2017, 7623145. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Ospelt, C.; Gay, R.E.; Gay, S.; Klein, K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014, 66, 40–48. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2019, 38, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Interactions between reactive oxygen species and autophagy: Special issue: Death mechanisms in cellular homeostasis. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 119041. [Google Scholar] [CrossRef]
- Vomero, M.; Barbati, C.; Colasanti, T.; Perricone, C.; Novelli, L.; Ceccarelli, F.; Spinelli, F.R.; Di Franco, M.; Conti, F.; Valesini, G.; et al. Autophagy and Rheumatoid Arthritis: Current Knowledges and Future Perspectives. Front. Immunol. 2018, 9, 1577. [Google Scholar] [CrossRef] [Green Version]
- Alsousi, A.A.; Igwe, O.J. Autophagy protects against redox-active trace metal-induced cell death in rabbit synovial fibroblasts through Toll-like receptor 4 activation. Exp. Cell Res. 2019, 374, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kang, S.W.; Byun, H.S.; Jeon, J.; Park, K.A.; Kang, K.; Seo, W.; Won, M.; Seok, J.H.; Han, M.D.; et al. Brazilin Limits Inflammatory Responses through Induction of Prosurvival Autophagy in Rheumatoid Fibroblast-Like Synoviocytes. PLoS ONE 2015, 10, e0136122. [Google Scholar] [CrossRef]
- Cao, W.; Zhang, J.; Wang, G.; Lu, J.; Wang, T.; Chen, X. Reducing-Autophagy Derived Mitochondrial Dysfunction during Resveratrol Promotes Fibroblast-Like Synovial Cell Apoptosis. Anat. Rec. 2018, 301, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, J.; Zhang, M.; Wei, S.; Li, R.; Gao, Y.; Peng, W.; Wu, C. Apoptosis Induction of Fibroblast-Like Synoviocytes Is an Important Molecular-Mechanism for Herbal Medicine along with its Active Components in Treating Rheumatoid Arthritis. Biomolecules 2019, 9, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Kitsis, R.N.; Molkentin, J.D. Apoptotic cell death “Nixed” by an ER-mitochondrial necrotic pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 9031–9032. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.-H.; Pan, C.-Y.; Chen, N.-F.; Yang, S.-N.; Hsieh, S.; Wen, Z.-H.; Chen, W.-F.; Wang, J.-W.; Lu, W.-H.; Kuo, H.-M. Piscidin-1 Induces Apoptosis via Mitochondrial Reactive Oxygen Species-Regulated Mitochondrial Dysfunction in Human Osteosarcoma Cells. Sci. Rep. 2020, 10, 5045. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chang, Y.; Wei, W. Emerging role of targeting macrophages in rheumatoid arthritis: Focus on polarization, metabolism and apoptosis. Cell Prolif. 2020, 53, e12854. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Knaus, U.G. Raising the ‘Good’ Oxidants for Immune Protection. Front. Immunol. 2021, 12, 698042. [Google Scholar] [CrossRef]
- Asquith, D.L.; Ballantine, L.E.; Nijjar, J.S.; Makdasy, M.K.; Patel, S.; Wright, P.B.; Reilly, J.H.; Kerr, S.; Kurowska-Stolarska, M.; Gracie, J.A.; et al. The liver X receptor pathway is highly upregulated in rheumatoid arthritis synovial macrophages and potentiates TLR-driven cytokine release. Ann. Rheum. Dis. 2013, 72, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun. Rev. 2019, 18, 102397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.-Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [Green Version]
- Di Benedetto, P.; Ruscitti, P.; Vadasz, Z.; Toubi, E.; Giacomelli, R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun. Rev. 2019, 18, 102369. [Google Scholar] [CrossRef]
- You, S.; Yoo, S.-A.; Choi, S.; Kim, J.-Y.; Park, S.-J.; Ji, J.D.; Kim, T.-H.; Kim, K.-J.; Cho, C.-S.; Hwang, D.; et al. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc. Natl. Acad. Sci. USA 2014, 111, 550–555. [Google Scholar] [CrossRef] [Green Version]
- De Rycke, L.; Baeten, D.; Foell, D.; Kruithof, E.; Veys, E.M.; Roth, J.; De Keyser, F. Differential expression and response to anti-TNFalpha treatment of infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J. Pathol. 2005, 206, 17–27. [Google Scholar] [CrossRef]
- Zeisbrich, M.; Yanes, R.E.; Zhang, H.; Watanabe, R.; Li, Y.; Brosig, L.; Hong, J.; Wallis, B.B.; Giacomini, J.C.; Assimes, T.L.; et al. Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann. Rheum. Dis. 2018, 77, 1053–1062. [Google Scholar] [CrossRef]
