Abstract
Skin aging is one of the most evident signs of human aging. Modification of the skin during the life span is characterized by fine lines and wrinkling, loss of elasticity and volume, laxity, rough-textured appearance, and pallor. In contrast, photoaged skin is associated with uneven pigmentation (age spot) and is markedly wrinkled. At the cellular and molecular level, it consists of multiple interconnected processes based on biochemical reactions, genetic programs, and occurrence of external stimulation. The principal cellular perturbation in the skin driving senescence is the alteration of oxidative balance. In chronological aging, reactive oxygen species (ROS) are produced mainly through cellular oxidative metabolism during adenosine triphosphate (ATP) generation from glucose and mitochondrial dysfunction, whereas in extrinsic aging, loss of redox equilibrium is caused by environmental factors, such as ultraviolet radiation, pollution, cigarette smoking, and inadequate nutrition. During the aging process, oxidative stress is attributed to both augmented ROS production and reduced levels of enzymatic and non-enzymatic protectors. Apart from the evident appearance of structural change, throughout aging, the skin gradually loses its natural functional characteristics and regenerative potential. With aging, the skin immune system also undergoes functional senescence manifested as a reduced ability to counteract infections and augmented frequency of autoimmune and neoplastic diseases. This review proposes an update on the role of oxidative stress in the appearance of the clinical manifestation of skin aging, as well as of the molecular mechanisms that underline this natural phenomenon sometimes accelerated by external factors.
Keywords:
skin; photoprotection; epidermis; dermis; inflammation; UV; melanocyte; skin cancer; aging 1. Introduction
1.1. Human Skin Tissue Structure and Biology
The skin is the largest organ of the human body and is composed of three compartments: the epidermis, the dermis, and the deeper subcutaneous fat tissue (also referred to as hypodermis) (Figure 1).
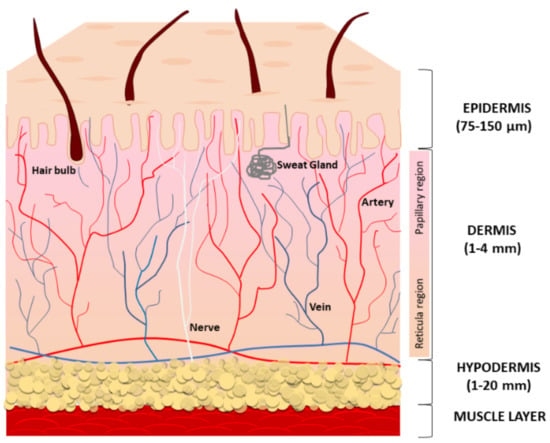
Figure 1.
A schematic representation of the structure and functions of the skin. Human skin is composed of three layers: the epidermis (the top layer), the dermis (the middle layer), and the hypodermis (the bottom fatty layer).
The main function of the epidermis is to provide a protective barrier against microbes, environmental pollution, and ultraviolet (UV) radiation. The epidermis is avascular and mostly composed of multiple layers of keratinocytes at different differentiation levels. Immune cells infiltrating the epidermis, especially Langerhans cells, counteract infections. Melanocytes located at the basal layer of the epidermis produce and distribute the melanin pigment to surrounding keratinocytes, ensuring the absorption of a broad spectrum of solar irradiation wavelengths and consequent protection from UV radiation [1], whereas Merkel cells serve as a touch-perception receptor [2]. The multilayered stratum corneum (the outermost layer) consists of anucleated, densely keratinized keratinocytes and lipid-laden extracellular matrix (ECM), to prevent excess trans-epidermal water loss. Lipids consist of neutral molecules such as sterols, free fatty acids, triglycerides, highly nonpolar species, and sphingolipids [3]. Keratinocyte precursors proliferate symmetrically and asymmetrically. The asymmetric division generates two daughter cells with non-identical fates; one daughter remains a progenitor while the other commits to cell terminal differentiation. During the differentiation, keratinocytes slowly migrate towards the surface, finally becoming fully differentiated corneocyte cells that are continuously lost from the skin surface. Like other epithelial structures with an intense cell turnover, normal epidermal homeostasis firmly depends on staminal precursors, and keratinocyte aging is due to changes in stem cell numbers and functions [4]. Desquamation rates of the stratum corneum and the resultant renewal of the keratinocyte layer have been calculated at 40–56 days in middle-aged humans [5]. Thus, it has been suggested that the impact of a senescent phenotype is limited in keratinocytes. Accordingly, in the epidermis from the sun-protected areas of young and aged donors, p16INK4a-positive cells are mainly melanocytes and less frequently keratinocytes [6]. In the epidermis, melanocytes are highly differentiated cells rarely replaced during normal adult skin homeostasis and their turnover occurs only by stimulation as in wounding and exposure to UV [7]. Melanocyte precursors reside in the hair bulge and in the epidermis to pigment the hair and skin [8,9,10]. Additionally, some melanocyte precursors have been demonstrated in the dermis [11,12].
The dermis, derived from mesoderm, underlies the epidermis and includes sebaceous glands, hair follicles, nerve endings, blood, and lymphatic vessels embedded within connective tissue, a fiber-dense ECM due to the intense secretory activity of sparse fibroblasts. The dermal compartment also includes a wide repertoire of immune cells, including macrophages, dendritic cells, natural killer cells, lymphocytes, and mast cells [13]. Collagen fibers confer resistance to the tissue and elastin keeps skin flexible. In addition to structural proteins, the relative abundance and the distribution of adhesion proteins (fibronectin, lamina, fibrillin, tenascin), glycosaminoglycans (hyaluronan, heparin-sulfate, chondroitin-sulfate), and proteoglycans (versican, aggrecan, neurocan) define the biological properties of the dermis. This includes the retention and consequent availability of soluble factors, particularly growth factors and second small messengers [14,15,16]. Further, the physical properties of ECM may affect adult mesenchymal stem cell (MSC) proliferation and differentiation potential [17,18]. The specific architecture of ECM might contribute to MSCs’ fate via direct physical interaction with these cells. Moreover, the loss of stem cell properties that coincides with the spontaneous differentiation may be due to the response of stem cells to growth factors, which in turn are influenced by the microenvironment [19]. Histologically, many age-dependent changes affect the structural components of the connective tissue. Low biosynthesis, increased degradation, and the accumulation of unfunctional disorganized collagen and elastin fibers impair the tissue integrity during intrinsic aging, whereas exacerbated expression of collagen and elastin proteolytic enzymes in sun-exposed areas leads to the accumulation of partially fragmented elastin fibers, causing the typical solar elastosis of extrinsic aging [20]. The hypodermis, the bottom layer of cutaneous tissue, regulates body temperature and makes protection for blood vessels, nerves, muscles, and bones. It is a well-vascularized connective tissue prevalently composed of adipose tissue that forms a layer of variable thickness depending on its location in the body and scarce collagen fibers. Adipose tissue functions in thermal insulation and energy storage, whereas mesenchymal stem cells are key players in wound healing and re-epithelialization [21]. The depletion of stem cell reserves can compromise the ability to restore spontaneous tissue repair. Wound healing slows with age, thus older adults frequently have chronic wounds, with a significant impact on the quality of life of patients and their families [22]. Abnormal repeated requirements of tissue repair, such as in the case of disease-specific tissue dysfunction or chronic oxidative stress, might lead to premature consumption of the stem cell reservoir and consequent gain of senescent cells. The intense secretory activity of subcutaneous adipose tissue includes repair-inducing activators of fibroblasts and stem cells during wounds and very important signals to modulate hair follicle physiology, while bacteria-sensing adipocytes produce antimicrobial peptides, supporting innate immune responses in the skin [21,23]. Adipose-derived stem cells play a key role in protecting skin from oxidative damage and inflammation by the secretion of bioactive molecules and antioxidant factors [24,25].
1.2. Skin Antioxidant Defense System
To scavenge reactive oxygen species (ROS), cutaneous cells utilize a conspicuous apparatus of small antioxidant molecules and endogenous enzymes. Ubiquinol (coenzyme Q10) is a lipid-soluble intracellular and extracellular radical scavenger that protects mitochondria and key cutaneous proteins. CoQ10 also inhibits the expression of some metalloproteinases (MMPs), such as collagenase, preserving the collagen content of the skin [26]. Vitamin E is implicated in membrane stabilization, preventing lipid peroxidation and oxidation of unsaturated fatty acids [27,28]. In the skin, vitamin E level is strongly sensitive to UV-induced depletion [29], and levels of vitamin E also decrease with age [30], suggesting that impairment in its detoxification activity might be involved in both natural and photo-accelerated aging. Vitamin C acts by removing free radicals and repairing oxidized vitamin E [31]. Moreover, in the skin, vitamin C is implicated in procollagen synthesis and collagen cross-linking [32,33,34]. The function of superoxide dismutase (SOD) is to catalyze the breakdown of superoxide radical anion (O2) into hydrogen peroxide (H2O2) [35]. In mammals, three different isoforms of SOD exert non-overlapping functions. The isoform that utilizes Cu/Zn as cofactors (SOD1) localizes in the cytoplasm and the nucleus [36], the isoform that binds Mn (SOD2) localizes in mitochondria, and SOD3, which also binds Cu/Zn, has been detected mainly in the extracellular space [37]. In vivo studies in mice evidenced that all three SODs impact skin aging [38,39,40]. In animal models, SOD2 deletion corresponds to the more dramatic phenotypes with the thinner epidermis, atrophy of the dermal connective tissue, reduced complexity of the extracellular fiber network, and a smaller amount of the subcutaneous fat tissue, all of which have been described as major characteristics [39,41]. Interestingly, Velarde et al. demonstrated an age-dependent effect of SOD2 depletion in mouse skin. In old mice, SOD2 deficiency delayed wound closure and reduced epidermal thickness due to exhaustion of premature epidermal stem cells, whereas in young animals SOD2 deficiency stimulated wound closure, sustaining epidermal differentiation, despite the induction of cellular senescence in keratinocytes [42]. In human senescent skin, fibroblasts that develop a growth arrest, and morphological and functional changes, demonstrated an adaptive upregulation of the SOD2 at mRNA and protein levels due to increased ROS concentrations [43,44]. An important enzyme that detoxifies hydrogen peroxide (H2O2) is the peroxisomal localized catalase (Cat) [45]. In the aged human dermis, Cat activity is lower with consequent elevation of H2O2 concentration [43]. By contrast, a parallel increase in ROS and Cat activity has been observed in the epidermis [46]. Another supporter of the antioxidant capability of the cell is the tripeptide glutathione (GSH). The GSH acts as a scavenger because of its thiol functional group. During the reaction, GSH is oxidized by reactive oxygen radicals and makes a dimer with another GSH (GSSG). GSH can be retaken in a reducing enzymatic reaction by the glutathione reductase consuming NADPH [47]. The mouse model demonstrated that not only the absolute amount of the oxidized GSSG but also the GSSG:GSH increases in the dermis during aging [48]. In addition to its role as an antioxidant, GSH is also a cofactor for many metabolic processes. In humans, all eight glutathione peroxidases (GPXs) are known to reduce hydrogen peroxide in water and stop lipid peroxidation [45]. Overall, the endogenous antioxidant capacity (enzymatic and non-enzymatic) of the skin is lowered with age, and the aged skin is more vulnerable to external factors, especially UV radiation, pollution, and microorganisms [49]. Since the epidermis is more exposed to external stimuli than the dermis, the ROS load is higher in the epidermis compared to the dermis [50]. Correspondingly, defensive enzymes and non-enzymatic antioxidants are present in higher concentrations in the epidermis than in the dermis [51]. Particularly, small antioxidants such as vitamins C and E, glutathione, and ubiquinol, and defensive enzymes such as Cat and SODs are concentrated in deeper layers of the stratum corneum [29,52,53]. From the biological point of view, this might correspond to more accurate protection of epidermal stem cells that mostly reside at the dermal–epidermal junction. On the other hand, the production of ROS in the epidermis occurs in the deepest layers, especially at the basal layer, since in the final phase of the differentiation process, keratinocytes of the stratum corneum lose their nuclei and organelles [54]. Moreover, the promelanogenic effect of solar radiation promotes the formation of free radicals related to the melanin biosynthetic pathway at the dermal–epidermal junction [55]. Particularly, in fair-skinned individuals, pheomelanin is responsible for free radical generation in melanocytes even in absence of UV [56,57]. Likewise, carriers of melanocortin 1 receptor (MC1R) variants presenting a shift in melanin synthesis from eumelanin to pheomelanin and consequent elevation of reactive oxygen species are at increased melanoma risk, independent of their sun exposure [58].
1.3. A Brief Introduction to Skin Aging
Skin aging is a multifactorial biological process macroscopically manifested by modification of its appearance due to the progressive decline of physiological functionality (Figure 2).
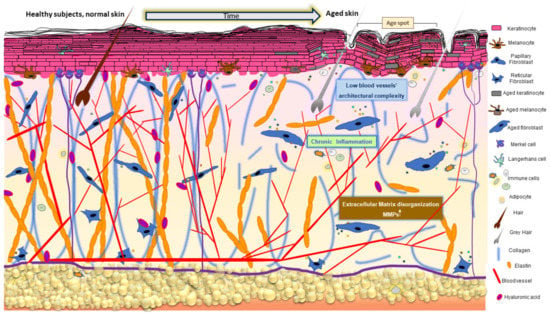
Figure 2.
Age-related changes in the skin. Skin aging results in cumulative detrimental effects characterized by abnormal ECM organization, pigmentary changes, loss of subcutaneous fat, hair greying, minored hair density, decreased sebaceous gland function, and low-grade chronic inflammation. Cellular and molecular events reviewed in the text describe the impact of oxidative disequilibrium on these time-dependent and/or extrinsically accelerated tissue transformations.
Fine wrinkles, tissue atrophy with minor elasticity, and remarkable dryness often accompanied by pruritus are the most common phenotypic changes in aging observed in all skin areas [59]. However, they diversify among different anatomical regions and within diverse ethnical groups [60,61]. The subcutaneous adipose tissue is decreased in some body areas, especially the face, shins, hands, and feet, explaining the visible volume reduction, while in other body areas, peculiarly the abdomen in males and the thighs in females, it is augmented. Anatomical differences emerged by the comparative analysis of facial and abdominal adipocyte gene expression profiles, suggesting a possible implication in the diverse modification of subcutaneous tissue of these body areas during aging [62]. Sebaceous glands progressively increase in size, but their secretory output is attenuated in aged individuals [63]. There is a progressive decline in the density of hair follicles, and the hair shaft diameter is frequently smaller [64]. At the cellular level, aging is characterized by the accumulation of senescent cells in both the epidermis and the dermis and by a significant depletion of stem/progenitor cells [65]. Since MSCs do not escape the deleterious effects of natural aging, their propensity to senesce is firstly determined by intrinsic factors [66]. Aging affects MSCs from a quantitative (stem cells exhaustion) and a qualitative point of view, since advanced age subcutaneous MSCs lose their osteogenic potential and in turn augment the adipogenic potential [67]. In line with this idea, Orciani and collaborators demonstrated that MSCs isolated from the skin do not have an efficient antioxidant defense system, but their integrity is preserved by the surrounding microenvironment of the niche [68]. Increased intracellular ROS and lower SOD activity characterizes aged MSCs in both undifferentiated and differentiated conditions. Diabetic subcutaneous MSCs displayed lower proliferation activity, upregulation of pluripotent staminal markers, and a propensity for neurogenic differentiation. At the same time, normal MSCs cultured in a hyperglycemic milieu showed an ROS-dependent similar phenotype characterized by low proliferation and migration, senescent-prone phenotype, and a relatively immature state with an inclination to neuron-like differentiation [69]. This study demonstrated that metabolic dysfunction, frequently arising in the elderly, might impact skin aging and its reparative potential.
Replicative senescence is principally the result of repeated cell division that induces gradual shortening of telomeres [70]. Although cellular replication is a major contributor to telomere dysfunction, it has been largely documented that telomere attrition is accelerated when cells are exposed to mild, eventually chronic stress, leading to reduced replicative capacity and a phenotype similar to replicative senescence [71,72,73]. The physiological elimination of senescent cells is mainly modulated by the immune system, but the mechanisms involved are not yet completely elucidated. In addition, tissue degeneration may help senescent cells to escape from immune clearance [74]. In the complicated context of organismal aging, it is not known to what extent continuous proliferation contributes to regulating the number of senescent cells, and whether age-related impairment of the immune system function contributes to the accumulation of senescent cells in old individuals [75]. Furthermore, senescence in fibroblasts matches with resistance to apoptosis caused by UV radiation [76], a characteristic that could influence the persistence of these cells. In a restricted number of clinical trials, pharmacological treatments, termed “senolytics”, have been tested to remove senescent cells from the body [77,78,79]. The prevalent mechanism of action of senolytic therapies involves the induction of apoptosis in senescent cells or the stimulation of immune cells to clear senescent cells. Pharmacological clearance of senescent cells markedly enhances health span [80,81,82]. Due to the discovery of practicable “therapeutic” intervention, it has been proposed to consider aging as a real “disease” [83]. Cell-specific functional diversity impacts senescence differently [84,85]. Though one might expect cells with a rapid turnover to be senescence prone, this does not seem to be the case for keratinocytes, since keratinocytes, damaged or not, are physiologically quickly replaced by new differentiating cells. Instead, fibroblasts could progress through the senescence program and accumulate functional defects, impacting tissue integrity due to their limited proliferation rate. The fact that the senescence-associated phenotype is mostly referred to as long-living post-mitotic cells underlies that elements other than those associated with replication play a relevant role in the acquisition of the senescent phenotype [86]. A plethora of stresses can provoke premature cellular senescence, including UV radiation, mitochondria dysfunction, oxidative stress, DNA damage, epigenetic alteration, and expression of some oncogenes [87,88]. Thus far, cellular senescence is not only a time-defined process: each cell experiences senescence conforming to its proliferation speed and its history. Therefore, in real life, a tissue that underwent exclusively intrinsic aging does not exist. Since skin is the interface between the body and the external environment, it is constantly or intermittently in contact with extrinsic stimulation (e.g., ultraviolet light exposure, pollution, smoking, chemotherapy, radiotherapy, cosmetics, microbial insults, trauma) in addition to intrinsic factors (e.g., time, genetic factors, hormones, comorbidities) that can impact the senescence of its cells and on the overall decline of tissue function. Thus, it is considered the organ of choice for aging studies [89,90,91]. Moreover, due to the skin’s accessibility, it is a useful model to test translational approaches in the regenerative medicine field [92,93]. The neologism of “exposome” has been recently introduced to explain the complex exposures we face throughout our lives, and encompasses air pollution, climate factors, infections, the food we ingest, the objects we touch, and the psychological stresses [94]. Exposomes exacerbate tissue damage, accelerating the aging process [95]. UV radiation is the most potent extrinsic driver of age-related change in the skin, known as “photoaging”. Photoaging accounts for approximately 80% of facial aging [96]. Except for pigmentation, which presents the opposite feature in chronological aging (hypopigmentation) [85,97,98] and in photoaging (hyperpigmentation) [99,100], intrinsic and extrinsic skin senescence demonstrated several types of overlapping pathogenic molecular signaling. The common feature of both types of cutaneous aging is the generation of ROS, impacting DNA, protein, and lipid damage and the disorganization of the ECM [101]. During naturally occurring aging, collagen and elastic fibers are partially degraded but form a wider-mashed network. During extrinsic aging, the dermis strongly loses collagen type I (Col-I), III, and VII expression [102]. Additionally, the migration of neutrophils after inflammation or UV exposure strongly accelerates collagen and elastin degradation due to their intense production of MMPs and elastases [103]. In mice, a comparative analysis of the gene expression profile of young and old animals revealed that most of the differentially regulated genes encompassed those induced by oxidative damage and associated with energy metabolism, mitochondrial function, and turnover [104]. Furthermore, the relevance of redox equilibrium in the aging process is supported by the proof that human skin fibroblasts from progeria patients, a premature aging syndrome, show a significant reduction in SOD2, Cat, and GPX expression and activity at the basal level. Moreover, these cells also demonstrated impaired scavenger activity under chronic oxidative stress conditions [105].