- Siouti, E.; Andreakos, E. The many facets of macrophages in rheumatoid arthritis. Biochem. Pharm. 2019, 165, 152–169. [Google Scholar] [CrossRef]
- Qiu, J.; Wu, B.; Goodman, S.B.; Berry, G.J.; Goronzy, J.J.; Weyand, C.M. Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 652771. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.W.; Choi, Y.S.; Kim, H.W.; Shin, S.; Ha, Y.-J.; Kang, E.H.; Park, J.W.; Park, J.K.; Shin, K.; Song, Y.W.; et al. Extracellular pyruvate kinase M2 promotes osteoclastogenesis and is associated with radiographic progression in early rheumatoid arthritis. Sci. Rep. 2022, 12, 4024. [Google Scholar] [CrossRef] [PubMed]
- Hagert, C.; Sareila, O.; Kelkka, T.; Jalkanen, S.; Holmdahl, R. The Macrophage Mannose Receptor Regulate Mannan-Induced Psoriasis, Psoriatic Arthritis, and Rheumatoid Arthritis-Like Disease Models. Front. Immunol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Shimamura, T.; Hamuro, J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002, 14, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, I.J.; Liu, S.-C.; Su, C.-M.; Wang, Y.-H.; Tsai, C.-H.; Tang, C.-H. Implications of Angiogenesis Involvement in Arthritis. Int. J. Mol. Sci. 2018, 19, 2012. [Google Scholar] [CrossRef] [Green Version]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [Green Version]
- Colville-Nash, P.R.; Scott, D.L. Angiogenesis and rheumatoid arthritis: Pathogenic and therapeutic implications. Ann. Rheum. Dis. 1992, 51, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Leblond, A.; Allanore, Y.; Avouac, J. Targeting synovial neoangiogenesis in rheumatoid arthritis. Autoimmun. Rev. 2017, 16, 594–601. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Su, C.-M.; Hsu, C.-J.; Huang, C.-C.; Wang, S.-W.; Liu, S.-C.; Chen, W.-C.; Fuh, L.-J.; Tang, C.-H. CCN1 Promotes VEGF Production in Osteoblasts and Induces Endothelial Progenitor Cell Angiogenesis by Inhibiting miR-126 Expression in Rheumatoid Arthritis. J. Bone Min. Res. 2017, 32, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.-H.; Chen, C.-J.; Gong, C.-L.; Liu, S.-C.; Chen, P.-C.; Huang, C.-C.; Hu, S.-L.; Wang, S.-W.; Tang, C.-H. CXCL13/CXCR5 axis facilitates endothelial progenitor cell homing and angiogenesis during rheumatoid arthritis progression. Cell Death Dis. 2021, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spanò, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef]
- Mason, J.C. Cytoprotective pathways in the vascular endothelium. Do they represent a viable therapeutic target? Vasc. Pharm. 2016, 86, 41–52. [Google Scholar] [CrossRef]
- Murdaca, G.; Spanò, F.; Cagnati, P.; Puppo, F. Free radicals and endothelial dysfunction: Potential positive effects of TNF-α inhibitors. Redox Rep. 2013, 18, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Barvitenko, N.; Skverchinskaya, E.; Lawen, A.; Matteucci, E.; Saldanha, C.; Uras, G.; Manca, A.; Aslam, M.; Pantaleo, A. Pleiotropic and Potentially Beneficial Effects of Reactive Oxygen Species on the Intracellular Signaling Pathways in Endothelial Cells. Antioxidants 2021, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Manea, A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010, 342, 325–339. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharm. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, J.; Kim, S.-J.; Furuta, E.; Caliceti, C.; Chen, G.-F.; McKinney, R.D.; Kuhr, F.; Levitan, I.; Fukai, T.; Ushio-Fukai, M. Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H724–H732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-L.; Zhang, Q.; Zhao, R.; Medford, R.M. Superoxide, H2O2, and iron are required for TNF-alpha-induced MCP-1 gene expression in endothelial cells: Role of Rac1 and NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1001–H1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenevier-Gobeaux, C.; Simonneau, C.; Lemarechal, H.; Bonnefont-Rousselot, D.; Poiraudeau, S.; Rannou, F.; Anract, P.; Borderie, D. Hypoxia induces nitric oxide synthase in rheumatoid synoviocytes: Consequences on NADPH oxidase regulation. Free Radic. Res. 2012, 46, 628–636. [Google Scholar] [CrossRef]
- Biniecka, M.; Connolly, M.; Gao, W.; Ng, C.T.; Balogh, E.; Gogarty, M.; Santos, L.; Murphy, E.; Brayden, D.; Veale, D.J.; et al. Redox-mediated angiogenesis in the hypoxic joint of inflammatory arthritis. Arthritis Rheumatol. 2014, 66, 3300–3310. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Leblond, A.; Pezet, S.; Cauvet, A.; Casas, C.; Pires Da Silva, J.; Hervé, R.; Clavel, G.; Dumas, S.; Cohen-Kaminsky, S.; Bessis, N.; et al. Implication of the deacetylase sirtuin-1 on synovial angiogenesis and persistence of experimental arthritis. Ann. Rheum. Dis. 2020, 79, 891–900. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Fang, W.-J.; Pan, S.; Zhang, S.-S.; Li, D.-F.; Wang, Z.-F.; Chen, W.-G.; Yin, Q.; Zuo, J. Regulation of Sirt1 on energy metabolism and immune response in rheumatoid arthritis. Int. Immunopharmacol. 2021, 101, 108175. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Kaihara, K.; Nakagawa, S.; Arai, Y.; Inoue, H.; Tsuchida, S.; Fujii, Y.; Kamada, Y.; Kishida, T.; Mazda, O.; Takahashi, K. Sustained Hypoxia Suppresses Joint Destruction in a Rat Model of Rheumatoid Arthritis via Negative Feedback of Hypoxia Inducible Factor-1α. Int. J. Mol. Sci. 2021, 22, 3898. [Google Scholar] [CrossRef]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, S.-S.; Zong, M.; Fan, S.-S.; Lu, T.-B.; Gong, R.-H.; Sun, L.-S.; Fan, L.-Y. Glucose-6-Phosphate Isomerase (G6PI) Mediates Hypoxia-Induced Angiogenesis in Rheumatoid Arthritis. Sci. Rep. 2017, 7, 40274. [Google Scholar] [CrossRef]
- Ma, Q.; Reiter, R.J.; Chen, Y. Role of melatonin in controlling angiogenesis under physiological and pathological conditions. Angiogenesis 2020, 23, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yang, H.-L.; Gu, C.-J.; Liu, Y.-K.; Shao, J.; Zhu, R.; He, Y.-Y.; Zhu, X.-Y.; Li, M.-Q. Melatonin restricts the viability and angiogenesis of vascular endothelial cells by suppressing HIF-1α/ROS/VEGF. Int. J. Mol. Med. 2019, 43, 945–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turunen, S.; Huhtakangas, J.; Nousiainen, T.; Valkealahti, M.; Melkko, J.; Risteli, J.; Lehenkari, P. Rheumatoid arthritis antigens homocitrulline and citrulline are generated by local myeloperoxidase and peptidyl arginine deiminases 2, 3 and 4 in rheumatoid nodule and synovial tissue. Arthritis Res. Ther. 2016, 18, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, H.L.; Lyon, M.; Chapman, E.A.; Moots, R.J.; Edwards, S.W. Rheumatoid Arthritis Synovial Fluid Neutrophils Drive Inflammation Through Production of Chemokines, Reactive Oxygen Species, and Neutrophil Extracellular Traps. Front. Immunol. 2020, 11, 584116. [Google Scholar] [CrossRef]
- Cecchi, I.; Arias de la Rosa, I.; Menegatti, E.; Roccatello, D.; Collantes-Estevez, E.; Lopez-Pedrera, C.; Barbarroja, N. Neutrophils: Novel key players in Rheumatoid Arthritis. Current and future therapeutic targets. Autoimmun. Rev. 2018, 17, 1138–1149. [Google Scholar] [CrossRef]
- Sur Chowdhury, C.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014, 16, R122. [Google Scholar] [CrossRef] [Green Version]
- Strzepa, A.; Pritchard, K.A.; Dittel, B.N. Myeloperoxidase: A new player in autoimmunity. Cell. Immunol. 2017, 317, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef]
- Stamp, L.K.; Khalilova, I.; Tarr, J.M.; Senthilmohan, R.; Turner, R.; Haigh, R.C.; Winyard, P.G.; Kettle, A.J. Myeloperoxidase and oxidative stress in rheumatoid arthritis. Rheumatology 2012, 51, 1796–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [Green Version]
- Fresneda Alarcon, M.; McLaren, Z.; Wright, H.L. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 2021, 12, 649693. [Google Scholar] [CrossRef] [PubMed]
- Spengler, J.; Lugonja, B.; Ytterberg, A.J.; Zubarev, R.A.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Kienhöfer, D.; Hahn, J.; Stoof, J.; Csepregi, J.Z.; Reinwald, C.; Urbonaviciute, V.; Johnsson, C.; Maueröder, C.; Podolska, M.J.; Biermann, M.H.; et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight 2017, 2, e92920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareila, O.; Kelkka, T.; Pizzolla, A.; Hultqvist, M.; Holmdahl, R. NOX2 complex-derived ROS as immune regulators. Antioxid. Redox Signal. 2011, 15, 2197–2208. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.-C.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Olsson, L.M.; Urbonaviciute, V.; Yang, M.; Bäckdahl, L.; Holmdahl, R. Association of NOX2 subunits genetic variants with autoimmune diseases. Free Radic. Biol. Med. 2018, 125, 72–80. [Google Scholar] [CrossRef]