At the low level, ROS are the first line of defense and are involved in various physiological functions; however, an excessive amount of ROS or an insufficient endogenous defense system can compromise intracellular redox homeostasis. A large amount of ROS activates mitogen-activated protein kinases (MAPKs) and critical transcription factors such as nuclear factor-κB (NF-κB), nuclear factor erythroid 2-like (Nrf2), and c-Jun-N-terminal kinase (JNK) and transcription factor activator protein 1 (AP-1) [106]. Nrf2 is one of the most important transcription factors in the cellular response to oxidative stress. Nrf2 cytoprotective action concerns mainly antioxidant enzymes such as glutathione S-transferase (GST), heme oxygenase-1 (HO-1), quinone reductase NAD(P)H (NQO1), Cat, SODs, UDP-glucuronosyltransferases (UGT), epoxide hydrolase (EPHX), γ-glutamylcysteine ligase (GCL), glutathione reductase (GR), and thioredoxin reductase (TrxR) under normal and critical circumstances [107]. For this reason, natural Nrf2 modulators received notable attention in dermatology [108]. AP-1 activation elevates the expression of MMP1, 3, and 9 in fibroblasts and keratinocytes [109,110]. AP-1 and NF-κB inhibit TGF-β, which is responsible for Col-I and connective tissue growth factor (CTGF) production [111,112]. ROS-triggered activation of NF-κB also drives an elevation of proinflammatory cytokines (IL1, IL6, and TNFα) [113], provoking localized phenotypic changes that are independent of the systemic immune system function [114,115]. Stress-induced senescence is associated with a marked pro-inflammatory secretory profile of dermal and epidermal cells [116]. Reduced ability to manage persistent inflammation contributes to the occurrence of skin inflammaging, immunosuppression, and skin cancers [117,118]. Since mitochondria are the main source of intracellular free radical production [119], dysfunctional mitochondria contribute to the aging process [120]. At the molecular level, a typical marker of aging is the low expression level of mitochondrial electron transport chain proteins [121]. A disturbance of reductive overload of the mitochondrial respiratory chain leads to uncontrolled ROS accumulation [122]. Characteristic features of the aged dermis and epidermis are the presence of damaged mitochondria, frequent mitochondrial DNA (mtDNA) deletions, elevated ROS levels, and oxidative stress [123]. Skin aging and stem cell senescence are characterized by lower mitochondrial complex I–IV activity [124]. A 4977-base-pair extended region of mtDNA, coding for genes of complexes I, IV, and V respiratory chain, is frequently deleted in the aged human skin [125]. This ″common deletion″ positively correlates with sun exposure and skin wrinkles [126]. Damaged mitochondria are selectively sequestered in double-membrane vesicles and cleared away by lysosome-dependent mitophagy (selective autophagy of mitochondria). Mitophagy is utilized under cellular stress conditions to preserve healthy mitochondria or to regulate homeostasis when an excess of mitochondria is present [127]. Older skin had a significantly fragmented mitochondrial network, indicating poor recycling and excessive mitophagy [128]. Signals of mitochondrial dysfunction are sensed by the mammalian target of rapamycin (mTOR) or AMP-dependent protein kinase (AMPK) and calmodulin [129,130]. Conditions that activate mTOR (deregulated autophagy, oxidative stress, or systemic inflammation) when the cell cycle is blocked lead the cell to a hypertrophic senescence state [131]. Further, ROS activates the PI3K/mTOR/S6K axis, leading to an increase in cell volume and protein content [132]. Thus, ROS can be linked to cell senescence, not only through damaged macromolecules but also through mTOR. Notably, the activation of mTOR, which promotes cell growth even though the cell cycle is blocked downstream, causes hypermitogenic senescence. Increased cellular function (hypertrophy, pro-inflammatory and hypersecretory phenotypes) in a context of reduced or absent proliferation resembles senescence caused by DNA damage, and it is considered a distinctive marker of hypermitogenic arrest [133]. Hypertrophy, hyperplasia, and hypermitogenic phenotype [134,135] by themselves may cause changes in skin appearance. In the mouse model, overexpression of the insulin-like growth factor II gene (IGF-2) in keratinocytes causes overgrowth of the skin, as denoted by wrinkling [136].
2. The Role of Oxidative Stress in Chronological Senescence of the Skin
In human skin, consistent with all other organs, physiological aging is the natural consequence of the passage of time. Skin that ages only by intrinsic factors virtually does not exist. Thus, it is generally considered that skin that exclusively undergoes intrinsic aging is usually present in body areas habitually unexposed to sunlight. Naturally, aged skin presents fine wrinkles, dryness, thinning, and augmented temperature sensitivity. During skin aging, sebocytes decrease the size and secretory activity with a significant repercussion in the surface lipid amount and skin hydration [137]. In menopausal women, a significantly higher pH of the hydro-lipid film surface and a decrease in sebum production have been observed, whereas measurements of trans-epidermal water loss showed a minimal variation in stratum corneum hydration [138]. Chronic itching is likewise common in the elderly due to dryness. However, it may be caused by the age-related alteration of touch to itch sensation due to dysfunctional Merkel cells [139]. During aging, keratinocytes acquire a typical shorter and fatter morphology, while corneocytes become bigger as a result of lower epidermal turnover [140]. In old skin, the stratum corneum is not replaced as quickly, so the skin appears rough and dry. Extreme skin dryness (xerosis) is more susceptible to irritant dermatitis [141]. As mitosis of keratinocyte precursors in the basal layer of the epidermis is slowed down, healing requires a longer time. Moreover, there is a reduction in the surface contact between the epidermis and dermis, which aggravates the delivery of oxygen and the nutrients in the epidermis [141]. The aging-associated anatomical change includes the decrease in the number and size of blood vessels, and low architectural complexity that explains not only the impaired nutrition supplementation but also the removal of metabolic debris and toxins [142]. Vascular permeability changes, especially in the superficial dermis, produce adaptative remodeling of the epidermis such as the decrease in several cell layers, thus reducing the thickness [143,144]. At the tegumentary level, dermis fibers undergo fragmentation processes and finally lysis. Elastic fiber degeneration is faster than that of collagen fibers [144]. The age-related decrease in sweat glands might be responsible for impaired metabolic waste removal and consequent accumulation of toxins on the skin [145]. With aging, hair becomes thinner on the scalp and terminal hair follicles are progressively miniaturized [146]. As can be easily observed, the natural hair color is lost due to a reduced transfer of melanin from follicular melanocytes to hair keratinocytes [146]. Intrinsic aging is mainly regulated by genetic factors affecting the entire body. Most of the genes associated with a younger appearance are directly or indirectly linked with protective factors such as DNA repair, response to oxidative stress, cell replication, protein metabolism, or ECM architecture [147]. Single nucleotide polymorphisms (SNPs) for genes responding to NAD(P)H dehydrogenase (NADPH), SOD2, SOD3, Nrf2, Cat, and GPX1 have been correlated to skin aging [147,148]. Since the activation of α-melanocytes hormone (α-MSH)-dependent intracellular signaling not only regulates melanogenesis but also cellular defense mechanisms in the dermis and epidermis, individuals carrying loss-of-function polymorphic variants of its receptor, the MC1R, present a reduced capacity to counteract oxidative stress and DNA damage [149,150,151,152,153]. Correspondingly, loss-of-signaling MC1R polymorphisms are linked to an increased risk of melanoma and other skin cancers [154,155,156]. Further, three different MC1R common polymorphisms, namely R151C, R142H, and D84E, demonstrated a significant correlation with photoaging [157]. In dermal fibroblasts, α-MSH modulates collagen metabolism by improving the orientation of the collagen fibers [158,159,160]. α-MSH may drive the healing into a more regenerative/less scarring pathway by counteraction the pro-fibrotic action of TGFβ [160,161]. Recently, a higher prevalence of coarse collagen, pixel, and vessel density in photo-exposed areas has been associated with MC1R polymorphic variants [162]. Apart from the genetic difference, in the epidermis, the production of proopiomelanocortin (POMC), the precursor of α-MSH, increases with age, whereas its receptor decreases, indicating that α-MSH-dependent intracellular signaling is deeply involved in the skin aging process [163]. Other genes involved in skin color regulation, including heterogeneous nuclear ribonucleoprotein (hnRNP), agouti signaling protein (RALY/ASIP), basonuclin 2 (BNC2), and interferon regulatory factor 4 (IRF4) have been associated with pigment spot and generally the aged appearance of the skin through pathways independent of the pigment production [164]. Regarding the dermis, SNPs of collagen type 1 alpha-2 gene (COL1A2), COL17A1, MMP3, MMP9, and MMP16 are linked with features of aging [147,148,165,166]. Further, genetic variants of TNF receptor superfamily member 6b (TNFRSF6B), TNFRSF8, IL6, and NOS1, involved in inflammation, have been correlated with wrinkle risk [147,148,166]. Altogether, genetic data confirm the involvement of oxidative stress and inflammation in aging propensity. Epigenetic changes also participate in the regulation of the homeostasis and regeneration of aged skin [167]. Skin samples from the elderly display increased heterogeneity of global methylation patterns that are characterized by reduced connectivity of gene expression networks probably mediated by methylation-dependent changes in transcription factor binding [168]. On the opposite, studies restricted to the epidermis showed very similar methylation patterns in the epidermis of young and old individuals, demonstrating a limited destabilization of the epigenome of this tissue compartment during aging [169]. Slowing the cell cycle coincides with the lengthening epidermal turnover rate resulting in less effectiveness in wound healing and desquamation in older adults [170]. Physiological changes in skin color due to aging alone are minimal. With the aging of the non-sun-exposed skin, pigment is light and homogenously distributed. An inverse relationship between age and the proliferative activity of melanocytes has been observed independently of skin phototype [97,98,171,172]. Thus, reduced skin pigmentation and tanning response after UV exposure make aged skin more vulnerable to solar radiation [173,174]. When occurring, the white spots in aged skin are usually stellate pseudo scars or idiopathic guttate hypomelanosis [173]. Recently, Victorelli and collaborators proposed a central role of senescent melanocytes in skin aging priming because their secretory activity diminishes basal keratinocyte proliferation and guides the epidermis to atrophy in in vitro 3D human epidermal equivalents [175]. This seems to be due to paracrine CXCR3-dependent mitochondrial ROS activation, which in turn induces telomere dysfunction in neighboring cells. Epidemiologic studies further evidenced age-related changes in melanocytic nevi [176]. During life, there is a gradual lessening in the number of common and atypical nevi [173,177]. In the skin, cells positive for senescence markers p16INK4a and senescence-associated lysosomal beta-galactosidase (SA-βgal) physiologically accumulate with age in the epidermis and the dermis [178,179,180]. Senescent cells are in a non-proliferative metabolically active state and constantly produce several pro-inflammatory mediators, proteases, and mitogenic factors in a state known as the “senescence-associated secretory phenotype” (SASP) [181]. SASP spreads senescence in neighboring cells, tissues, and organs, as it functions in an autocrine and paracrine manner [182,183]. Studying human in vitro skin equivalent, Adamus and collaborators demonstrated that the age of the keratinocyte’s donor strongly impacts the model’s quality [184]. For these cells, non-univocal results have been reported in the literature regarding the expression of the proliferation marker Ki-67 in skin biopsies since inverse correlation with p16INK4a was observed by some authors [184,185], whereas others reported no change [179] or an increase [186]. From a functional point of view, biological aging manifests as a decreased physiological reserve that oxidizes DNA, enzymes, and proteins. Structural and functional dermal changes are the major contributor to the skin aging appearance [187,188]. The rate of proliferation and the total number of fibroblasts, the most abundant cell type of the dermis, decreases progressively with age, suggesting that lower fibroblast cellularity could be responsible for the occurrence of age-related features, particularly for the decreased collagen production that has been estimated at 75% lower in people aged ≥80 years [180,189,190]. Studies at a proteomic level highlighted age-dependent dermal fibroblast secretion patterns including inflammatory regulators, mitogens, angiogenic factors, and MMPs. MMPs degrade matrix components, causing less effective epidermal anchorage, skin relaxation, and decreased interstitial fluid [191,192]. More in detail, ‘‘skin-aging-associated secreted proteins”, or SAASP, in addition to the classical SASP, are enriched in molecules involved in elastic fiber formation, glycosphingolipids, and sphingolipid metabolism. Apart from ECM organization, SAASP has been associated with the regulation of insulin-like growth factor (IGF) transport and uptake by IGF binding proteins (IGFBPs), alteration in adherent junctions’ interactions (N-cadherin and Cadherin-11), glucose and carbohydrate metabolism, and capability of glutathione synthesis and recycling. Specifically, a lower level of expression of glutathione-s-transferase (GST) in middle-aged and old fibroblasts explains the of loss detoxification capability in intrinsically aged skin [193]. Naturally, aged dermal fibroblasts with reduced mechanical force downregulate TGFβRII, thus impairing the TGFβ/SMAD signal transduction pathway [194]. Reduced contractile force and migratory potential of old fibroblasts have been proposed as biomarkers of dermal aging processes [195,196]. Abnormal TGFβ/SMAD3 signaling in turn results in repression of CTGF-dependent type I collagen synthesis as well as stimulation of MMP1-induced type I collagen degradation, leading to dermal thinning [112,197]. Reciprocal regulation of TGFβ and ROS strongly impact normal and pathological connective tissue remodeling [198,199]. Reduced levels of tissue inhibitors of metalloproteinases (TIMPs) are a consequence of natural aging [200]. Interestingly, Salzer and collaborators demonstrated that aged upper (papillary) dermal fibroblasts progressively acquire the characteristics of the lower (reticular) dermis, with a minor expression of ECM proteins and gain of adipogenic markers [201]. Since it has been proposed that reticular fibroblasts are in a more advanced stage of differentiation than papillary fibroblasts, it seems plausible that the transition from the papillar to reticular state is related to the relentless terminal differentiation process of post-mitotic cells [202,203]. Another recent study proposed a specific feature of chronological age-related partial loss of dermal fibroblasts described as “loss of cellular identity” [204]. In detail, old papillary fibroblasts presented fewer papillary and more reticular gene expression profiles, while the reticular counterpart presented a less decided reticular gene expression profile [205]. This observation is of interest since dermal ECM produced by papillary fibroblasts better supports epidermal longevity compared to reticular-generated ECM [206]. A comparative analysis of matched papillary and reticular fibroblasts revealed that intrinsic aging differentially impacts these two cell populations. Colony growth at low culture density and growth rate in mass is strongly reduced in papillary fibroblasts compared to reticular fibroblasts [203]. This could be explained by the more pronounced exposition of superficial dermis to external stimuli and the consequent impact on the redox equilibrium of the papillary dermis. A recent work pointed out the attention to the effect of intrinsic age on the differentiation capacity of a restricted population of fibroblasts localized within the conjunctival junctions that connect the dermis to the hypodermis, i.e., dermo–hypodermal junction fibroblasts [207]. These cells displayed distinct age-related features when compared to papillary and reticular fibroblasts, presenting attenuated osteogenic differentiation potential, no adipocyte differentiation capacity, and unmodified chondrocyte differentiation capacity [207]. Intrinsic aging also reduces subcutaneous fat with accompanying increased vessel disorganization, loss of cellularity, and vascularity [208]. The lack of microvascular organization in the skin has retained the cause of age-related deficits in the diffusive transport capacity of the skin vasculature [142]. During chronological aging, the body undergoes a continuous increase in systemic low-grade inflammation, a process known as ″inflammaging″ [209,210]. Such chronic aging-related inflammatory response is marked by an increase in systemic levels of IL1, IL6, and IL33, as well as TNF, IFNγ, and GM-CSF [211,212]. The relationship between the senescence of skin-resident immune cells and inflammaging has not been fully understood yet. In aged skin, decreased self-renewal capacity of in situ Langerhans cell progenitors causes a reduced number of their mature counterparts. Additionally, the lower migration propensity of these cells in geriatric individuals contributes to impaired skin barrier integrity [212,213]. In addition, Langerhans cells in aged skin express a low amount of human beta-defensin-3, which is an important antimicrobial peptide produced in response to microbial infection or skin dysbiosis [214]. Thus far, the lessening of skin barrier function facilitates pathogens and pollution invasion driving low-grade chronic inflammation. Thus, when not locally restricted, the presence of inflammatory markers in senile skin might mirror a more complicated dysfunction of the immune system. Inflammaging has a widely recognized role in most common age-related diseases such as Alzheimer’s Disease, Parkinson’s Disease, heart diseases, multiple sclerosis, atherosclerosis, cancer, type II diabetes, and many others [215,216]. Finally, a defective epidermal barrier resulting from aged skin is also linked to age-related alternations to the gut and skin microbiome consisting of millions of bacteria, fungi, and viruses [217,218]. Investigations regarding the skin microbiome and its association with common clinical skin aging parameters including pigmentation, wrinkles, and texture demonstrated a clear age-dependent microbial signature attributing a causative effect of the altered skin microbiome in skin aging promotion [219].