- Winter, S.; Hultqvist Hopkins, M.; Laulund, F.; Holmdahl, R. A Reduction in Intracellular Reactive Oxygen Species Due to a Mutation in NCF4 Promotes Autoimmune Arthritis in Mice. Antioxid. Redox Signal. 2016, 25, 983–996. [Google Scholar] [CrossRef]
- Baruah, S.; Murthy, S.; Keck, K.; Galvan, I.; Prichard, A.; Allen, L.-A.H.; Farrelly, M.; Klesney-Tait, J. TREM-1 regulates neutrophil chemotaxis by promoting NOX-dependent superoxide production. J. Leukoc. Biol. 2019, 105, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Lo, P.-C.; Yen, C.-L.; Nigrovic, P.A.; Chao, W.-C.; Wang, W.-Z.; Hsu, G.C.; Tsai, Y.-S.; Shieh, C.-C. Redox Regulation of Pro-IL-1β Processing May Contribute to the Increased Severity of Serum-Induced Arthritis in NOX2-Deficient Mice. Antioxid. Redox Signal. 2015, 23, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.-C.; Wu, S.-Y.; Huang, Y.-F.; Lo, P.-C.; Chan, T.-Y.; Chen, C.-A.; Wu, C.-H.; Hsu, C.-C.; Yen, C.-L.; Chen, P.-C.; et al. NOX2-Deficient Neutrophils Facilitate Joint Inflammation Through Higher Pro-Inflammatory and Weakened Immune Checkpoint Activities. Front. Immunol. 2021, 12, 743030. [Google Scholar] [CrossRef]
- Lopes, F.; Coelho, F.M.; Costa, V.V.; Vieira, É.L.M.; Sousa, L.P.; Silva, T.A.; Vieira, L.Q.; Teixeira, M.M.; Pinho, V. Resolution of neutrophilic inflammation by H2O2 in antigen-induced arthritis. Arthritis Rheum. 2011, 63, 2651–2660. [Google Scholar] [CrossRef]
- van Dalen, S.C.M.; Kruisbergen, N.N.L.; Walgreen, B.; Helsen, M.M.A.; Slöetjes, A.W.; Cremers, N.A.J.; Koenders, M.I.; van de Loo, F.A.J.; Roth, J.; Vogl, T.; et al. The role of NOX2-derived reactive oxygen species in collagenase-induced osteoarthritis. Osteoarthr. Cartil. 2018, 26, 1722–1732. [Google Scholar] [CrossRef] [Green Version]
- Kruisbergen, N.N.L.; Di Ceglie, I.; van Gemert, Y.; Walgreen, B.; Helsen, M.M.A.; Slöetjes, A.W.; Koenders, M.I.; van de Loo, F.A.J.; Roth, J.; Vogl, T.; et al. Nox2 Deficiency Reduces Cartilage Damage and Ectopic Bone Formation in an Experimental Model for Osteoarthritis. Antioxidants 2021, 10, 1660. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Yau, A.C.Y.; Holmdahl, R. Regulation of T Cell Function by Reactive Nitrogen and Oxygen Species in Collagen-Induced Arthritis. Antioxid. Redox Signal. 2020, 32, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. The immunology of rheumatoid arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Schönland, S.O.; Lopez, C.; Widmann, T.; Zimmer, J.; Bryl, E.; Goronzy, J.J.; Weyand, C.M. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc. Natl. Acad. Sci. USA 2003, 100, 13471–13476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Fujii, H.; Mohan, S.V.; Goronzy, J.J.; Weyand, C.M. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 2013, 210, 2119–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in the development of rheumatoid arthritis. Immunol. Rev. 2020, 294, 177–187. [Google Scholar] [CrossRef]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy 2014, 10, 382–383. [Google Scholar] [CrossRef] [Green Version]
- Weyand, C.M.; Shen, Y.; Goronzy, J.J. Redox-sensitive signaling in inflammatory T cells and in autoimmune disease. Free Radic. Biol. Med. 2018, 125, 36–43. [Google Scholar] [CrossRef]
- Weyand, C.M.; Wu, B.; Goronzy, J.J. The metabolic signature of T cells in rheumatoid arthritis. Curr. Opin. Rheumatol. 2020, 32, 159–167. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 331ra338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Wen, Z.; Li, Y.; Matteson, E.L.; Hong, J.; Goronzy, J.J.; Weyand, C.M. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat. Immunol. 2017, 18, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Goronzy, J.J.; Weyand, C.M. Metabolic Fitness of T Cells in Autoimmune Disease. Immunometabolism 2020, 2, e200017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Li, Y.; Goronzy, J.J.; Weyand, C.M. DNA damage, metabolism and aging in pro-inflammatory T cells: Rheumatoid arthritis as a model system. Exp. Gerontol. 