3. The Role of Oxidative Stress in Extrinsic Aging of the Skin
In addition to the physiological mechanism of aging, exposure to UV light and environmental pollutants accelerate the acquisition of the aged phenotype. UV light is the major extrinsic agent responsible for skin aging. Premature photoaged skin typically presents with increased thickness of the epidermis, irregular pigmentation, and dermal connective tissue damage including the typical solar elastosis, laxity, dullness, roughness, and alteration of the vascular system [220]. The primary modification of the dermis during photo-accelerated aging regards structural components: collagen, elastin, and glycosaminoglycans. In older skin, the collagen network looks disorganized, and the ratio of collagen type III (Col-III) to I has been shown to increase due to less Col-I production [221]. Dissimilar to chronologic skin aging, MMP-induced collagen degradation and elastin degeneration are key mechanisms in photoaging [222]. In addition, the reduction in fibrillin structures and Col-VII involved in the bond between epidermis and dermis contribute to wrinkling formation [223]. Photoaging occurs principally due to UVA and UVB irradiation, which based on their distinct physical properties, induces different partially overlapping biological responses including abnormal ROS accumulation and/or DNA damage in both the epidermal and dermal compartment [224]. The fact that photoaging is largely due to oxidative disequilibrium is confirmed by the efficacy of topical antioxidants in UV-induced damage prevention [53,225]. Overall, depending on the cell type and the intensity of the stress, cells can either transiently block the cell cycle and repair the damage before restarting cell proliferation or enter apoptosis if the sensed damage is too serious. Therefore, sub-cytotoxic acute or chronic stress can induce premature senescence. Several pieces of evidence revealed that under the same conditions, human fibroblasts predominantly respond via senescence, while epithelial cells prefer to exert apoptosis [185,226]. Together with the cell-specific turnover rate, this partially explains the preferential detection of the senescent markers in the dermal compartment compared to the epidermal one. The magnitude of photodamage depends on constitutional factors, e.g., skin phototype (skin color, capacity to tan) and frequency and/or intensity of sunlight exposure [227]. The main cause of sunburn is UVB radiation. Skin UVB exposure results in the enhancement of NF-kB signaling. This signaling pathway is responsible for the augmented secretion of inflammatory mediators (IL1, IL3, IL6, IL8, IL7, IL10, and TNFα) by keratinocytes [228]. Exposure to UV light also activates lipoxygenase and cyclooxygenase pathways (LOX and COX-2), resulting in the production of leukotrienes and prostaglandins [228,229]. Prostaglandin E-2 (PGE-2) has a prominent role in the development of skin damage associated with both intrinsic and extrinsic aging. Studies focused on full-thickness sun-protected skin showed that both PGES-1 (PGE Synthase-1) and COX-2 expression progressively increased in dermal fibroblasts with age [230]. Additional studies confirmed higher COX-2 enzyme expression in chronologically aged skin and photoaged skin compared to young skin [231]. In addition, inflammatory cells such as lymphocytes, eosinophils, mast cells, and mononuclear cells are incremented [232]. This complex cascade of inflammatory events might determine local and potentially also systemic immunosuppression, which may not only undermine the control of dysplastic and neoplastic skin lesions but also favor immuno-pathological and infectious skin diseases [233]. Lower energy UVA rays penetrate deeply to the dermis, generating ROS that exacerbate the UVB-dependent mutagenic risk [234,235]. If not repaired, DNA mutations persist through subsequent cell subdivisions causing precancerous lesions (actinic keratosis) and skin cancers (basal cell carcinomas BCC, squamous cell carcinomas SCC, and melanoma).
UV radiation stimulates keratinocytes and fibroblasts to secrete numerous cytokines that are responsible for the proliferation of melanocytes and transient melanogenesis activation [236,237,238,239]. Fibroblasts isolated from habitually sun-exposed skin produce a greater amount of promelanogenic factors, such as hepatocyte growth factor (HGF), stem cell factor (SCF), and keratinocyte growth factor (KGF), whereas keratinocytes secrete α-MSH, SCF, prostaglandins, endothelin (ET), KGF, granulocyte-macrophage colony-stimulating factor (GM-CSF), and basic fibroblast growth factor (bFGF) [239,240,241]. The pigmentation response is an adaptation that prevents further DNA damage. Consequently, photoaged skin is characterized by irregular areas of pigmentation and hyperpigmented lesions [242,243]. Clinically, these dyspigmentations are known as solar lentigines (SL). Hyperpigmentation of this nature occurs due to changes in melanin synthesis, distribution, and turnover maintained by the focally increased number of melanocytes in the epidermis [244]. However, since decreased melanin removal rather than increased melanin production is considered the cause of hyperpigmented age spots, keratinocytes play a central role in this type of hyperpigmentation [245]. Melanin-loaded keratinocytes in SL show a lower proliferative capacity, altered differentiation, and are resistant to apoptosis. In addition, enhanced expression of p16INK4a and enlarged cell body size in SL suggest that keratinocytes are in a senescent condition [245]. In line with the idea that intrinsic factors could make the difference in the impact of extrinsic insult responses, darker skin types produce fewer radicals in the UV light compared to light skin types, whereas no differences were observed in the visible and infrared light [117]. This is mostly due to the prominent presence of pheomelanin in lighter skin amplifying UV-induced ROS formation [246]. Thus, in the dermatology practice, skin-type-specific sun protection is desired [247]. Apart from UV light, other radiation sources significantly impact skin aging. Infrared radiation, visible light, and artificial light promote ROS formation [248]. Infrared radiation causes photoaging and erythema, whereas visible light and artificial light stimulate inflammatory factors and hyperpigmentation [248]. Extensive clinical data demonstrated that outdoor (smog, ozone, particulate matter, etc.) and indoor (tobacco, solid fuel) pollution act in synergy with UV light in premature skin aging appearance [249,250]. A correlation between the number of years and packs of cigarettes smoked and the degree of skin aging has been documented [251]. Smokers have less skin elasticity and a reduced amount of collagen due to the low level of collagen synthesis [249]. Smoking promotes keratinocyte dysplasia and roughness of the cutis, and a dose-dependent relationship between wrinkling and smoking has been demonstrated [89,252]. In vitro exposure to tobacco extract induced the MMP1 expression by activating the aryl hydrocarbon receptor signaling pathway in human keratinocytes and fibroblasts [253]. An in vivo study confirmed a higher level of MMP1 expression in the dermis of smokers compared to non-smokers, resulting in collagen and elastin breakdown [254]. Due to an increase in ROS, reactive nitrogen species (RNS), DNA damage, protein, lipid oxidation, and altered mitochondria homeostasis, pesticides are a potential risk for skin aging and carcinogenesis [255,256]. Among exposome factors, dicarbonyl compounds such as glyoxal and methylglyoxal displayed the capacity to trigger oxidative stress-induced senescence in fibroblasts and keratinocytes [257,258,259]. These dicarbonyl compounds come from environmental exposure or food consumption [260]. However, age and metabolic diseases, such as diabetes, compromise the detoxifying activity of glyoxalases, leading to the accumulation of toxic glycation end products [261]. Accordingly, antioxidant exogenous supplementation with dietary antioxidants and/or cutaneous treatment with antioxidant molecules have been proven beneficial to improve photoaging [262]. However, the penetration in the stratum corneum of some antioxidants might be challenging. Several anti-aging strategies attempt to reduce (or avoid) exposure to UV and to reinforce the antioxidant capacity of the skin. The combination of both strategies is desired, since sun protection does not offer an appropriate protection to resist UVA-induced ROS. Numerous lines of evidence support the hypothesis that antioxidant compounds such as ascorbic acid, polyphenols, tocopherols, and other natural substances limit the concentration of free radicals, attenuating oxidative stress and slowing down the process of aging [263]. Ascorbic acid (vit C) directly scavenges ROS generated by UV radiation and promotes the biosynthesis of elastin and collagens [264]. In addition, phytochemicals such as resveratrol, quercetin, and green tea extract act as antioxidants to scavenge free radicals or ROS and have been reported to be effective in decreasing or retarding the progression of the aging process [265]. In addition, some phytochemicals act as anti-inflammatory agents by inhibiting the production of inflammatory mediators and cytokines and as stimulators of fibroblasts [266]. More recently, oral administration of bioactive collagen peptides to prevent skin aging has attracted more and more attention. Different formulations demonstrated efficacy in increasing skin hydration, elastin, pro-collagen I, and fibrillin, reducing wrinkle width [267,268] and increasing dermal matrix synthesis. Similarly, in preclinical animal models, oral supplementation plant polysaccharides extracted from Tremella fuciformis and Sargassum fusiforme enhanced SOD, Cat, and GPX activity significantly and decreased ROS and malondialdehyde levels, improving skin damage following UV exposure [264,269]
4. ROS and Hair Greying
One of the simplest indicators of aging in humans is hair greying or ″canities″. This fascinating phenomenon, characterized by loss of pigment production and deposition within the hair shafts [270], has attracted the attention of researchers for years and has led to a better comprehension of the intricate relationship between genetic, metabolic, neuroendocrine, and oxidative factors in its manifestation. Hair greying occurs in all individuals independent of sun exposure and results from the progressive loss of hair follicular melanocytes with age. This could result from numerous impaired processes, including follicular melanocyte death, migration or differentiation failure of melanocyte stem cells, or melanocyte stem cell depletion or death. Of course, when speaking about aging, the “free radical” theory is one of the most widely experimented with (Figure 3).
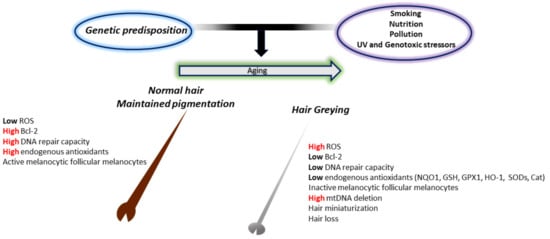
Figure 3.
The involvement of oxidative stress in the greying process. In young and healthy subjects, melanocytes in pigmented hair follicles can deal with the low endogenous oxidative stress caused by the melanin biosynthetic pathway. This is due to the adequate presence of endogenous antioxidant levels and the high DNA repair activity. In old subjects, however, reduced production of endogenous antioxidants and repair enzymes induce deleterious oxidative stress damage in pigment-producing cells. This results in the accumulation of senescent inactive melanocytes around the dermal papilla and at the outer root sheath, no melanocyte stem cells at the bulge area, and consequent a non-pigmented hair shaft.
Excluding UV light, hair follicles (HFs) can be exposed to other sources of ROS such as intrinsic metabolic by-products [271] and extrinsic agents (inflammatory process, smoke, drugs, poor nutrition). Indeed, the frequent 4977 bp mitochondrial DNA deletion (a marker of oxidative stress) is more frequent in greying HFs than in matched pigmented follicles, and melanocyte death by oxidative stress is increased in greying follicles [272]. The presence of highly vacuolated melanocytes within the HF, a cellular appearance that is ROS induced, corroborates the ″free radical theory of hair greying″. Some grey hair melanosomes were identified within auto-phagolysosomes, suggesting the removal of damaged melanosomes [273]. To cope with oxidative stress, the HF possesses an extremely elaborate antioxidant system [274]. A progressive failing of this system has been described in studies confronting prematurely grey hair follicles to pigmented ones, evidencing a downregulation of Cat, GPX1, and SODs in the former. These are accompanied by a 20-fold reduced expression of genes involved in melanogenesis such as Tyrosinase (TYR), Tyrosinase-Related Protein-1 (TYRP1), Microphthalmia Transcription Factor (MITF), Paired Box-3 (PAX3), POMC, KIT Proto-Oncogene, Receptor Tyrosine Kinase (KIT), and SRY-Box Transcription Factor 10 (SOX10) [275]. In a study of aging, Kauser et al. have also shown that follicular melanocytes age more than their epidermal counterparts, highlighting lower expression of Cat in follicular melanocytes [276]. In the same study, TRP-2 was significantly lowered in the older epidermal melanocytes, but at the same time, it was strongly elevated in the aged follicular melanocytes [276]. Compellingly, TRP-2 depletion has not been confirmed in eyelashes and eyebrows of the same patient group, suggesting possible anatomical differences between hair follicles of these locations. Lower antioxidant capacity due to depletion of TRP-2 from pigmented bulbar melanocytes suggests an additional antioxidant pathway that could affect hair greying. The intrinsic Cat deficiency could justify the high concentration of H2O2, which has been described in greying HFs in several reports [272,277]. Of note, the inadequate activity of Cat activity has been depicted in vitiligo, a common depigmentation disorder characterized by local or diffused destruction of melanocytes in the skin [278,279,280,281] sometimes presenting loss of hair pigmentation during disease progression [282]. Furthermore, Wood et al. have reported the inhibition of Tyrosinase, the key enzyme of the melanin biosynthetic pathway, by elevated levels of H2O2 [283]. This observation, coupled with the intrinsic ROS generation characteristic of melanogenesis [284], could justify an age-dependent melanin synthesis defect in the HF and, thus, hair greying. Accordingly, amelanotic melanocytes at the outer root sheath are somewhat less affected by oxidative damage and survive for a long time even within the white, aging hair follicles [285]. Another hypothesis has considered a possible “bleaching” phenomenon of the melanin pigment induced by H2O2 [286], even though bleached melanosomes have not yet been reported in hair follicles [287]. Of course, an obvious source of ROS is UV radiation, and Lu et al. have shown premature HF melanogenesis termination after UVB irradiation [288]. Altogether, these observations documented a high susceptibility to oxidative stress of HF melanocytes, implying an important role in the hair greying phenomenon. Ex vivo cultured human hair follicles demonstrated ROS-induced hair growth retardation, suggesting that oxidative disequilibrium intersects with diverse aspects of HF biology [289]. Oxidative stress and the aging process of HF are associated with reduced expression of the protective factor Bcl-2 [271]. Bcl-2 has a critical role in melanocyte maintenance, with knockout mice displaying accelerated greying [290]. By increasing Bcl-2 expression, melanocyte populations in the hair bulb and melanocyte stem cells in the bulge could be maintained, preventing hair greying. Finally, the compromised redox equilibrium has also been linked to the pathogenesis of androgenetic alopecia, a common heritable, androgen, and age-dependent process that results in large reductions in scalp hair density [291,292]. However, it is important to consider that ROS has an ambivalent function in hair biology. One key example of ROS relevance in HF physiology is the mitochondrial ROS-dependent activation of β-catenin and Notch signaling during HF development [293]. Based on this, photobiomodulation therapy, effective treatment for hair loss, benefits ROS-dependent stimulation of the Akt/GSK3β/β-catenin signaling pathway to drive quiescent hair follicle stem cells and alleviate HF atrophy [294].
5. Contribution of ROS in Common Age-Related Skin Diseases
5.1. Evidence of the Impact of ROS and Age-Related Skin Alteration on Tissue Vulnerability
Skin aging also represents a health risk, resulting in skin fragility. Age-dependent changes affecting both the adaptative and innate immune response have been well documented in older humans [295]. The immunosenescence consists of three main events: reduced immune response; increased production of autoantibodies; and inflammation (chronic, sterile, low-grade inflammation). The decrease in cutaneous immune functions facilitates a series of bacterial infections including cellulitis (particularly of the lower legs), erysipelas, necrotizing fasciitis, impetigo, folliculitis, and furunculosis [296,297]. Fungal and viral infections are also more frequent in the elderly [296,298]. Opposite, chronic, low-grade inflammation typical of elderly individuals could serve as a stimulus for the onset of autoimmunity [299]. Moreover, the possible release of self-antigens by damaged keratinocytes that contribute to the production of antibodies against neo-antigens might be enhanced by skin barrier impairment [297]. Accordingly, Bullous Pemphigoid, a subepidermal autoimmune blistering disease, is common in elderly patients [300,301]. Low antioxidant capacity/redox disequilibrium has been proven in Bullous Pemphigoid as well as in other non-age-related autoimmune skin blistering diseases [302]. Sporadically, epidermal injury following UVA and psoralen UVA (PUVA) therapies has been reported as a causative factor in attraction of autoantibodies [303,304]. Benign mucous membrane pemphigoid, paraneoplastic pemphigoid, and pemphigus Vulgaris are also more prevalent in the elderly [305]. Moreover, evidence of oxidative-stress-mediated pathogenic mechanisms has been reported for pemphigus Vulgaris [306,307,308] (Figure 4).
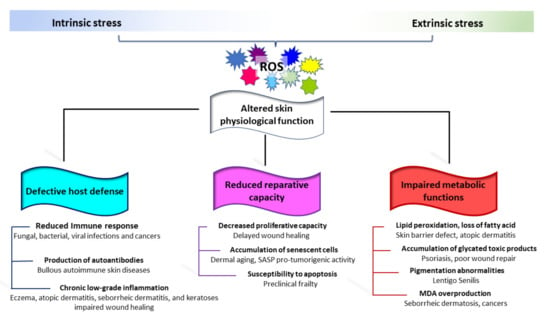
Figure 4.
Schematic representation of the involvement of oxidative stress in common age-related skin diseases. Progressive structural and functional degeneration of the skin leaves it prone to a wide variety of very common cutaneous diseases. The onset of most of these clinical conditions involves extrinsic activation of cutaneous immune cells in a redox-dependent manner. This includes a reduced immune response, production of autoantibodies, and chronic low-grade inflammation. Structural, biochemical changes, accumulation of senescent cells, and age-related stem cell depletion impact the cutaneous repair capacity. Metabolic alterations in aged skin do not fully support the physiological function of the skin and its appendages.
Structural and functional degeneration of the skin leaves it prone to various common cutaneous diseases, including eczema [309], contact and allergic dermatitis [310], seborrheic dermatitis [305], seborrheic keratoses (SK, a benign epithelial skin tumor) [311], and various forms of neoplasms. The onset of most of these clinical conditions is triggered by extrinsic activation of cutaneous immune cells in a redox-dependent manner [312,313,314]. Several age-associated characteristics of the cutaneous tissue might represent a hostile environment for wound healing. These include an abundance of ROS, persistent inflammation, and increased destruction of ECM components [315]. Further, age-associated deficits in microvascular function and poor staminal reservoir contribute to deficits in tissue healing in the older population [316]. Changes in the skin that occur in the elderly, especially dermal vascular changes, pH, and skin thickness, might directly or indirectly affect percutaneous penetration of drugs with a relevant consequence in pharmacotherapy. Albeit not linked to the classical idea of aging, several dermatological disorders are associated with the gain of senescent cells. For example, senescence-related acquired pathological pigmentary alteration including vitiligo and melasma have been described to be associated with premature senescence of the entire skin [241,317,318,319]. Harmed mitochondrial electron transport chain complex I activity in vitiligo cells, a high level of mitochondrial malate dehydrogenase activity, lower ATP production, and a diminished capacity to cope with stressful stimuli indicate an important role of mitochondrial defective functionality in the pathogenesis of vitiligo [320,321]. In vitiligo melanocytes, chronic oxidative stress drives the acquisition of a pro-inflammatory premature senescent phenotype [281]. Similarly, melasma (melanotic hypermelanosis) is a chronic relapsing hyperpigmentary disease presenting evidence of oxidative stress, subclinical inflammation, and several senescence markers [322,323]. In melasma, senescent-associated markers have been documented exclusively in disease-involved skin areas, whereas in vitiligo patients, the presence of the senescence feature has been delineated as a diffuse trait of the epidermis and the dermis in both lesional and non-lesional skin [238,281,324], suggesting an intrinsic susceptibility to premature senescence of vitiligo patients. Accordingly, for vitiligo, disease risk has been partially attributed to polygenic variants [325].
5.2. Contribution of ROS in Age-Related Skin Cancers
In old skin, most serious conditions are related to forms of neoplasms, such as a precancerous lesion (including actinic keratosis (AK) and lentigo maligna) resulting from the focal proliferation of mutated cells, and tumors. Merkel cell carcinoma [326], non-melanoma skin cancers [327,328], and malignant melanoma [328,329] are more frequent in the elderly due to somatic mutations accumulated over time. In melanoma, age is also an important prognostic factor. Accordingly, multivariate analyses demonstrate that age and Breslow thickness are the strongest independent adverse prognostic factors [329,330]. Both melanoma and non-melanoma occur prevalently on sun-exposed areas of the skin and present a severe response to solar radiation. UV-induced skin carcinogenesis is a complex and continuous biological process caused by different wavelengths. The two most abundant UV-induced DNA lesions, cyclobutane pyrimidine dimer (CPD) and the pyrimidone photoproduct (6-4PP), are due to UVB [331]. The increment of ROS that in turn could damage DNA by the formation of 7,8-dihydro-8-oxyguanine (8-oxodG) is largely dependent on UVA [332]. It is widely documented that sunscreens are also an important aspect of photoprotection, confirmed by their efficacy in reducing photocarcinogenesis and photoaging [333,334]. Further, several topically or systemically administered compounds confer protection against oxidative stress in dermal and epidermal cells. Vitamin D and related metabolites have been found to facilitate keratinocyte survival after UVB exposure [335], enhancing DNA repair [336,337]. Nicotinamide (NAM), a water-soluble vitamin B3 derivate, enhances DNA repair and mitigates the UV-induced suppression of immunity at the cellular level, whereas in clinical trials NAM in both topical and oral forms decreased trans-epidermal water loss and the development of cancers [338]. Thompson et al. demonstrated that nicotinamide lowered levels of 8-oxo-7,8-dihydro-2′-deoxyguanosine and cyclobutane pyrimidine dimers (two products of photolesions) in keratinocytes after exposure to both types of UV rays [339].
Independent of sun exposition, during the aging process, the skin gradually loses the capacity to counteract redox disequilibrium. Importantly, an age-related decline in the expression and activity of DNA repair proteins has been demonstrated, indicating that an organism is naturally predisposed to accumulate genomic and mitochondrial DNA mutations over time, which could further increase exposure to other ROS sources, impacting cancer probability [340]. Yet, independent of UV exposure (photoaging), intrinsic aging is perhaps the most important cancer risk factor. Furthermore, the production of ROS is a common cellular event occurring during the exposure to many, if not all, modifiable cancer risk factors, and it is generally accepted that increased ROS levels have a tumor-promoting effect in the early stages of the tumorigenic process [341,342]. Thus, mitigation of oxidative skin damage by topical or oral delivery of extrinsic antioxidant supplements may have therapeutic benefits in preventing melanoma and non-melanoma skin cancers.
Deregulated ROS in cancer are involved in cell cycle progression and proliferation, survival, apoptosis, intracellular adhesion, and cell migration. Nevertheless, from the tumor-centric point of view, senescence is a potent barrier to tumor progression. Melanocytic nevi are considered an excellent in vivo example of senescence protection of precancerous cells. Melanocytes in the nevi display features of oncogene-induced senescence, including intact telomeres, high p16INK4a expression, and SA-β-galactosidase activity [343,344,345,346,347]. This phenotype is mostly referred to as the mutational activation of BRAF and/or N-RAS, present in up to 81% of melanocytic nevi [348]. Apart from its function in cell cycle regulation, p16INK4a is involved in the prevention of ROS accumulation [349,350]. p16INK4a regulates mitochondrial biogenesis, dynamics, and function [349,350]. The central role of p16INK4a in melanocyte biology is demonstrated by the fact that individuals with familial CDKN2A gene (encoding p16INK4a and p14ARF) deficiency (also known as Leiden syndrome or Familial Atypical Multiple Mole Melanoma syndrome) have a characteristic accumulation of nevi and an increased melanoma risk [351]. Nevi remain growth arrested for a long time and infrequently develop into melanomas [352,353]. Thus, primary melanomas are considered a paradigm of senescence evasion. A decrease or loss of the tensin homolog (PTEN) and consequent activation of the PI3K/AKT signaling pathway may abolish senescence status, facilitating the melanocytic nevi progression into dysplastic nevi or melanoma [354]. A similar situation has been described for the keratinocyte lineage. Sasaki et al. showed ROS-induced senescence in normal epidermal keratinocytes might be triggered by upregulation of p16INK4a through demethylation in its promoter region. Since this genomic regulation has been not found in SCC cell lines, a key role in counteracting malignant transformation of normal human epidermal keratinocytes has been proposed [355]. However, the presence of p16INK4a-positive senescent cells in both benign (SK) and potentially malignant age-related skin lesions (AK) underlines the dual nature of senescence in cancer [356]. In keratinocytes, due to the renewal process regulated mainly by the proliferation/apoptosis ratio, reduced death of damaged cells causes cancer onset. At the molecular level, in keratinocyte carcinomas, p53 is the critical regulator of cell fate. P53 plays a critical role in supporting the DNA repair process and restoring genome stability. In genetically unstable cells, p53 can induce apoptosis, maintaining tissue homeostasis and tumor suppression [357]. However, loss-of-function of tumor suppressor p53 mutation, which is very frequent in the skin due to the repeated UV insult, impairs the physiological DNA repair machinery activity leading to the accumulation of further oncogenic mutations and expansion of pre-neoplastic clones [358]. Consistent with the early involvement of p53 in skin carcinogenesis, a wide number of studies documented p53 mutation in AK [359]. Interestingly, mutations at particular p53 codons are present in sun-exposed normal human skin at different frequencies depending on genetic background and lifestyle [360,361] and are considered a valid predictor of risk for basal cell carcinoma [362].
In the contest of skin cancer, pre-existing ECM alterations occurring in both aging and photoaging acquire a prominent interest in disease onset, progression, and invasion. The accumulation of senescent fibroblasts in habitually sun-exposed skin might direct stroma modification onto a tumor-promoting one [363,364]. Meanwhile, several reports have consistently suggested a mechanistic link between the aging microenvironment and neoplastic disease progress in several tissues, including the skin [365]. Aging-related fibrosis and the resulting increase in tissue stiffness have also been suggested to fuel carcinogenesis [366,367], possibly via alterations in the mechanical force balance between ECM, cell, and cytoskeleton [368]. The microenvironment surrounding benign nevi and melanomas displays greater stiffness than healthy skin, suggesting that physical assets of the tissue impact melanoma onset or progression [369]. Instead, in the contest of melanoma, modification of dermal ECM architecture and compromission of basement membrane integrity can facilitate the invasion of tumor cells. Accordingly, copious production of MMPs has been frequently reported in melanoma lesions [370]. Several studies have investigated the expression and activity of antioxidant enzymes in skin cancer cell lines, demonstrating a disequilibrium of the redox state within these cells [371]. Sander et al. reported that in human melanoma and non-melanoma skin cancer the natural redox balance is perturbed, leading to the accumulation of lipid peroxides and the formation of α, β-unsaturated aldehydes, including malondialdehyde (MDA) [372], which was shown to be mutagenic and carcinogenic [373]. MDA can combine with free amino groups of proteins, resulting in MDA-modified protein adducts, which in turn can be used as a measure for oxidative-stress-induced lipid peroxidation [374]. Intriguingly, significantly elevated MDA levels were found in SCC [375]. Primary and metastatic melanoma has a better antioxidant status than other skin tumors, including non-melanoma skin cancers [376]. Higher activity of Cat and SODs explains increased resistance of melanoma cells to oxidative stress compared to normal melanocytes and melanocytic nevi, suggesting that the acquisition of a robust antioxidant network is prominent for melanoma development [377,378]. A significant increase in plasma MDA and Cat activity but a simultaneous low SOD activity has been recorded in melanoma patients [379]. The elevation of both intracellular and extracellular ROS by tumor cells plays an important role in driving tumorigenesis by shaping the tumor microenvironment [380].
A particular feature of tumor stroma is the increased number of fibroblasts, pathologically activated and referred to as cancer-associated fibroblasts (CAFs). Due to the extraordinary tumor stroma paracrine dialogue, fibroblasts progressively acquire a molecular signature that partially overlaps with the stress-induced premature senescence one. This includes the release of molecules critically involved in metabolic and immune reprogramming of the tumor stroma with an effect on angiogenesis and resistance to therapy [381,382]. Extensive data demonstrated that ROS exerts a pivotal role in the process of fibroblast activation. It is known that ROS can be transferred from cancer cells to neighboring fibroblasts. ROS affects CAF’s features by promoting the conversion of fibroblasts to myofibroblasts that contribute to the tumor progression and spreading processes [383]. ROS activates CAFs that in turn enhance tumorigenesis by activating signaling pathways crucial for tumor cell proliferation and epithelial to mesenchymal transition [383]. As for other cancer types, the secretory profile of fibroblasts residing within the tumor margins or infiltrating melanoma substantially overlaps with that of senescent dermal fibroblasts [384,385]. It is important to underline that in contrast to the aging process that is characterized by a reduced number of dermal fibroblasts, skin neoplasms are permeated by the ratio of augmented fibroblasts/myofibroblasts, and that the presence of a large number of myofibroblasts in the tumor microenvironment has been associated with an elevated risk of invasion, metastasis, and a poor prognosis [364,386]. Paracrine TGFβ release by tumor cells causes an increase in NAD(P)H oxidase activity and triggers ROS-dependent fibroblast conversion into myofibroblasts by alpha-smooth muscle actin (α-SMA) gene transcription [387]. This crosstalk between tumor cells and CAFs could be abrogated with the addition of Cat [388]. Conversely, the decrease in oxidative stress in the tissue surrounding a tumor can slow down cancer growth and counteract its metastatic potential. Undeniably, SOD2 in CAFs can act as tumor suppressors [389].
6. Conclusions
Aging, as a broad term, encompasses several visible cutaneous phenomena such as skin wrinkling, atrophy, hair greying, and xerosis. Furthermore, since the skin gradually loses its structural and functional characteristics, skin aging is associated with several other processes such as viral, bacterial, and fungal infections, autoimmune blistering diseases, eczema, contact dermatitis, seborrheic dermatitis, and both melanoma and non-melanoma skin cancer. In this review, we provided an overview of the role of ROS in the appearance or the worsening of several age-related signs. We highlighted the importance of the former in both intrinsic and extrinsic aging. Indeed, chronic perturbation of redox equilibrium in the skin induces senescence and persistent inflammation. Overall, the understanding of the molecular mechanisms implicated in ROS formation and contrast is important to prevent physiologic skin aging and to compensate for functional deficits implicated in premature skin aging. However, the major challenging point is the definition of preventive strategies capable of sustaining the cutaneous intrinsic defense network. According to the minor capacity to preserve redox equilibrium, the elderly population needs to be more effectively treated to avoid an overabundance of ROS. On the other hand, pharmacological targeting of specific signaling pathways implicated in antioxidant/oxidant imbalance might represent a therapeutic opportunity for specific pathophysiological conditions frequently associated with aging, such as autoimmune disorders, chronic inflammation, and a multiple sequential skin cancer setting.