2018, 105, 118–127. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Y.; Jin, K.; Wen, Z.; Cao, W.; Wu, B.; Wen, R.; Tian, L.; Berry, G.J.; Goronzy, J.J.; et al. The DNA Repair Nuclease MRE11A Functions as a Mitochondrial Protector and Prevents T Cell Pyroptosis and Tissue Inflammation. Cell Metab. 2019, 30, 477–492.e6. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Y.; Hohensinner, P.; Ju, J.; Wen, Z.; Goodman, S.B.; Zhang, H.; Goronzy, J.J.; Weyand, C.M. Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity 2016, 45, 903–916. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, S.-Q.; Li, C.; Lykken, E.; Jiang, S.; Wong, E.; Gong, Z.; Tao, Z.; Zhu, B.; Wan, Y.; et al. MicroRNA-23a Curbs Necrosis during Early T Cell Activation by Enforcing Intracellular Reactive Oxygen Species Equilibrium. Immunity 2016, 44, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Jahraus, B.; Balta, E.; Ziegler, J.D.; Hübner, K.; Blank, N.; Niesler, B.; Wabnitz, G.H.; Samstag, Y. Sulforaphane Inhibits Inflammatory Responses of Primary Human T-Cells by Increasing ROS and Depleting Glutathione. Front. Immunol. 2018, 9, 2584. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Chávez, M.D.; Tse, H.M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front. Immunol. 2021, 12, 703972. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Anquetil, F.; Clavel, C.; Ndongo-Thiam, N.; Offer, G.; Miossec, P.; Pasquali, J.-L.; Sebbag, M.; Serre, G. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann. Rheum. Dis. 2015, 74, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154. [Google Scholar] [CrossRef]
- Humby, F.; Lewis, M.; Ramamoorthi, N.; Hackney, J.A.; Barnes, M.R.; Bombardieri, M.; Setiadi, A.F.; Kelly, S.; Bene, F.; DiCicco, M.; et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann. Rheum. Dis. 2019, 78, 761–772. [Google Scholar] [CrossRef]
- Rivellese, F.; Humby, F.; Bugatti, S.; Fossati-Jimack, L.; Rizvi, H.; Lucchesi, D.; Lliso-Ribera, G.; Nerviani, A.; Hands, R.E.; Giorli, G.; et al. B Cell Synovitis and Clinical Phenotypes in Rheumatoid Arthritis: Relationship to Disease Stages and Drug Exposure. Arthritis Rheumatol. 2020, 72, 714–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alivernini, S.; Kurowska-Stolarska, M.; Tolusso, B.; Benvenuto, R.; Elmesmari, A.; Canestri, S.; Petricca, L.; Mangoni, A.; Fedele, A.L.; Di Mario, C.; et al. MicroRNA-155 influences B-cell function through PU.1 in rheumatoid arthritis. Nat. Commun. 2016, 7, 12970. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. B Cells in Rheumatoid Arthritis: Pathogenic Mechanisms and Treatment Prospects. Front. Immunol. 2021, 12, 750753. [Google Scholar] [CrossRef]
- Khmaladze, I.; Saxena, A.; Nandakumar, K.S.; Holmdahl, R. B-cell epitope spreading and inflammation in a mouse model of arthritis is associated with a deficiency in reactive oxygen species production. Eur. J. Immunol. 2015, 45, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Yang, H.-Y.; Lai, J.-H. Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis. Int. J. Mol. Sci. 2020, 21, 4015. [Google Scholar] [CrossRef]
- Corsiero, E.; Pratesi, F.; Prediletto, E.; Bombardieri, M.; Migliorini, P. NETosis as Source of Autoantigens in Rheumatoid Arthritis. Front. Immunol. 2016, 7, 485. [Google Scholar] [CrossRef]
- Corsiero, E.; Bombardieri, M.; Carlotti, E.; Pratesi, F.; Robinson, W.; Migliorini, P.; Pitzalis, C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann. Rheum. Dis. 2016, 75, 1866–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behl, T.; Upadhyay, T.; Singh, S.; Chigurupati, S.; Alsubayiel, A.M.; Mani, V.; Vargas-De-La-Cruz, C.; Uivarosan, D.; Bustea, C.; Sava, C.; et al. Polyphenols Targeting MAPK Mediated Oxidative Stress and Inflammation in Rheumatoid Arthritis. Molecules 2021, 26, 6570. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Sultana, F.; Neog, M.K.; Rasool, M. Withaferin-A, a steroidal lactone encapsulated mannose decorated liposomes ameliorates rheumatoid arthritis by intriguing the macrophage repolarization in adjuvant-induced arthritic rats. Colloids Surf. B Biointerfaces 2017, 155, 349–365. [Google Scholar] [CrossRef]