Author Contributions
B.B., F.P. and A.D. performed the literature research and drafted the manuscript. F.P. filled the reference section. S.C. summarized data, designed the figures, and performed technical editing of the original draft. B.B. took the lead in writing the manuscript. All authors provided critical feedback on the review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nielsen, K.P.; Zhao, L.; Stamnes, J.J.; Stamnes, K.; Moan, J. The importance of the depth distribution of melanin in skin for DNA protection and other photobiological processes. J. Photochem. Photobiol. B 2006, 82, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Boulais, N.; Misery, L. Merkel cells. J. Am. Acad. Dermatol. 2007, 57, 147–165. [Google Scholar] [CrossRef]
- Matsui, T.; Amagai, M. Dissecting the formation, structure and barrier function of the stratum corneum. Int. Immunol. 2015, 27, 269–280. [Google Scholar] [CrossRef]
- Eckhart, L.; Tschachler, E.; Gruber, F. Autophagic Control of Skin Aging. Front. Cell Dev. Biol. 2019, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Halprin, K.M. Epidermal “turnover time”—A re-examination. Br. J. Dermatol. 1972, 86, 14–19. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Gunn, D.A.; Adams, P.D.; Pawlikowski, J.S.; Griffiths, C.E.; van Heemst, D.; Slagboom, P.E.; Westendorp, R.G.; Maier, A.B. P16INK4a Positive Cells in Human Skin Are Indicative of Local Elastic Fiber Morphology, Facial Wrinkling, and Perceived Age. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1022–1028. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin melanocytes: Biology and development. Postepy Derm. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.C. The migratory pathway of neural crest cells into the skin of mouse embryos. Dev. Biol. 1973, 34, 39–46. [Google Scholar] [CrossRef]
- Birlea, S.A.; Costin, G.E.; Roop, D.R.; Norris, D.A. Trends in Regenerative Medicine: Repigmentation in Vitiligo Through Melanocyte Stem Cell Mobilization. Med. Res. Rev. 2017, 37, 907–935. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Fisher, D.E. Melanocyte stem cells as potential therapeutics in skin disorders. Expert Opin. Biol. Ther. 2014, 14, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fukunaga-Kalabis, M.; Yu, H.; Xu, X.; Kong, J.; Lee, J.T.; Herlyn, M. Human dermal stem cells differentiate into functional epidermal melanocytes. J. Cell Sci. 2010, 123, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Hoerter, J.D.; Bradley, P.; Casillas, A.; Chambers, D.; Denholm, C.; Johnson, K.; Weiswasser, B. Extrafollicular dermal melanocyte stem cells and melanoma. Stem Cells Int. 2012, 2012, 407079. [Google Scholar] [CrossRef] [PubMed]
- Tsepkolenko, A.; Tsepkolenko, V.; Dash, S.; Mishra, A.; Bader, A.; Melerzanov, A.; Giri, S. The regenerative potential of skin and the immune system. Clin. Cosmet. Investig. Dermatol. 2019, 12, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. FASEB J. 1997, 11, 51–59. [Google Scholar] [CrossRef]
- De Laporte, L.; Rice, J.J.; Tortelli, F.; Hubbell, J.A. Tenascin C promiscuously binds growth factors via its fifth fibronectin type III-like domain. PLoS ONE 2013, 8, e62076. [Google Scholar] [CrossRef] [PubMed]
- Briquez, P.S.; Hubbell, J.A.; Martino, M.M. Extracellular Matrix-Inspired Growth Factor Delivery Systems for Skin Wound Healing. Adv. Wound Care 2015, 4, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.; Pei, M. Age associated communication between cells and matrix: A potential impact on stem cell-based tissue regeneration strategies. Organogenesis 2014, 10, 289–298. [Google Scholar] [CrossRef]
- Kurtz, A.; Oh, S.J. Age related changes of the extracellular matrix and stem cell maintenance. Prev. Med. 2012, 54, S50–S56. [Google Scholar] [CrossRef] [PubMed]
- Asumda, F.Z. Age-associated changes in the ecological niche: Implications for mesenchymal stem cell aging. Stem. Cell Res. Ther. 2013, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Weihermann, A.C.; Lorencini, M.; Brohem, C.A.; de Carvalho, C.M. Elastin structure and its involvement in skin photoageing. Int. J. Cosmet. Sci. 2017, 39, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Juarez, C.F.; Plikus, M.V. Emerging nonmetabolic functions of skin fat. Nat. Rev. Endocrinol. 2018, 14, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Alam, W.; Hasson, J.; Reed, M. Clinical approach to chronic wound management in older adults. J. Am. Geriatr. Soc. 2021, 69, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Guerrero-Juarez, C.F.; Hata, T.; Bapat, S.P.; Ramos, R.; Plikus, M.V.; Gallo, R.L. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015, 347, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Bellei, B.; Migliano, E.; Tedesco, M.; Caputo, S.; Papaccio, F.; Lopez, G.; Picardo, M. Adipose tissue-derived extracellular fraction characterization: Biological and clinical considerations in regenerative medicine. Stem Cell Res. Ther. 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Mazini, L.; Malka, G.; Zeller, M.; Cottin, Y.; Vergely, C. The Crosstalk of Adipose-Derived Stem Cells (ADSC), Oxidative Stress, and Inflammation in Protective and Adaptive Responses. Int. J. Mol. Sci. 2020, 21, 9262. [Google Scholar] [CrossRef] [PubMed]
- Bliznakov, E.G. Aging, mitochondria, and coenzyme Q (10): The neglected relationship. Biochimie 1999, 81, 1131–1132. [Google Scholar] [PubMed]
- Krol, E.S.; Kramer-Stickland, K.A.; Liebler, D.C. Photoprotective actions of topically applied vitamin E. Drug Metab. Rev. 2000, 32, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Valacchi, G. Antioxidants and the response of skin to oxidative stress: Vitamin E as a key indicator. Skin Pharmacol. Appl. Skin Physiol. 2002, 15, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.J.; Traber, M.G.; Tsang, K.; Cross, C.E.; Packer, L. In vivo exposure to ozone depletes vitamins C and E and induces lipid peroxidation in epidermal layers of murine skin. Free Radic. Biol. Med. 1997, 23, 385–391. [Google Scholar] [CrossRef]
- Rhie, G.; Shin, M.H.; Seo, J.Y.; Choi, W.W.; Cho, K.H.; Kim, K.H.; Park, K.C.; Eun, H.C.; Chung, J.H. Aging- and photoaging-dependent changes of enzymic and nonenzymic antioxidants in the epidermis and dermis of human skin in vivo. J. Investig. Dermatol. 2001, 117, 1212–1217. [Google Scholar] [CrossRef]
- McArdle, F.; Rhodes, L.E.; Parslew, R.; Jack, C.I.; Friedmann, P.S.; Jackson, M.J. UVR-induced oxidative stress in human skin in vivo: Effects of oral vitamin C supplementation. Free Radic. Biol. Med. 2002, 33, 1355–1362. [Google Scholar] [CrossRef]
- Geesin, J.C.; Darr, D.; Kaufman, R.; Murad, S.; Pinnell, S.R. Ascorbic acid specifically increases type I and type III procollagen messenger RNA levels in human skin fibroblast. J. Investig. Dermatol. 1988, 90, 420–424. [Google Scholar] [CrossRef]
- Davidson, J.M.; LuValle, P.A.; Zoia, O.; Quaglino, D., Jr.; Giro, M. Ascorbate differentially regulates elastin and collagen biosynthesis in vascular smooth muscle cells and skin fibroblasts by pretranslational mechanisms. J. Biol. Chem. 1997, 272, 345–352. [Google Scholar] [CrossRef]
- Burke, K.E. Interaction of vitamins C and E as better cosmeceuticals. Dermatol. Ther. 2007, 20, 314–321. [Google Scholar] [CrossRef]
- Landis, G.N.; Tower, J. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 2005, 126, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Saxena, P.; Selvaraj, K.; Khare, S.K.; Chaudhary, N. Superoxide dismutase as multipotent therapeutic antioxidant enzyme: Role in human diseases. Biotechnol. Lett. 2022, 44, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun. 2009, 382, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Treiber, N.; Maity, P.; Singh, K.; Ferchiu, F.; Wlaschek, M.; Scharffetter-Kochanek, K. The role of manganese superoxide dismutase in skin aging. Dermatoendocrinology 2012, 4, 232–235. [Google Scholar] [CrossRef]
- Treiber, N.; Maity, P.; Singh, K.; Kohn, M.; Keist, A.F.; Ferchiu, F.; Sante, L.; Frese, S.; Bloch, W.; Kreppel, F.; et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 2011, 10, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Demaria, M.; Melov, S.; Campisi, J. Pleiotropic age-dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10407–10412. [Google Scholar] [CrossRef]
- Shin, M.H.; Rhie, G.E.; Kim, Y.K.; Park, C.H.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. H2O2 accumulation by catalase reduction changes MAP kinase signaling in aged human skin in vivo. J. Investig. Dermatol. 2005, 125, 221–229. [Google Scholar] [CrossRef]
- Lu, C.Y.; Lee, H.C.; Fahn, H.J.; Wei, Y.H. Oxidative damage elicited by imbalance of free radical scavenging enzymes is associated with large-scale mtDNA deletions in aging human skin. Mutat. Res. 1999, 423, 11–21. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant enzyme activity in human stratum corneum shows seasonal variation with an age-dependent recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; Ayer, A.; Breitenbach, M.; Dawes, I.W. Oxidative stresses and ageing. Subcell. Biochem. 2012, 57, 13–54. [Google Scholar]
- Lopez-Torres, M.; Shindo, Y.; Packer, L. Effect of age on antioxidants and molecular markers of oxidative damage in murine epidermis and dermis. J. Investig. Dermatol. 1994, 102, 476–480. [Google Scholar] [CrossRef]
- Poljšak, B.; Dahmane, R. Free radicals and extrinsic skin aging. Dermatol. Res. Pract. 2012, 2012, 135206. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Alia, A.; Backendorf, C. ROS quenching potential of the epidermal cornified cell envelope. J. Investig. Dermatol. 2011, 131, 1435–1441. [Google Scholar] [CrossRef]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Investig. Dermatol. 1994, 102, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.U.; Thiele, J.J.; Cross, C.E.; Packer, L. Vitamin C, uric acid, and glutathione gradients in murine stratum corneum and their susceptibility to ozone exposure. J. Investig. Dermatol. 1999, 113, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Woodby, B.; Penta, K.; Pecorelli, A.; Lila, M.A.; Valacchi, G. Skin Health from the Inside Out. Annu. Rev. Food Sci. Technol. 2020, 11, 235–254. [Google Scholar] [CrossRef]
- Parrado, C.; Mercado-Saenz, S.; Perez-Davo, A.; Gilaberte, Y.; Gonzalez, S.; Juarranz, A. Environmental Stressors on Skin Aging. Mechanistic Insights. Front. Pharmacol. 2019, 10, 759. [Google Scholar] [CrossRef]
- Sarangarajan, R.; Apte, S.P. The polymerization of melanin: A poorly understood phenomenon with egregious biological implications. Melanoma Res. 2006, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Edwards, C.; Gaskell, S.; Pearse, A.; Marks, R. Melanin content and distribution in the surface corneocyte with skin phototypes. Br. J. Dermatol. 1996, 135, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Wendt, J.; Rauscher, S.; Burgstaller-Muehlbacher, S.; Fae, I.; Fischer, G.; Pehamberger, H.; Okamoto, I. Human Determinants and the Role of Melanocortin-1 Receptor Variants in Melanoma Risk Independent of UV Radiation Exposure. JAMA Dermatol. 2016, 152, 776–782. [Google Scholar] [CrossRef]
- Zhang, S.; Duan, E. Fighting against Skin Aging: The Way from Bench to Bedside. Cell Transplant. 2018, 27, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Makrantonaki, E.; Nikolakis, G. When the skin is in the center of interest: An aging issue. Clin. Dermatol. 2019, 37, 296–305. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Bekou, V.; Zouboulis, C.C. Genetics and skin aging. Dermatoendocrinology 2012, 4, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Chon, S.H.; Pappas, A. Differentiation and characterization of human facial subcutaneous adipocytes. Adipocyte 2014, 4, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.B. Physiology of sweat gland function: The roles of sweating and sweat composition in human health. Temperature 2019, 6, 211–259. [Google Scholar] [CrossRef]
- Buffoli, B.; Rinaldi, F.; Labanca, M.; Sorbellini, E.; Trink, A.; Guanziroli, E.; Rezzani, R.; Rodella, L.F. The human hair: From anatomy to physiology. Int. J. Dermatol. 2014, 53, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Demaria, M. Targeting Senescent Cells: Possible Implications for Delaying Skin Aging: A Mini-Review. Gerontology 2016, 62, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Fathi, E.; Charoudeh, H.N.; Sanaat, Z.; Farahzadi, R. Telomere shortening as a hallmark of stem cell senescence. Stem Cell Investig. 2019, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kornicka, K.; Marycz, K.; Tomaszewski, K.A.; Marędziak, M.; Śmieszek, A. The Effect of Age on Osteogenic and Adipogenic Differentiation Potential of Human Adipose Derived Stromal Stem Cells (hASCs) and the Impact of Stress Factors in the Course of the Differentiation Process. Oxid. Med. Cell. Longev. 2015, 2015, 309169. [Google Scholar] [CrossRef] [PubMed]
- Orciani, M.; Gorbi, S.; Benedetti, M.; Di Benedetto, G.; Mattioli-Belmonte, M.; Regoli, F.; Di Primio, R. Oxidative stress defense in human-skin-derived mesenchymal stem cells versus human keratinocytes: Different mechanisms of protection and cell selection. Free Radic. Biol. Med. 2010, 49, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.C.; Hsieh, T.Y.; Lai, H.S.; Young, T.H. High glucose-induced reactive oxygen species generation promotes stemness in human adipose-derived stem cells. Cytotherapy 2016, 18, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Saretzki, G.; Döcke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J.; Hwang, Y.C.; Kang, C.M.; Choy, H.E.; Park, S.C. Reduction of UV-induced cell death in the human senescent fibroblasts. Mol. Cells 2000, 10, 415–422. [Google Scholar] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Melo Pereira, S.; Ribeiro, R.; Logarinho, E. Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies. Int. J. Mol. Sci. 2019, 20, 938. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 2019, 18, e12841. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Disease or not, aging is easily treatable. Aging 2018, 10, 3067–3078. [Google Scholar] [CrossRef]
- Tuttle, C.S.L.; Waaijer, M.E.C.; Slee-Valentijn, M.S.; Stijnen, T.; Westendorp, R.; Maier, A.B. Cellular senescence and chronological age in various human tissues: A systematic review and meta-analysis. Aging Cell 2020, 19, e13083. [Google Scholar] [CrossRef]
- Bellei, B.; Picardo, M. Premature cell senescence in human skin: Dual face in chronic acquired pigmentary disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Sahin, E.; Depinho, R.A. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 2010, 464, 520–528. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef]
- Krutmann, J.; Schikowski, T.; Morita, A.; Berneburg, M. Environmentally-Induced (Extrinsic) Skin Aging: Exposomal Factors and Underlying Mechanisms. J. Investig. Dermatol. 2021, 141, 1096–1103. [Google Scholar] [CrossRef]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Bellei, B.; Migliano, E.; Picardo, M. Therapeutic potential of adipose tissue-derivatives in modern dermatology. Exp. Dermatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Garcovich, S. Adipose-Derived Mesenchymal Stem Cells (AD-MSCs) against Ultraviolet (UV) Radiation Effects and the Skin Photoaging. Biomedicines 2021, 9, 532. [Google Scholar] [CrossRef]
- Passeron, T.; Krutmann, J.; Andersen, M.L.; Katta, R.; Zouboulis, C.C. Clinical and biological impact of the exposome on the skin. J. Eur. Acad. Dermatol. Venereol. 2020, 34 (Suppl. S4), 4–25. [Google Scholar] [CrossRef]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef] [PubMed]
- Friedman, O. Changes associated with the aging face. Facial Plast. Surg. Clin. N. Am. 2005, 13, 371–380. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Blog, F.B.; Szabo, G. Effects of aging and chronic sun exposure on melanocytes in human skin. J. Investig. Dermatol. 1979, 73, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.J. The lives of pigment cells. Dermatol. Clin. 1986, 4, 407–418. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.M.; Kwon, S.; Park, T.J.; Kang, H.Y. Senescent fibroblasts in melasma pathophysiology. Exp. Dermatol. 2019, 28, 719–722. [Google Scholar] [CrossRef]
- Kang, H.Y.; Lee, J.W.; Papaccio, F.; Bellei, B.; Picardo, M. Alterations of the pigmentation system in the aging process. Pigment Cell Melanoma Res. 2021, 34, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Rittié, L.; Fisher, G.J. Natural and sun-induced aging of human skin. Cold Spring Harb. Perspect. Med. 2015, 5, a015370. [Google Scholar] [CrossRef]
- El-Domyati, M.; Medhat, W.; Abdel-Wahab, H.M.; Moftah, N.H.; Nasif, G.A.; Hosam, W. Forehead wrinkles: A histological and immunohistochemical evaluation. J. Cosmet. Dermatol. 2014, 13, 188–194. [Google Scholar] [CrossRef]
- Labat-Robert, J.; Fourtanier, A.; Boyer-Lafargue, B.; Robert, L. Age dependent increase of elastase type protease activity in mouse skin. Effect of UV-irradiation. J. Photochem. Photobiol. B 2000, 57, 113–118. [Google Scholar] [CrossRef]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Li, S.; Jiang, X.; Oberley, L.W. Altered levels of primary antioxidant enzymes in progeria skin fibroblasts. Biochem. Biophys. Res. Commun. 1999, 257, 163–167. [Google Scholar] [CrossRef]
- Chung, J.H.; Kang, S.; Varani, J.; Lin, J.; Fisher, G.J.; Voorhees, J.J. Decreased extracellular-signal-regulated kinase and increased stress-activated MAP kinase activities in aged human skin in vivo. J. Investig. Dermatol. 2000, 115, 177–182. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef]
- Boo, Y.C. Natural Nrf2 Modulators for Skin Protection. Antioxidants 2020, 9, 812. [Google Scholar] [CrossRef]
- Fisher, G.J.; Datta, S.C.; Talwar, H.S.; Wang, Z.Q.; Varani, J.; Kang, S.; Voorhees, J.J. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature 1996, 379, 335–339. [Google Scholar] [CrossRef]
- Kohl, E.; Steinbauer, J.; Landthaler, M.; Szeimies, R.M. Skin ageing. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 873–884. [Google Scholar] [CrossRef]
- Quan, T.; He, T.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces Smad7 via induction of transcription factor AP-1 in human skin fibroblasts. J. Biol. Chem. 2005, 280, 8079–8085. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xu, Y.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces CYR61/CCN1, a mediator of collagen homeostasis, through activation of transcription factor AP-1 in human skin fibroblasts. J. Investig. Dermatol. 2010, 130, 1697–1706. [Google Scholar] [CrossRef]
- Haustead, D.J.; Stevenson, A.; Saxena, V.; Marriage, F.; Firth, M.; Silla, R.; Martin, L.; Adcroft, K.F.; Rea, S.; Day, P.J.; et al. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-κB. Sci. Rep. 2016, 6, 26846. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef] [PubMed]
- Meinke, M.C.; Busch, L.; Lohan, S.B. Wavelength, dose, skin type and skin model related radical formation in skin. Biophys. Rev. 2021, 13, 1091–1100. [Google Scholar] [CrossRef]
- Okayama, Y. Oxidative stress in allergic and inflammatory skin diseases. Curr. Drug Targets Inflamm. Allergy 2005, 4, 517–519. [Google Scholar] [CrossRef]
- Kandola, K.; Bowman, A.; Birch-Machin, M.A. Oxidative stress—A key emerging impact factor in health, ageing, lifestyle and aesthetics. Int. J. Cosmet. Sci. 2015, 37 (Suppl. S2), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cedikova, M.; Pitule, P.; Kripnerova, M.; Markova, M.; Kuncova, J. Multiple roles of mitochondria in aging processes. Physiol. Res. 2016, 65, S519–S531. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, K.; Hanna, R.; Birch-Machin, M.A. What is the role of mitochondrial dysfunction in skin photoaging? Exp. Dermatol. 2018, 27, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Yang, J.H.; Lee, H.C.; Wei, Y.H. Photoageing-associated mitochondrial DNA length mutations in human skin. Arch. Dermatol. Res. 1995, 287, 641–648. [Google Scholar] [PubMed]
- Kaneko, N.; Vierkoetter, A.; Kraemer, U.; Sugiri, D.; Matsui, M.; Yamamoto, A.; Krutmann, J.; Morita, A. Mitochondrial common deletion mutation and extrinsic skin ageing in German and Japanese women. Exp. Dermatol. 2012, 21 (Suppl. S1), 26–30. [Google Scholar] [CrossRef] [PubMed]
- Mellem, D.; Sattler, M.; Pagel-Wolff, S.; Jaspers, S.; Wenck, H.; Rübhausen, M.A.; Fischer, F. Fragmentation of the mitochondrial network in skin in vivo. PLoS ONE 2017, 12, e0174469. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Albensi, B.C. The nuclear factor kappa B (NF-κB) signaling pathway is involved in ammonia-induced mitochondrial dysfunction. Mitochondrion 2021, 57, 63–75. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell senescence, rapamycin and hyperfunction theory of aging. Cell Cycle 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. The role of cellular senescence in skin aging. J. Investig. Dermatol. Symp. Proc. 1998, 3, 1–5. [Google Scholar]
- Blagosklonny, M.V. Aging and immortality: Quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle 2006, 5, 2087–2102. [Google Scholar] [CrossRef]
- Ward, A.; Bates, P.; Fisher, R.; Richardson, L.; Graham, C.F. Disproportionate growth in mice with Igf-2 transgenes. Proc. Natl. Acad. Sci. USA 1994, 91, 10365–10369. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Zouboulis, C.C. German National Genome Research Network 2 The skin as a mirror of the aging process in the human organism—State of the art and results of the aging research in the German National Genome Research Network 2 (NGFN-2). Exp. Gerontol. 2007, 42, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Luebberding, S.; Krueger, N.; Kerscher, M. Age-related changes in skin barrier function–Quantitative evaluation of 150 female subjects. Int. J. Cosmet. Sci. 2013, 35, 183–190. [Google Scholar] [CrossRef]
- Feng, J.; Luo, J.; Yang, P.; Du, J.; Kim, B.S.; Hu, H. Piezo2 channel-Merkel cell signaling modulates the conversion of touch to itch. Science 2018, 360, 530–533. [Google Scholar] [CrossRef]
- Ale, I.S.; Maibach, H.I. Irritant contact dermatitis. Rev. Environ. Health 2014, 29, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.J. Introduction to skin aging. J. Tissue Viability 2017, 26, 37–46. [Google Scholar] [CrossRef]
- Chung, J.H.; Eun, H.C. Angiogenesis in skin aging and photoaging. J. Dermatol. 2007, 34, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Pierzga, J.M.; Frymoyer, A.; Kenney, W.L. Delayed distribution of active vasodilation and altered vascular conductance in aged skin. J. Appl. Physiol. 2003, 94, 1045–1053. [Google Scholar] [CrossRef]
- Bonta, M.; Daina, L.; Muţiu, G. The process of ageing reflected by histological changes in the skin. Rom. J. Morphol. Embryol. 2013, 54, 797–804. [Google Scholar]
- Dufour, A.; Candas, V. Ageing and thermal responses during passive heat exposure: Sweating and sensory aspects. Eur. J. Appl. Physiol. 2007, 100, 19–26. [Google Scholar] [CrossRef]
- Fernandez-Flores, A.; Saeb-Lima, M.; Cassarino, D.S. Histopathology of aging of the hair follicle. J. Cutan. Pathol. 2019, 46, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Flood, K.S.; Houston, N.A.; Savage, K.T.; Kimball, A.B. Genetic basis for skin youthfulness. Clin. Dermatol. 2019, 37, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Naval, J.; Alonso, V.; Herranz, M.A. Genetic polymorphisms and skin aging: The identification of population genotypic groups holds potential for personalized treatments. Clin. Cosmet. Investig. Dermatol. 2014, 7, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.C.; Wakamatsu, K.; Ito, S.; Kadekaro, A.L.; Kobayashi, N.; Groden, J.; Kavanagh, R.; Takakuwa, T.; Virador, V.; Hearing, V.J.; et al. Human melanocortin 1 receptor variants, receptor function and melanocyte response to UV radiation. J. Cell Sci. 2002, 115, 2349–2355. [Google Scholar] [CrossRef]
- Castejón-Griñán, M.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Chen, J.; Yang, J.; Chen, S.; Jameson, J.; Swope, V.B.; Cheng, T.; Kadakia, M.; Abdel-Malek, Z. Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol. Cancer Res. 2012, 10, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Henri, P.; Beaumel, S.; Guezennec, A.; Poumès, C.; Stoebner, P.E.; Stasia, M.J.; Guesnet, J.; Martinez, J.; Meunier, L. MC1R expression in HaCaT keratinocytes inhibits UVA-induced ROS production via NADPH oxidase- and cAMP-dependent mechanisms. J. Cell Physiol. 2012, 227, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Mosby, N.; Yang, J.; Xu, A.; Abdel-Malek, Z.; Kadekaro, A.L. alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment. Cell Melanoma Res. 2009, 22, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Fargnoli, M.C.; Spica, T.; Sera, F.; Pellacani, G.; Chiarugi, A.; Seidenari, S.; Carli, P.; Chimenti, S.; Peris, K. Re: MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J. Natl. Cancer Inst. 2006, 98, 144–145. [Google Scholar] [CrossRef]
- Li, X.; Mao, W.; Chen, J.; Goding, C.R.; Cui, R.; Xu, Z.X.; Miao, X. The protective role of MC1R in chromosome stability and centromeric integrity in melanocytes. Cell Death Discov. 2021, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Fargnoli, M.C.; Gandini, S.; Maisonneuve, P.; Liu, F.; Kayser, M.; Nijsten, T.; Han, J.; Kumar, R.; Gruis, N.A.; et al. M-SKIP Study Group MC1R gene variants and non-melanoma skin cancer: A pooled-analysis from the M-SKIP project. Br. J. Cancer 2015, 113, 354–363. [Google Scholar] [CrossRef]
- Elfakir, A.; Ezzedine, K.; Latreille, J.; Ambroisine, L.; Jdid, R.; Galan, P.; Hercberg, S.; Gruber, F.; Malvy, D.; Tschachler, E.; et al. Functional MC1R-gene variants are associated with increased risk for severe photoaging of facial skin. J. Investig. Dermatol. 2010, 130, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Kokot, A.; Sindrilaru, A.; Schiller, M.; Sunderkötter, C.; Kerkhoff, C.; Eckes, B.; Scharffetter-Kochanek, K.; Luger, T.A.; Böhm, M. Alpha-Melanocyte-Stimulating Hormone Suppresses Bleomycin-Induced Collagen Synthesis and Reduces Tissue Fibrosis in a Mouse Model of Scleroderma: Melanocortin Peptides as a Novel Treatment Strategy for Scleroderma? Arthritis Rheum. 2009, 60, 592–603. [Google Scholar] [CrossRef]
- Böhm, M.; Luger, T.A. Melanocortins in fibroblast biology--current update and future perspective for dermatology. Exp. Dermatol. 2004, 13 (Suppl. S4), 16–21. [Google Scholar] [CrossRef]
- De Souza, K.S.; Cantaruti, T.A.; Azevedo, G.M., Jr.; Galdino, D.A.; Rodrigues, C.M.; Costa, R.A.; Vaz, N.M.; Carvalho, C.R. Improved cutaneous wound healing after intraperitoneal injection of alpha-melanocyte-stimulating hormone. Exp. Dermatol. 2015, 24, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Dinparastisaleh, R.; Mirsaeidi, M. Antifibrotic and Anti-Inflammatory Actions of α-Melanocytic Hormone: New Roles for an Old Player. Pharmaceuticals 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Ciardo, S.; De Pace, B.; De Carvalho, N.; Peccerillo, F.; Manfredini, M.; Farnetani, F.; Chester, J.; Kaleci, S.; Manganelli, M.; et al. The influence of MC1R on dermal morphological features of photo-exposed skin in women revealed by reflectance confocal microscopy and optical coherence tomography. Exp. Dermatol. 2019, 28, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Pain, S.; Dezutter, C.; Reymermier, C.; Vogelgesang, B.; Delay, E.; André, V. Age-related changes in pro-opiomelanocortin (POMC) and related receptors in human epidermis. Int. J. Cosmet. Sci. 2010, 32, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.C.; Hamer, M.A.; Gunn, D.A.; Deelen, J.; Lall, J.S.; van Heemst, D.; Uh, H.W.; Hofman, A.; Uitterlinden, A.G.; Griffiths, C.E.M.; et al. A Genome-Wide Association Study Identifies the Skin Color Genes IRF4, MC1R, ASIP, and BNC2 Influencing Facial Pigmented Spots. J. Investig. Dermatol. 2015, 135, 1735–1742. [Google Scholar] [CrossRef]
- Gao, W.; Tan, J.; Hüls, A.; Ding, A.; Liu, Y.; Matsui, M.S.; Vierkötter, A.; Krutmann, J.; Schikowski, T.; Jin, L.; et al. Genetic variants associated with skin aging in the Chinese Han population. J. Dermatol. Sci. 2017, 86, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kang, S.; Lee, W.J. Menopause, Ultraviolet Exposure, and Low Water Intake Potentially Interact with the Genetic Variants Related to Collagen Metabolism Involved in Skin Wrinkle Risk in Middle-Aged Women. Int. J. Environ. Res. Public Health 2021, 18, 2044. [Google Scholar] [CrossRef] [PubMed]
- Orioli, D.; Dellambra, E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells 2018, 7, 268. [Google Scholar] [CrossRef]
- Bormann, F.; Rodríguez-Paredes, M.; Hagemann, S.; Manchanda, H.; Kristof, B.; Gutekunst, J.; Raddatz, G.; Haas, R.; Terstegen, L.; Wenck, H.; et al. Reduced DNA methylation patterning and transcriptional connectivity define human skin aging. Aging Cell 2016, 15, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, G.; Hagemann, S.; Aran, D.; Söhle, J.; Kulkarni, P.P.; Kaderali, L.; Hellman, A.; Winnefeld, M.; Lyko, F. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenet. Chromatin 2013, 6, 36. [Google Scholar] [CrossRef]
- Baumann, L. Skin ageing and its treatment. J. Pathol. 2007, 211, 241–251. [Google Scholar] [CrossRef]
- Szabo, G. The number of melanocytes in human epidermis. Br. Med. J. 1954, 1, 1016–1017. [Google Scholar] [CrossRef]
- Fitzpatrick, T.B. Hypomelanosis. South. Med. J. 1964, 57, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.P. Pigmentary changes of the ageing skin. Br. J. Dermatol. 1990, 122 (Suppl. S35), 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shlivko, I.L.; Petrova, G.A.; Zor’kina, M.V.; Tchekalkina, O.E.; Firsova, M.S.; Ellinsky, D.O.; Agrba, P.D.; Kamensky, V.A.; Donchenko, E.V. Complex assessment of age-specific morphofunctional features of skin of different anatomic localizations. Skin Res. Technol. 2013, 19, e85–e92. [Google Scholar] [CrossRef]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Maize, J.C.; Foster, G. Age-related changes in melanocytic naevi. Clin Exp. Dermatol. 1979, 4, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Chikeka, I.; Hornyak, T.J. Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence. Dermatol. Clin. 2017, 35, 85–93. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Parish, W.E.; Strongitharm, B.H.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.; Sedivy, J.M.; Westendorp, R.G.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Adamus, J.; Aho, S.; Meldrum, H.; Bosko, C.; Lee, J.M. p16INK4A influences the aging phenotype in the living skin equivalent. J. Investig. Dermatol. 2014, 134, 1131–1133. [Google Scholar] [CrossRef]
- Gilhar, A.; Ullmann, Y.; Karry, R.; Shalaginov, R.; Assy, B.; Serafimovich, S.; Kalish, R.S. Ageing of human epidermis: The role of apoptosis, Fas and telomerase. Br. J. Dermatol. 2004, 150, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Giangreco, A.; Qin, M.; Pintar, J.E.; Watt, F.M. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 2008, 7, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, S.; Ray, S.; Tarone, R.E.; Kraemer, K.H.; Grossman, L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: Reduced DNA repair capacity and increased DNA mutability. Mutat. Res. 1996, 364, 117–123. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Dame, M.K.; Rittie, L.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Pathol. 2006, 168, 1861–1868. [Google Scholar] [CrossRef]
- Le Varlet, B.; Chaudagne, C.; Saunois, A.; Barré, P.; Sauvage, C.; Berthouloux, B.; Meybeck, A.; Dumas, M.; Bonté, F. Age-related functional and structural changes in human dermo-epidermal junction components. J. Investig. Dermatol. Symp. Proc. 1998, 3, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Langton, A.K.; Halai, P.; Griffiths, C.E.; Sherratt, M.J.; Watson, R.E. The impact of intrinsic ageing on the protein composition of the dermal-epidermal junction. Mech. Ageing Dev. 2016, 156, 14–16. [Google Scholar] [CrossRef]
- Waldera Lupa, D.M.; Kalfalah, F.; Safferling, K.; Boukamp, P.; Poschmann, G.; Volpi, E.; Götz-Rösch, C.; Bernerd, F.; Haag, L.; Huebenthal, U.; et al. Characterization of Skin Aging-Associated Secreted Proteins (SAASP) Produced by Dermal Fibroblasts Isolated from Intrinsically Aged Human Skin. J. Investig. Dermatol. 2015, 135, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Shao, Y.; He, T.; Qin, Z.; Perry, D.; Voorhees, J.J.; Quan, T. Reduction of fibroblast size/mechanical force down-regulates TGF-β type II receptor: Implications for human skin aging. Aging Cell 2016, 15, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Smith, M.J.; Siow, R.C.M.; Liu, K.K. Ageing modulates human dermal fibroblast contractility: Quantification using nano-biomechanical testing. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118972. [Google Scholar] [CrossRef]
- Brun, C.; Jean-Louis, F.; Oddos, T.; Bagot, M.; Bensussan, A.; Michel, L. Phenotypic and functional changes in dermal primary fibroblasts isolated from intrinsically aged human skin. Exp. Dermatol. 2016, 25, 113–119. [Google Scholar] [CrossRef]
- Yuan, W.; Varga, J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad. J. Biol. Chem. 2001, 276, 38502–38510. [Google Scholar] [CrossRef]
- He, T.; Quan, T.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative exposure impairs TGF-β pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age 2014, 36, 9623. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Herrick, S.E.; Tarnuzzer, R.W.; Horan, M.A.; Schultz, G.S.; Ferguson, M.W. Human ageing impairs injury-induced in vivo expression of tissue inhibitor of matrix metalloproteinases (TIMP)-1 and -2 proteins and mRNA. J. Pathol. 1997, 183, 169–176. [Google Scholar] [CrossRef]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Stephan-Otto Attolini, C.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef]
- Bayreuther, K.; Rodemann, H.P.; Hommel, R.; Dittmann, K.; Albiez, M.; Francz, P.I. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc. Natl. Acad Sci. USA 1988, 85, 5112–5116. [Google Scholar] [CrossRef]
- Mine, S.; Fortunel, N.O.; Pageon, H.; Asselineau, D. Aging alters functionally human dermal papillary fibroblasts but not reticular fibroblasts: A new view of skin morphogenesis and aging. PLoS ONE 2008, 3, e4066. [Google Scholar] [CrossRef] [PubMed]
- Rorteau, J.; Chevalier, F.P.; Bonnet, S.; Barthélemy, T.; Lopez-Gaydon, A.; Martin, L.S.; Bechetoille, N.; Lamartine, J. Maintenance of Chronological Aging Features in Culture of Normal Human Dermal Fibroblasts from Old Donors. Cells 2022, 11, 858. [Google Scholar] [CrossRef]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef]