- Kalashnikova, I.; Chung, S.-J.; Nafiujjaman, M.; Hill, M.L.; Siziba, M.E.; Contag, C.H.; Kim, T. Ceria-based nanotheranostic agent for rheumatoid arthritis. Theranostics 2020, 10, 11863–11880. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.Y.; Song, S.Y.; Go, S.-H.; Sohn, H.S.; Baik, S.; Soh, M.; Kim, K.; Kim, D.; Kim, H.-C.; et al. Synergistic Oxygen Generation and Reactive Oxygen Species Scavenging by Manganese Ferrite/Ceria Co-decorated Nanoparticles for Rheumatoid Arthritis Treatment. ACS Nano 2019, 13, 3206–3217. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, L.; Wang, Z.; Liu, P.; Liu, X.; Ding, J.; Zhou, W. Targeted silver nanoparticles for rheumatoid arthritis therapy via macrophage apoptosis and Re-polarization. Biomaterials 2021, 264, 120390. [Google Scholar] [CrossRef]
- Esalatmanesh, K.; Jamali, A.; Esalatmanesh, R.; Soleimani, Z.; Khabbazi, A.; Malek Mahdavi, A. Effects of N-acetylcysteine supplementation on disease activity, oxidative stress, and inflammatory and metabolic parameters in rheumatoid arthritis patients: A randomized double-blind placebo-controlled trial. Amino Acids 2022, 54, 433–440. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef] [Green Version]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultqvist, M.; Olsson, L.M.; Gelderman, K.A.; Holmdahl, R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009, 30, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Holmdahl, R.; Sareila, O.; Olsson, L.M.; Bäckdahl, L.; Wing, K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 2016, 269, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Olofsson, P.; Wallner, F.K.; Holmdahl, R. Pharmacological Potential of NOX2 Agonists in Inflammatory Conditions. Antioxid. Redox Signal. 2015, 23, 446–459. [Google Scholar] [CrossRef]
- Doering, M.; Ba, L.A.; Lilienthal, N.; Nicco, C.; Scherer, C.; Abbas, M.; Zada, A.A.P.; Coriat, R.; Burkholz, T.; Wessjohann, L.; et al. Synthesis and selective anticancer activity of organochalcogen based redox catalysts. J. Med. Chem. 2010, 53, 6954–6963. [Google Scholar] [CrossRef]
- Zhang, K.; Gao, S.; Guo, J.; Ni, G.; Chen, Z.; Li, F.; Zhu, X.; Wen, Y.; Guo, Y. Hypericin-photodynamic therapy inhibits proliferation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes cell line MH7A. Iran. J. Basic Med. Sci. 2018, 21, 130–137. [Google Scholar] [CrossRef]
- Jeong, M.; Cho, J.; Shin, J.I.; Jeon, Y.J.; Kim, J.H.; Lee, S.J.; Kim, E.S.; Lee, K. Hempseed oil induces reactive oxygen species- and C/EBP homologous protein-mediated apoptosis in MH7A human rheumatoid arthritis fibroblast-like synovial cells. J. Ethnopharmacol. 2014, 154, 745–752. [Google Scholar] [CrossRef]
- Li, J.; Pang, J.; Liu, Z.; Ge, X.; Zhen, Y.; Jiang, C.C.; Liu, Y.; Huo, Q.; Sun, Y.; Liu, H. Shikonin induces programmed death of fibroblast synovial cells in rheumatoid arthritis by inhibiting energy pathways. Sci. Rep. 2021, 11, 18263. [Google Scholar] [CrossRef]
- Pu, L.; Meng, Q.; Li, S.; Liu, B.; Li, F. Icariin arrests cell cycle progression and induces cell apoptosis through the mitochondrial pathway in human fibroblast-like synoviocytes. Eur. J. Pharmacol. 2021, 912, 174585. [Google Scholar] [CrossRef]
- Shang, C.H.; Zhang, Q.Q.; Zhou, J.H. Oridonin Inhibits Cell Proliferation and Induces Apoptosis in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Inflammation 2016, 39, 873–880. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Q.; Wang, S.; Li, T.; Xiao, Z.; Lan, W.; Huang, G.; Cai, X. α-Mangostin, A Natural Xanthone, Induces Apoptosis and ROS Accumulation in Human Rheumatoid Fibroblast-Like Synoviocyte MH7A Cells. Curr. Mol. Med. 2017, 17, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Li, J.; Zhang, C.; Zhao, L.; Guo, L.; Xu, T.; Jin, J.; Wu, M.; Xia, Y. α-Mangostin promotes apoptosis of human rheumatoid arthritis fibroblast-like synoviocytes by reactive oxygen species-dependent activation of ERK1/2 mitogen-activated protein kinase. J. Cell. Biochem. 2019, 120, 14986–14994. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.C.; Kim, C.; Lee, J.M.; Cho, W.S.; Lee, S.G.; Jeong, M.; Cho, J.; Lee, K. Apigenin-induced apoptosis is mediated by reactive oxygen species and activation of ERK1/2 in rheumatoid fibroblast-like synoviocytes. Chem.