- Janson, D.; Rietveld, M.; Mahé, C.; Saintigny, G.; El Ghalbzouri, A. Differential effect of extracellular matrix derived from papillary and reticular fibroblasts on epidermal development in vitro. Eur. J. Dermatol. 2017, 27, 237–246. [Google Scholar] [CrossRef]
- Haydont, V.; Neiveyans, V.; Perez, P.; Busson, É.; Lataillade, J.; Asselineau, D.; Fortunel, N.O. Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling. Cells 2020, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Characteristics of the Aging Skin. Adv. Wound Care 2013, 2, 5–10. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed]
- Salvioli, S.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Bacalini, M.G.; Garagnani, P.; Giuliani, C.; Fontanesi, E.; Ostan, R.; et al. Immune system, cell senescence, aging and longevity--inflamm-aging reappraised. Curr. Pharm. Des. 2013, 19, 1675–1679. [Google Scholar]
- Chen, B.; Yang, J.; Song, Y.; Zhang, D.; Hao, F. Skin Immunosenescence and Type 2 Inflammation: A Mini-Review With an Inflammaging Perspective. Front. Cell Dev. Biol. 2022, 10, 835675. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, T.G.; Kim, S.H.; Park, J.Y.; Lee, M.; Lee, J.W.; Lee, S.H.; Lee, M.G. Epidermal Barrier Function Is Impaired in Langerhans Cell-Depleted Mice. J. Investig. Dermatol. 2019, 139, 1182–1185. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Dearman, R.J.; Kimber, I.; Griffiths, C.E.M. Langerhans cells express human β-defensin 3: Relevance for immunity during skin ageing. Br. J. Dermatol. 2018, 179, 1170–1171. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.G.; Markofski, M.M.; Carrillo, A.E. Elevated Inflammatory Status and Increased Risk of Chronic Disease in Chronological Aging: Inflamm-aging or Inflamm-inactivity? Aging Dis. 2019, 10, 147–156. [Google Scholar] [CrossRef]
- Szántó, M.; Dózsa, A.; Antal, D.; Szabó, K.; Kemény, L.; Bai, P. Targeting the gut-skin axis-Probiotics as new tools for skin disorder management? Exp. Dermatol. 2019, 28, 1210–1218. [Google Scholar] [CrossRef]
- Boyajian, J.L.; Ghebretatios, M.; Schaly, S.; Islam, P.; Prakash, S. Microbiome and Human Aging: Probiotic and Prebiotic Potentials in Longevity, Skin Health and Cellular Senescence. Nutrients 2021, 13, 4550. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 16748. [Google Scholar] [CrossRef] [PubMed]
- Lavker, R.M.; Gerberick, G.F.; Veres, D.; Irwin, C.J.; Kaidbey, K.H. Cumulative effects from repeated exposures to suberythemal doses of UVB and UVA in human skin. J. Am. Acad. Dermatol. 1995, 32, 53–62. [Google Scholar] [CrossRef]
- Oikarinen, A. The aging of skin: Chronoaging versus photoaging. Photodermatol. Photoimmunol. Photomed. 1990, 7, 3–4. [Google Scholar]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-degrading metalloproteinases in photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Contet-Audonneau, J.L.; Jeanmaire, C.; Pauly, G. A histological study of human wrinkle structures: Comparison between sun-exposed areas of the face, with or without wrinkles, and sun-protected areas. Br. J. Dermatol. 1999, 140, 1038–1047. [Google Scholar] [CrossRef]
- Gilchrest, B.A. Photoaging. J. Investig. Dermatol. 2013, 133, E2–E6. [Google Scholar] [CrossRef]
- Oresajo, C.; Pillai, S.; Manco, M.; Yatskayer, M.; McDaniel, D. Antioxidants and the skin: Understanding formulation and efficacy. Dermatol. Ther. 2012, 25, 252–259. [Google Scholar] [CrossRef]
- Georgakopoulou, E.; Evangelou, K.; Havaki, S.; Townsend, P.; Kanavaros, P.; Gorgoulis, V.G. Apoptosis or senescence? Which exit route do epithelial cells and fibroblasts preferentially follow? Mech. Ageing Dev. 2016, 156, 17–24. [Google Scholar] [CrossRef]
- Gupta, V.; Sharma, V.K. Skin typing: Fitzpatrick grading and others. Clin. Dermatol. 2019, 37, 430–436. [Google Scholar] [CrossRef]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef]
- Schwarz, T.; Luger, T.A. Effect of UV irradiation on epidermal cell cytokine production. J. Photochem. Photobiol. B 1989, 4, 1–13. [Google Scholar] [CrossRef]
- Li, Y.; Lei, D.; Swindell, W.R.; Xia, W.; Weng, S.; Fu, J.; Worthen, C.A.; Okubo, T.; Johnston, A.; Gudjonsson, J.E.; et al. Age-Associated Increase in Skin Fibroblast-Derived Prostaglandin E2 Contributes to Reduced Collagen Levels in Elderly Human Skin. J. Investig. Dermatol. 2015, 135, 2181–2188. [Google Scholar] [CrossRef]
- Habib, M.A.; Salem, S.A.; Hakim, S.A.; Shalan, Y.A. Comparative immunohistochemical assessment of cutaneous cyclooxygenase-2 enzyme expression in chronological aging and photoaging. Photodermatol. Photoimmunol. Photomed. 2014, 30, 43–51. [Google Scholar] [CrossRef]
- Bosset, S.; Bonnet-Duquennoy, M.; Barré, P.; Chalon, A.; Kurfurst, R.; Bonté, F.; Schnébert, S.; Le Varlet, B.; Nicolas, J.F. Photoageing shows histological features of chronic skin inflammation without clinical and molecular abnormalities. Br. J. Dermatol. 2003, 149, 826–835. [Google Scholar] [CrossRef]
- Hossain, M.R.; Ansary, T.M.; Komine, M.; Ohtsuki, M. Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. Int. J. Mol. Sci. 2021, 22, 3970. [Google Scholar] [CrossRef]
- Ikehata, H. Mechanistic considerations on the wavelength-dependent variations of UVR genotoxicity and mutagenesis in skin: The discrimination of UVA-signature from UV-signature mutation. Photochem. Photobiol. Sci. 2018, 17, 1861–1871. [Google Scholar] [CrossRef]
- Sarkar, S.; Gaddameedhi, S. Solar ultraviolet-induced DNA damage response: Melanocytes story in transformation to environmental melanomagenesis. Environ. Mol. Mutagen. 2020, 61, 736–751. [Google Scholar] [CrossRef]
- Imokawa, G. Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment. Cell Res. 2004, 17, 96–110. [Google Scholar] [CrossRef]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key regulatory role of dermal fibroblasts in pigmentation as demonstrated using a reconstructed skin model: Impact of photo-aging. PLoS ONE 2014, 9, e114182. [Google Scholar] [CrossRef]
- Bastonini, E.; Bellei, B.; Filoni, A.; Kovacs, D.; Iacovelli, P.; Picardo, M. Involvement of non-melanocytic skin cells in vitiligo. Exp. Dermatol. 2019, 28, 667–673. [Google Scholar] [CrossRef]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential expression of melanoma-associated growth factors in keratinocytes and fibroblasts by ultraviolet A and ultraviolet B radiation. Br. J. Dermatol. 2005, 153, 733–739. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.H.; Kim, E.K. Repeated exposure of human fibroblasts to UVR induces secretion of stem cell factor and senescence. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1577–1580. [Google Scholar] [CrossRef]
- Kovacs, D.; Cardinali, G.; Aspite, N.; Cota, C.; Luzi, F.; Bellei, B.; Briganti, S.; Amantea, A.; Torrisi, M.R.; Picardo, M. Role of fibroblast-derived growth factors in regulating hyperpigmentation of solar lentigo. Br. J. Dermatol. 2010, 163, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Monestier, S.; Gaudy, C.; Gouvernet, J.; Richard, M.A.; Grob, J.J. Multiple senile lentigos of the face, a skin ageing pattern resulting from a life excess of intermittent sun exposure in dark-skinned caucasians: A case-control study. Br. J. Dermatol. 2006, 154, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Bastiaens, M.; Hoefnagel, J.; Westendorp, R.; Vermeer, B.J.; Bouwes Bavinck, J.N. Solar lentigines are strongly related to sun exposure in contrast to ephelides. Pigment Cell Res. 2004, 17, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Hearing, V.J. Modifying skin pigmentation—Approaches through intrinsic biochemistry and exogenous agents. Drug Discov. Today Dis. Mech. 2008, 5, e189–e199. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Park, J.Y.; Kim, S.J.; Kang, H.Y. Characteristics of keratinocytes in facial solar lentigo with flattened rete ridges: Comparison with melasma. Clin. Exp. Dermatol. 2015, 40, 489–494. [Google Scholar] [CrossRef]
- Morgan, A.M.; Lo, J.; Fisher, D.E. How does pheomelanin synthesis contribute to melanomagenesis?: Two distinct mechanisms could explain the carcinogenicity of pheomelanin synthesis. Bioessays 2013, 35, 672–676. [Google Scholar] [CrossRef]
- Egambaram, O.P.; Kesavan Pillai, S.; Ray, S.S. Materials Science Challenges in Skin UV Protection: A Review. Photochem. Photobiol. 2020, 96, 779–797. [Google Scholar] [CrossRef]
- Furukawa, J.Y.; Martinez, R.M.; Morocho-Jácome, A.L.; Castillo-Gómez, T.S.; Pereda-Contreras, V.J.; Rosado, C.; Velasco, M.V.R.; Baby, A.R. Skin impacts from exposure to ultraviolet, visible, infrared, and artificial lights—A review. J. Cosmet. Laser Ther. 2021, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.; Bastiaens, M.T.; Bajdik, C.D.; Willemze, R.; Westendorp, R.G.; Bouwes Bavinck, J.N. Leiden Skin Cancer Study Effect of smoking and sun on the aging skin. J. Investig. Dermatol. 2003, 120, 548–554. [Google Scholar] [CrossRef]
- McDaniel, D.; Farris, P.; Valacchi, G. Atmospheric skin aging-Contributors and inhibitors. J. Cosmet. Dermatol. 2018, 17, 124–137. [Google Scholar] [CrossRef]
- Ortiz, A.; Grando, S.A. Smoking and the skin. Int. J. Dermatol. 2012, 51, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Leow, Y.H.; Maibach, H.I. Cigarette smoking, cutaneous vasculature, and tissue oxygen. Clin. Dermatol. 1998, 16, 579–584. [Google Scholar] [CrossRef]
- Ono, Y.; Torii, K.; Fritsche, E.; Shintani, Y.; Nishida, E.; Nakamura, M.; Shirakata, Y.; Haarmann-Stemmann, T.; Abel, J.; Krutmann, J.; et al. Role of the aryl hydrocarbon receptor in tobacco smoke extract-induced matrix metalloproteinase-1 expression. Exp. Dermatol. 2013, 22, 349–353. [Google Scholar] [CrossRef]
- Lahmann, C.; Bergemann, J.; Harrison, G.; Young, A.R. Matrix metalloproteinase-1 and skin ageing in smokers. Lancet 2001, 357, 935–936. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Yang, Y.; Ares, I.; Martínez, M.; Lopez-Torres, B.; Martínez-Larrañaga, M.R.; Wang, X.; Anadón, A.; Martinez, M.A. Neonicotinoids: Mechanisms of systemic toxicity based on oxidative stress-mitochondrial damage. Arch. Toxicol. 2022, 96, 1493–1520. [Google Scholar] [CrossRef]
- Sule, R.O.; Condon, L.; Gomes, A.V. A Common Feature of Pesticides: Oxidative Stress-The Role of Oxidative Stress in Pesticide-Induced Toxicity. Oxid. Med. Cell. Longev. 2022, 2022, 5563759. [Google Scholar] [CrossRef] [PubMed]
- Berge, U.; Behrens, J.; Rattan, S.I. Sugar-induced premature aging and altered differentiation in human epidermal keratinocytes. Ann. N. Y. Acad. Sci. 2007, 1100, 524–529. [Google Scholar] [CrossRef]
- Sejersen, H.; Rattan, S.I. Dicarbonyl-induced accelerated aging in vitro in human skin fibroblasts. Biogerontology 2009, 10, 203–211. [Google Scholar] [CrossRef]
- Halkoum, R.; Salnot, V.; Capallere, C.; Plaza, C.; L’honoré, A.; Pays, K.; Friguet, B.; Nizard, C.; Petropoulos, I. Glyoxal Induces Senescence in Human Keratinocytes through Oxidative Stress and Activation of the Protein Kinase B/FOXO3a/p27(KIP1) Pathway. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, M.; Gensberger-Reigl, S.; Henle, T.; Pischetsrieder, M. Food-derived 1,2-dicarbonyl compounds and their role in diseases. Semin. Cancer Biol. 2018, 49, 1–8. [Google Scholar] [CrossRef]
- Radjei, S.; Friguet, B.; Nizard, C.; Petropoulos, I. Prevention of dicarbonyl-mediated advanced glycation by glyoxalases: Implication in skin aging. Biochem. Soc. Trans. 2014, 42, 518–522. [Google Scholar] [CrossRef]
- Saewan, N.; Jimtaisong, A. Natural products as photoprotection. J. Cosmet. Dermatol. 2015, 14, 47–63. [Google Scholar] [CrossRef]
- Michalak, M. Plant-Derived Antioxidants: Sig.gnificance in Skin Health and the Ageing Process. Int. J. Mol. Sci. 2022, 23, 585. [Google Scholar] [CrossRef] [PubMed]
- Pinnell, S.R. Regulation of collagen biosynthesis by ascorbic acid: A review. Yale J. Biol. Med. 1985, 58, 553–559. [Google Scholar]
- Petruk, G.; Del Giudice, R.; Rigano, M.M.; Monti, D.M. Antioxidants from Plants Protect against Skin Photoaging. Oxid. Med. Cell. Longev. 2018, 2018, 1454936. [Google Scholar] [CrossRef] [PubMed]
- Forni, C.; Facchiano, F.; Bartoli, M.; Pieretti, S.; Facchiano, A.; D’Arcangelo, D.; Norelli, S.; Valle, G.; Nisini, R.; Beninati, S.; et al. Beneficial Role of Phytochemicals on Oxidative Stress and Age-Related Diseases. Biomed. Res. Int. 2019, 2019, 8748253. [Google Scholar] [CrossRef]
- Wen, L.; Gao, Q.; Ma, C.; Ge, Y.; You, L.; Liu, R.H.; Fu, X.; Liu, D. Effect of polysaccharides from Tremella fuciformis on UV-induced photoaging. J. Funct. Foods 2016, 20, 400–410. [Google Scholar] [CrossRef]
- Ye, Y.; Ji, D.; You, L.; Zhou, L.; Zhao, Z.; Brennan, C. Structural properties and protective effect of Sargassum fusiforme polysaccharides against ultraviolet B radiation in hairless Kun Ming mice. J. Funct. Foods 2018, 43, 8–16. [Google Scholar] [CrossRef]
- Hinek, A.; Kim, H.J.; Wang, Y.; Wang, A.; Mitts, T.F. Sodium L-ascorbate enhances elastic fibers deposition by fibroblasts from normal and pathologic human skin. J. Dermatol. Sci. 2014, 75, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Choi, G.I.; Kim, E.K.; Choi, Y.J.; Sohn, K.C.; Lee, Y.; Kim, C.D.; Yoon, T.J.; Sohn, H.J.; Han, S.H.; et al. Hair greying is associated with active hair growth. Br. J. Dermatol. 2011, 165, 1183–1189. [Google Scholar] [CrossRef]
- Jadkauskaite, L.; Coulombe, P.A.; Schäfer, M.; Dinkova-Kostova, A.T.; Paus, R.; Haslam, I.S. Oxidative stress management in the hair follicle: Could targeting NRF2 counter age-related hair disorders and beyond? Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Arck, P.C.; Overall, R.; Spatz, K.; Liezman, C.; Handjiski, B.; Klapp, B.F.; Birch-Machin, M.A.; Peters, E.M. Towards a “free radical theory of graying”: Melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. FASEB J. 2006, 20, 1567–1569. [Google Scholar] [CrossRef]
- Sato, S.; Kukita, A.; Jimbow, K. Electron microscopic studies of dendritic cells in the human gray and white matrix during anagen. In Pigment Cell Mechanisms in Pigmentation; McGovern, V.J., Russel, P., Eds.; Karger: Basel, Switzerland, 1973; Volume 1, pp. 20–26. [Google Scholar]
- Trüeb, R.M. The impact of oxidative stress on hair. Int. J. Cosmet. Sci. 2015, 37 (Suppl. S2), 25–30. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Luo, L.F.; Liu, X.M.; Zhou, Q.; Xu, S.Z.; Lei, T.C. Premature graysing as a consequence of compromised antioxidant activity in hair bulb melanocytes and their precursors. PLoS ONE 2014, 9, e93589. [Google Scholar]
- Kauser, S.; Westgate, G.E.; Green, M.R.; Tobin, D.J. Human hair follicle and epidermal melanocytes exhibit striking differences in their aging profile which involves catalase. J. Investig. Dermatol. 2011, 131, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; Decker, H.; Hartmann, H.; Chavan, B.; Rokos, H.; Spencer, J.D.; Hasse, S.; Thornton, M.J.; Shalbaf, M.; Paus, R.; et al. Senile hair graying: H2O2-mediated oxidative stress affects human hair color by blunting methionine sulfoxide repair. FASEB J. 2009, 23, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Gibbons, N.C.; Zothner, C.; Abou Elloof, M.M.; Wood, J.M. Hydrogen peroxide-mediated oxidative stress disrupts calcium binding on calmodulin: More evidence for oxidative stress in vitiligo. Biochem. Biophys. Res. Commun. 2007, 360, 70–75. [Google Scholar] [CrossRef]
- Maresca, V.; Roccella, M.; Roccella, F.; Camera, E.; Del Porto, G.; Passi, S.; Grammatico, P.; Picardo, M. Increased sensitivity to peroxidative agents as a possible pathogenic factor of melanocyte damage in vitiligo. J. Investig. Dermatol. 1997, 109, 310–313. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.J.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc. 1999, 4, 91–96. [Google Scholar] [CrossRef]
- Bellei, B.; Pitisci, A.; Ottaviani, M.; Ludovici, M.; Cota, C.; Luzi, F.; Dell’Anna, M.L.; Picardo, M. Vitiligo: A possible model of degenerative diseases. PLoS ONE 2013, 8, e59782. [Google Scholar] [CrossRef]
- Gan, E.Y.; Cario-André, M.; Pain, C.; Goussot, J.F.; Taïeb, A.; Seneschal, J.; Ezzedine, K. Follicular vitiligo: A report of 8 cases. J. Am. Acad. Dermatol. 2016, 74, 1178–1184. [Google Scholar] [CrossRef]
- Wood, J.M.; Chavan, B.; Hafeez, I.; Schallreuter, K.U. Regulation of tyrosinase by tetrahydropteridines and H2O. Biochem. Biophys. Res. Commun. 2004, 325, 1412–1417. [Google Scholar] [CrossRef]
- Koga, S.; Nakano, M.; Tero-Kubota, S. Generation of superoxide during the enzymatic action of tyrosinase. Arch. Biochem. Biophys. 1992, 292, 570–575. [Google Scholar] [CrossRef]
- Seiberg, M. Age-induced hair greying—The multiple effects of oxidative stress. Int. J. Cosmet. Sci. 2013, 35, 532–538. [Google Scholar] [CrossRef]
- Paus, R. A neuroendocrinological perspective on human hair follicle pigmentation. Pigment Cell Melanoma Res. 2011, 24, 89–106. [Google Scholar] [CrossRef]
- O′Sullivan, J.D.B.; Nicu, C.; Picard, M.; Chéret, J.; Bedogni, B.; Tobin, D.J.; Paus, R. The biology of human hair greying. Biol. Rev. Camb. Philos. Soc. 2021, 96, 107–128. [Google Scholar] [CrossRef]
- Lu, Z.; Fischer, T.W.; Hasse, S.; Sugawara, K.; Kamenisch, Y.; Krengel, S.; Funk, W.; Berneburg, M.; Paus, R. Profiling the response of human hair follicles to ultraviolet radiation. J. Investig. Dermatol. 2009, 129, 1790–1804. [Google Scholar] [CrossRef]