-Biol. Interact. 2009, 182, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.I.; Jeon, Y.J.; Lee, S.; Lee, Y.G.; Kim, J.B.; Kwon, H.C.; Kim, S.H.; Kim, I.; Lee, K.; Han, Y.S. Apoptotic and Anti-Inflammatory Effects of Eupatorium japonicum Thunb. in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. BioMed Res. Int. 2018, 2018, 1383697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.N.; Luo, Y.H.; Meng, L.Q.; Piao, X.J.; Wang, Y.; Wang, J.R.; Wang, H.; Zhang, Y.; Li, J.Q.; Xu, W.T.; et al. Cryptotanshinone induces reactive oxygen species-mediated apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes. Int. J. Mol. Med. 2019, 43, 1067–1075. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Wang, C.; Cui, Z.; Guo, P.; Meng, Q.; Shi, X.; Gao, Y.; Yang, G.; Han, Z. β-Elemene induces apoptosis of human rheumatoid arthritis fibroblast-like synoviocytes via reactive oxygen species-dependent activation of p38 mitogen-activated protein kinase. Pharmacol. Rep. PR 2016, 68, 7–11. [Google Scholar] [CrossRef]
- Zuo, J.; Dou, D.Y.; Wang, H.F.; Zhu, Y.H.; Li, Y.; Luan, J.J. Reactive oxygen species mediated NF-κB/p38 feedback loop implicated in proliferation inhibition of HFLS-RA cells induced by 1,7-dihydroxy-3,4-dimethoxyxanthone. Biomed. Pharmacother. 2017, 94, 1002–1009. [Google Scholar] [CrossRef]
- Li, Y.; Dai, Y.; Liu, M.; Pan, R.; Luo, Y.; Xia, Y.; Xia, X.J.D.D.R. Scopoletin induces apoptosis of fibroblast-like synoviocytes from adjuvant arthritis rats by a mitochondrial-dependent pathway. Drug Dev. Res. 2009, 70, 378–385. [Google Scholar] [CrossRef]
- Chen, H.; Pan, J.; Wang, J.D.; Liao, Q.M.; Xia, X.R. Suberoylanilide Hydroxamic Acid, an Inhibitor of Histone Deacetylase, Induces Apoptosis in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Inflammation 2016, 39, 39–46. [Google Scholar] [CrossRef]
- Huang, M.; Zeng, S.; Qiu, Q.; Xiao, Y.; Shi, M.; Zou, Y.; Yang, X.; Xu, H.; Liang, L. Niclosamide induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes. Int. Immunopharmacol. 2016, 31, 45–49. [Google Scholar] [CrossRef]
- Yan, C.; Kong, D.; Ge, D.; Zhang, Y.; Zhang, X.; Su, C.; Cao, X. Mitomycin C induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes via a mitochondrial-mediated pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 35, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Li, J.; Dong, X.; He, X.; Niu, X.; Liu, C.; Zhong, G.; Bauer, R.; Yang, D.; Lu, A. Anti-oxidative and TNF-α suppressive activities of puerarin derivative (4AC) in RAW264.7 cells and collagen-induced arthritic rats. Eur. J. Pharmacol. 2011, 666, 242–250. [Google Scholar] [CrossRef] [PubMed]
- He, Y.H.; Zhou, J.; Wang, Y.S.; Xiao, C.; Tong, Y.; Tang, J.C.O.; Chan, A.S.C.; Lu, A.P. Anti-inflammatory and anti-oxidative effects of cherries on Freund’s adjuvant-induced arthritis in rats. Scand. J. Rheumatol. 2006, 35, 356–358. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracts/Monomers/Chemicals | Cells | Tissues/Animal Model | Dose/Concentration | Results | Potential Pathways | References |
|---|---|---|---|---|---|---|
| Hypericin photodynamic therapy | MH7A | / | 0.25–4 μM | Induced apoptosis; increased intracellular ROS; cleaved caspase 9, increased PARP; decreased NF-κB. | Mitochondrial apoptosis pathway, the death receptor pathway, NF-κB pathways | [196] |
| Hempseed oil | MH7A | / | 1–2% | Promoted apoptosis; Increased intracellular ROS; up-regulating CHOP, GRP94 and GRP78; activated PARP. | ER stress-mediated apoptosis | [197] |
| Brazilin | RAFLS | / | 10, 25 μg/mL | Increased autophagosome, LC3-II, intracellular ROS; inhibited NF-κB, reduced IL-6, IL-8. | Autophagic, NF-κB pathways | [72] |
| Shikonin | RAFLS | AA | 2, 2.5, 3 μM; 1, 2 mg/kg/day, i g | Increased ROS, LC3-II/LC3-I, apoptosis; decreased ATP, TNF-α, IL-6, IL-1β, IL-8, IL-17A, and IL-10; upregulating Bax, caspase 3; downregulating Bcl-2. | Mitochondrial apoptosis pathway and PI3K-AKT-mTOR pathways | [198] |
| Icariin | RAFLS | / | 0.1, 0.5, 1, 2.5, 5 μM | Inhibited migration, proliferation; induced G2/M phase arrest, apoptosis; increased Bax, activated- caspase 3, cleaved-PARP, Cyt-C, ROS; decreased Bcl-2, p p65, IκBα; MCMP (Δψm). | G2/M phase arrest; mitochondrial apoptosis pathway and NF-κB pathway | [199] |
| Oridonin | RAFLS | / | 5, 10, 25, 40 μM | Triggered cell apoptosis; increased caspase 3, caspase 9, PARP, Cyt-C, ROS; inhibited p ERK1/2, p JNK1/2, MCMP (Δψm). | Mitochondrial apoptosis pathway | [200] |
| α-Mangostin | MH7A/RAFLS | / | 10–100 μM | Promoted apoptosis; increased Cyt-C, ROS, caspase 3, caspase 9, p ERK1/2. Decreased MCMP (Δψm). | Mitochondrial apoptosis pathway, ERK1/2 signaling pathway | [201,202] |
| Apigenin | RAFLS | / | 100 μM | Induced apoptotic pathway; activated MAPK, ERK1/2, caspase 3, caspase 7; increased intracellular ROS. | ERK1/2 signaling pathway, apoptosis pathway | [203] |
| Eupatorium japonicum Thunb | RAFLS | / | 37.5 μg/mL | Induced apoptosis, ATF4, CHOP; decreased NF-κB, p38, IL-1β, MMP-9. | ER stress-mediated apoptosis, NF-κB pathway | [204] |
| Cryptotanshinone | MH7A/RAFLS | / | 5 μM | Increased ROS; downregulated Bcl 2, p Akt p STAT3; upregulated Bad, caspase 3, PARP, p p38, p c Jun N-terminal kinase. | Akt, MAPK, STAT3 pathways, mitochondrial apoptosis pathway | [205] |
| β-Elemene | RAFLS | / | 10–200 μg/mL | Promoted apoptosis; decreased MCMP (Δψm); increased Cyt-C, ROS, caspase 9, caspase 3, p p38 MAPK. | Mitochondrial apoptosis pathway, MAPK pathway | [206] |
| 1,7-Dihydroxy-3,4-dimethoxyxanthone | RAFLS | / | 8.7, 17.4, 34.7 μM | Upregulated GADD45α, p-p38; increased apoptosis, ROS; inhibited NF-κB. | NF-κB/p38 pathway, apoptosis pathway | [207] |
| Scopoletin | rFLS (AIA) | / | 250, 500, 1000 µM | Upregulated Bax, caspase 3; decreased MCMP (Δψm), Bcl-2, NF-κB. | Mitochondrial apoptosis pathway, NF-κB pathway | [208] |
| Resveratrol | RAFLS | / | 40, 80, 160, 320 μM | Downregulated Bcl-2, Atg5, LC3B; increased ROS; released Ca2+. | Mitochondrial apoptosis pathway and autophagy | [73] |
| Sulforaphane | RA-T cell | / | 0.5, 1, 2.5, 5, 10 μM | Inhibited CD25/CD69, proliferation; increased intracellular ROS, decreased GSH, p STAT3, RORγt, IL-17A, IL-17F, IL-22. | STAT3 signaling | [169] |
| Extracts/Monomers/Chemicals | Cells | Tissues/Animal Model | Dose/Concentration | Results | Potential Pathways | References |
|---|---|---|---|---|---|---|
| Suberoylanilide hydroxamic acid | RAFLS | / | 0.5–10 μM | Induced apoptotic pathway; increased intracellular ROS, total IκBα; decreased p IκBα, NF-κB p65, Bcl-xL, Mcl-1. | Apoptosis pathway, NF-κB pathways | [209] |
| Niclosamide | RAFLS | / | 0.25, 0.5, 1 μM | Induced apoptotic pathway; increased intracellular ROS, Bax, Cyt-C, caspase 9, caspase 3; Decreased p Akt, Bcl-2. | Mitochondrial apoptosis pathway, Akt pathways | [210] |
| Mitomycin C | RAFLS | / | 10, 25, 50, 100 μg/mL | Induced apoptosis; increased intracellular ROS, Cyt-C, Bax/Bcl-2, caspase 9, caspase 3, PARP; decreased MCMP (Δψm). | Mitochondrial apoptosis pathway | [211] |
| Menadione | CD4 naive (CD4+CD45RO−) T cell | Human synovial tissue-NSG chimaera | 3 μM; 10 mg/kg/day, i g | Increased ROS, p ATM, T-bet, RORγ; decreased IFN-γ, IL-17, TNF-α, IL-1β, IL-6, RANKL; inhibited spontaneous hypermobility. | ATM signaling | [161] |
| Buthionine sulfoximine | / | Human synovial tissue-NSG chimaera | 1000 mg/kg/day, i g | Increased intracellular ROS; decreased IFN-γ, IL-17, TNF-α, IL-1β, IL-6, RANKL, GSH, T-bet, RORγ; inhibited spontaneous hypermobility | ATM signaling | [161] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Fan, D.; Cao, X.; Ye, Q.; Wang, Q.; Zhang, M.; Xiao, C. The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment. Antioxidants 2022, 11, 1153. https://doi.org/10.3390/antiox11061153
Wang X, Fan D, Cao X, Ye Q, Wang Q, Zhang M, Xiao C. The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment. Antioxidants. 2022; 11(6):1153. https://doi.org/10.3390/antiox11061153
Chicago/Turabian StyleWang, Xing, Danping Fan, Xiaoxue Cao, Qinbin Ye, Qiong Wang, Mengxiao Zhang, and Cheng Xiao. 2022. "The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment" Antioxidants 11, no. 6: 1153. https://doi.org/10.3390/antiox11061153
APA StyleWang, X., Fan, D., Cao, X., Ye, Q., Wang, Q., Zhang, M., & Xiao, C. (2022). The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment. Antioxidants, 11(6), 1153. https://doi.org/10.3390/antiox11061153