- Haslam, I.S.; Jadkauskaite, L.; Szabó, I.L.; Staege, S.; Hesebeck-Brinckmann, J.; Jenkins, G.; Bhogal, R.K.; Lim, F.L.; Farjo, N.; Farjo, B.; et al. Oxidative Damage Control in a Human (Mini-) Organ: Nrf2 Activation Protects against Oxidative Stress-Induced Hair Growth Inhibition. J. Investig. Dermatol. 2017, 137, 295–304. [Google Scholar] [CrossRef]
- Van Neste, D.; Tobin, D.J. Hair cycle and hair pigmentation: Dynamic interactions and changes associated with aging. Micron 2004, 35, 193–200. [Google Scholar] [CrossRef]
- Zhou, C.; Li, X.; Wang, C.; Zhang, J. Alopecia Areata: An Update on Etiopathogenesis, Diagnosis, and Management. Clin. Rev. Allergy Immunol. 2021, 61, 403–423. [Google Scholar] [CrossRef]
- Upton, J.H.; Hannen, R.F.; Bahta, A.W.; Farjo, N.; Farjo, B.; Philpott, M.P. Oxidative stress-associated senescence in dermal papilla cells of men with androgenetic alopecia. J. Investig. Dermatol. 2015, 135, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, J.E.; Baris, O.R.; Reuter, K.; Kobayashi, K.; Weiland, D.; Vidali, S.; Tobin, D.J.; Niemann, C.; Wiesner, R.J.; Paus, R. Mitochondrial function in murine skin epithelium is crucial for hair follicle morphogenesis and epithelial-mesenchymal interactions. J. Investig. Dermatol. 2015, 135, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zou, Z.; Chang, H.; Shen, Q.; Liu, L.; Xing, D. Photobiomodulation therapy for hair regeneration: A synergetic activation of β-CATENIN in hair follicle stem cells by ROS and paracrine WNTs. Stem Cell Rep. 2021, 16, 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Dupuis, G.; Witkowski, J.M.; Larbi, A. The Role of Immunosenescence in the Development of Age-Related Diseases. Rev. Investig. Clin. 2016, 68, 84–91. [Google Scholar]
- Laube, S. Skin infections and ageing. Ageing Res. Rev. 2004, 3, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M.; Vafaie, J.; Scheinfeld, N.S. Skin infections in the elderly. Dermatol. Clin. 2004, 22, 51–61. [Google Scholar] [CrossRef]
- Stevens, N.E.; Cowin, A.J.; Kopecki, Z. Skin Barrier and Autoimmunity-Mechanisms and Novel Therapeutic Approaches for Autoimmune Blistering Diseases of the Skin. Front. Immunol. 2019, 10, 1089. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Fania, L.; Sinagra, J.L.M.; Salemme, A.; Di Zenzo, G. Bullous Pemphigoid: Trigger and Predisposing Factors. Biomolecules 2020, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Deotto, M.L.; Spiller, A.; Sernicola, A.; Alaibac, M. Bullous pemphigoid: An immune disorder related to aging (Review). Exp. Ther. Med. 2022, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Dey-Rao, R.; Seiffert-Sinha, K.; Sinha, A.A. Increased oxidative stress in pemphigus vulgaris is related to disease activity and HLA-association. Autoimmunity 2016, 49, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Perl, S.; Rappersberger, K.; Födinger, D.; Anegg, B.; Hönigsmann, H.; Ortel, B. Bullous pemphigoid induced by PUVA therapy. Dermatology 1996, 193, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Pfau, A.; Hohenleutner, U.; Hohenleutner, S.; Eckert, F.; Landthaler, M. UV-A-provoked localized bullous pemphigoid. Acta Derm. Venereol. 1994, 74, 314–316. [Google Scholar] [PubMed]
- Farage, M.A.; Miller, K.W.; Berardesca, E.; Maibach, H.I. Clinical implications of aging skin: Cutaneous disorders in the elderly. Am. J. Clin. Dermatol. 2009, 10, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Y.; Ucmak, D.; Selek, S.; Dertlioğlu, S.B.; Sula, B.; Bozkus, F.; Turan, E. Oxidative stress index may play a key role in patients with pemphigus vulgaris. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, M.H.; Djalali, M.; Daneshpazhooh, M.; Zarei, M.; Eshraghian, M.R.; Derakhshanian, H.; Chams-Davatchi, C. Evaluation of antioxidant enzyme activity and antioxidant capacity in patients with newly diagnosed pemphigus vulgaris. Clin. Exp. Dermatol. 2015, 40, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jedličková, H.; Cai, Y.; Rehman, A.; Gammon, L.; Ahmad, U.S.; Uttagomol, J.; Parkinson, E.K.; Fortune, F.; Wan, H. Oxidative Stress-Mediated YAP Dysregulation Contributes to the Pathogenesis of Pemphigus Vulgaris. Front. Immunol. 2021, 12, 649502. [Google Scholar] [CrossRef] [PubMed]
- Tétart, F.; Joly, P. Eczema in elderly people. Eur. J. Dermatol. 2020, 30, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Katoh, N.; Tennstedt, D.; Abellan van Kan, G.; Saint Aroman, M.; Loir, A.; Bacqueville, D.; Duprat, L.; Guiraud, B.; Bessou-Touya, S.; Duplan, H. Gerontodermatology: The fragility of the epidermis in older adults. J. Eur. Acad. Dermatol. Venereol. 2018, 32 (Suppl. S4), 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Vogt, T. Seborrheic keratosis. J. Dtsch. Dermatol. Ges. 2008, 6, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Pambianchi, E.; Pecorelli, A.; Woodby, B.; Messano, N.; Therrien, J.P.; Lila, M.A.; Valacchi, G. Redox regulation of cutaneous inflammasome by ozone exposure. Free Radic. Biol. Med. 2020, 152, 561–570. [Google Scholar] [CrossRef]
- Corsini, E.; Galbiati, V.; Nikitovic, D.; Tsatsakis, A.M. Role of oxidative stress in chemical allergens induced skin cells activation. Food Chem. Toxicol. 2013, 61, 74–81. [Google Scholar] [CrossRef]
- Nakashima, H.; Fujimoto, M.; Asashima, N.; Watanabe, R.; Kuwano, Y.; Yazawa, N.; Maruyama, N.; Okochi, H.; Kumanogoh, A.; Tamaki, K. Serum chemokine profile in patients with bullous pemphigoid. Br. J. Dermatol. 2007, 156, 454–459. [Google Scholar] [CrossRef]
- Chen, D.; Hao, H.; Fu, X.; Han, W. Insight into Reepithelialization: How Do Mesenchymal Stem Cells Perform? Stem. Cells Int. 2016, 2016, 6120173. [Google Scholar] [CrossRef]
- Ding, X.; Kakanj, P.; Leptin, M.; Eming, S.A. Regulation of the Wound Healing Response during Aging. J. Investig. Dermatol. 2021, 141, 1063–1070. [Google Scholar] [CrossRef]
- Kamenisch, Y.; Berneburg, M. Progeroid syndromes and UV-induced oxidative DNA damage. J. Investig. Dermatol. Symp. Proc. 2009, 14, 8–14. [Google Scholar] [CrossRef]
- Goorochurn, R.; Viennet, C.; Tissot, M.; Locatelli, F.; Granger, C.; Varin-Blank, N.; Humbert, P.; Le Roy, C. Differential morphological and functional features of fibroblasts explanted from solar lentigo. Br. J. Dermatol. 2017, 177, e109–e111. [Google Scholar] [CrossRef]
- Kovacs, D.; Bastonini, E.; Ottaviani, M.; Cota, C.; Migliano, E.; Dell′Anna, M.L.; Picardo, M. Vitiligo Skin: Exploring the Dermal Compartment. J. Investig. Dermatol. 2018, 138, 394–404. [Google Scholar] [CrossRef]
- Dell′Anna, M.L.; Ottaviani, M.; Albanesi, V.; Vidolin, A.P.; Leone, G.; Ferraro, C.; Cossarizza, A.; Rossi, L.; Picardo, M. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J. Investig. Dermatol. 2007, 127, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Dell′Anna, M.L.; Ottaviani, M.; Kovacs, D.; Mirabilii, S.; Brown, D.A.; Cota, C.; Migliano, E.; Bastonini, E.; Bellei, B.; Cardinali, G.; et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci. Rep. 2017, 7, 13663. [Google Scholar] [CrossRef]
- Seçkin, H.Y.; Kalkan, G.; Baş, Y.; Akbaş, A.; Önder, Y.; Özyurt, H.; Sahin, M. Oxidative stress status in patients with melasma. Cutan. Ocul. Toxicol. 2014, 33, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Arámbula, A.; Torres-Álvarez, B.; Cortés-García, D.; Fuentes-Ahumada, C.; Castanedo-Cázares, J.P. CD4, IL-17, and COX-2 Are Associated With Subclinical Inflammation in Malar Melasma. Am. J. Dermatopathol. 2015, 37, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, T.H.; Park, T.J.; Kang, H.Y. p16(ink4a) Positivity of Melanocytes in Non-Segmental Vitiligo. Diagnostics 2020, 10, 878. [Google Scholar] [CrossRef]
- Spritz, R.A.; Santorico, S.A. The Genetic Basis of Vitiligo. J. Investig. Dermatol. 2021, 141, 265–273. [Google Scholar] [CrossRef]
- Patel, P.; Hussain, K. Merkel cell carcinoma. Clin. Exp. Dermatol. 2021, 46, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Tsatsou, F.; Trakatelli, M.; Patsatsi, A.; Kalokasidis, K.; Sotiriadis, D. Extrinsic aging: UV-mediated skin carcinogenesis. Dermatoendocrinology 2012, 4, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; Colloca, G.; Sollena, P.; Andrea, B.; Balducci, L.; Cho, W.C.; Bernabei, R.; Peris, K. Skin Cancer Epidemics in the Elderly as An Emerging Issue in Geriatric Oncology. Aging Dis. 2017, 8, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Ribero, S.; Stucci, L.S.; Marra, E.; Marconcini, R.; Spagnolo, F.; Orgiano, L.; Picasso, V.; Queirolo, P.; Palmieri, G.; Quaglino, P.; et al. Effect of Age on Melanoma Risk, Prognosis and Treatment Response. Acta Derm. Venereol. 2018, 98, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Buzaid, A.C.; Soong, S.J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A., Jr.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar] [CrossRef]
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 2001, 63, 88–102. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.L.; Lim, H.W.; Mohammad, T.F. Sunscreens and Photoaging: A Review of Current Literature. Am. J. Clin. Dermatol. 2021, 22, 819–828. [Google Scholar] [CrossRef] [PubMed]
- González, S.; Aguilera, J.; Berman, B.; Calzavara-Pinton, P.; Gilaberte, Y.; Goh, C.L.; Lim, H.W.; Schalka, S.; Stengel, F.; Wolf, P.; et al. Expert Recommendations on the Evaluation of Sunscreen Efficacy and the Beneficial Role of Non-filtering Ingredients. Front. Med. 2022, 9, 790207. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Dixon, K.M.; Deo, S.S.; Holliday, C.J.; Slater, M.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J. Investig. Dermatol. 2007, 127, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Gordon-Thomson, C.; Cole, L.; Stern, H.; Halliday, G.M.; Damian, D.L.; Reeve, V.E.; Mason, R.S. 1α,25-Dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. J. Steroid Biochem. Mol. Biol. 2013, 136, 131–138. [Google Scholar] [CrossRef]
- Gordon-Thomson, C.; Gupta, R.; Tongkao-on, W.; Ryan, A.; Halliday, G.M.; Mason, R.S. 1α,25 dihydroxyvitamin D3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photochem. Photobiol. Sci. 2012, 11, 1837–1847. [Google Scholar] [CrossRef]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28 (Suppl. S1), 15–22. [Google Scholar] [CrossRef]
- Thompson, B.C.; Halliday, G.M.; Damian, D.L. Nicotinamide enhances repair of arsenic and ultraviolet radiation-induced DNA damage in HaCaT keratinocytes and ex vivo human skin. PLoS ONE 2015, 10, e0117491. [Google Scholar] [CrossRef]
- Martin, J.E.; Sheaff, M.T. The pathology of ageing: Concepts and mechanisms. J. Pathol. 2007, 211, 111–113. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Margherita De Santi, M.; Schürfeld, K.; Santopietro, R.; Lalinga, A.V.; Fimiani, M.; Biagioli, M.; Brogi, M.; De Felice, C.; Luzi, P.; et al. Quantitative in situ evaluation of telomeres in fluorescence in situ hybridization-processed sections of cutaneous melanocytic lesions and correlation with telomerase activity. Br. J. Dermatol. 2002, 146, 399–408. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence--halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Reis-Filho, J.S.; da Rocha Dias, S.; Hayward, R.; Savage, K.; Delmas, V.; Larue, L.; Pritchard, C.; Marais, R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Goel, V.K.; Ibrahim, N.; Jiang, G.; Singhal, M.; Fee, S.; Flotte, T.; Westmoreland, S.; Haluska, F.S.; Hinds, P.W.; Haluska, F.G. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 2009, 28, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF mutations are common somatic events in melanocytic nevi. J. Investig. Dermatol. 2004, 122, 342–348. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Jung, J.; Liu, T.; Wilde, M.; Holmen, S.L.; Grossman, D. Familial melanoma-associated mutations in p16 uncouple its tumor-suppressor functions. J. Investig. Dermatol. 2013, 133, 1043–1051. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.; Ally, D.S.; Sheahan, M.D.; Clark, W.H., Jr.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef]
- Kuwata, T.; Kitagawa, M.; Kasuga, T. Proliferative activity of primary cutaneous melanocytic tumours. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 359–364. [Google Scholar] [CrossRef]
- Maldonado, J.L.; Timmerman, L.; Fridlyand, J.; Bastian, B.C. Mechanisms of cell-cycle arrest in Spitz nevi with constitutive activation of the MAP-kinase pathway. Am. J. Pathol. 2004, 164, 1783–1787. [Google Scholar] [CrossRef]
- Vredeveld, L.C.; Possik, P.A.; Smit, M.A.; Meissl, K.; Michaloglou, C.; Horlings, H.M.; Ajouaou, A.; Kortman, P.C.; Dankort, D.; McMahon, M.; et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012, 26, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kajiya, H.; Ozeki, S.; Okabe, K.; Ikebe, T. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16(INK4a). Biochem. Biophys. Res. Commun. 2014, 452, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nishioka, K. Enhanced expression of p16 in seborrhoeic keratosis; a lesion of accumulated senescent epidermal cells in G1 arrest. Br. J. Dermatol. 2003, 149, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Vaddavalli, P.L.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.M. The Role of p53 in Progression of Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef]
- Loureiro, J.B.; Abrantes, M.; Oliveira, P.A.; Saraiva, L. P53 in skin cancer: From a master player to a privileged target for prevention and therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188438. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; English, D.; Randell, P.L.; Nakkazawa, K.; Martel, N.; Armstrong, B.K.; Yamasaki, H. UV and skin cancer: Specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc. Natl. Acad. Sci. USA 1994, 91, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Ueda, M.; Nakazawa, H.; Ichihashi, M.; Dumaz, N.; Sarasin, A.; Yamasaki, H. Quantitative detection of ultraviolet-specific p53 mutations in normal skin from Japanese patients. Cancer Epidemiol. Biomarkers Prev. 1997, 6, 433–438. [Google Scholar] [PubMed]
- Ouhtit, A.; Nakazawa, H.; Armstrong, B.K.; Kricker, A.; Tan, E.; Yamasaki, H.; English, D.R. UV-radiation-specific p53 mutation frequency in normal skin as a predictor of risk of basal cell carcinoma. J. Natl. Cancer Inst. 1998, 90, 523–531. [Google Scholar] [CrossRef][Green Version]
- Alspach, E.; Fu, Y.; Stewart, S.A. Senescence and the pro-tumorigenic stroma. Crit. Rev. Oncog. 2013, 18, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Worrede, A.; Douglass, S.M.; Weeraratna, A.T. The dark side of daylight: Photoaging and the tumor microenvironment in melanoma progression. J. Clin. Investig. 2021, 131, e143763. [Google Scholar] [CrossRef]
- Vogel, V. Unraveling the Mechanobiology of Extracellular Matrix. Annu. Rev. Physiol. 2018, 80, 353–387. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E.; Cheri, S.; Fanti, M.; Marongiu, F. Aging and Cancer: The Waning of Community Bonds. Cells 2021, 10, 2269. [Google Scholar] [CrossRef]
- Ingber, D.E. Can cancer be reversed by engineering the tumor microenvironment? Semin. Cancer Biol. 2008, 18, 356–364. [Google Scholar] [CrossRef]
- Kirkpatrick, S.J.; Wang, R.K.; Duncan, D.D.; Kulesz-Martin, M.; Lee, K. Imaging the mechanical stiffness of skin lesions by in vivo acousto-optical elastography. Opt. Express 2006, 14, 9770–9779. [Google Scholar] [CrossRef]
- Berking, C.; Takemoto, R.; Schaider, H.; Showe, L.; Satyamoorthy, K.; Robbins, P.; Herlyn, M. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001, 61, 8306–8316. [Google Scholar]
- Godic, A.; Poljšak, B.; Adamic, M.; Dahmane, R. The role of antioxidants in skin cancer prevention and treatment. Oxid. Med. Cell. Longev. 2014, 2014, 860479. [Google Scholar] [CrossRef]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol. 2003, 148, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Duryee, M.J.; Clemens, D.L.; Opperman, P.J.; Thiele, G.M.; Duryee, L.M.; Garvin, R.P.; Anderson, D.R. Malondialdehyde-Acetaldehyde Modified (MAA) Proteins Differentially Effect the Inflammatory Response in Macrophage, Endothelial Cells and Animal Models of Cardiovascular Disease. Int. J. Mol. Sci. 2021, 22, 12948. [Google Scholar] [CrossRef]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Gadjeva, V.; Dimov, A.; Georgieva, N. Influence of therapy on the antioxidant status in patients with melanoma. J. Clin. Pharm. Ther. 2008, 33, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Bellei, B.; Migliano, E.; Picardo, M. A Framework of Major Tumor-Promoting Signal Transduction Pathways Implicated in Melanoma-Fibroblast Dialogue. Cancers 2020, 12, 3400. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Rebecca, V.; Fedorenko, I.V.; Messina, J.L.; Mathew, R.; Maria-Engler, S.S.; Basanta, D.; Smalley, K.S.; Anderson, A.R. Senescent fibroblasts in melanoma initiation and progression: An integrated theoretical, experimental, and clinical approach. Cancer Res. 2013, 73, 6874–6885. [Google Scholar] [CrossRef] [PubMed]
- Moinfar, F.; Beham, A.; Friedrich, G.; Deutsch, A.; Hrzenjak, A.; Luschin, G.; Tavassoli, F.A. Macro-environment of breast carcinoma: Frequent genetic alterations in the normal appearing skins of patients with breast cancer. Mod. Pathol. 2008, 21, 639–646. [Google Scholar] [CrossRef][Green Version]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Alili, L.; Sack, M.; Puschmann, K.; Brenneisen, P. Fibroblast-to-myofibroblast switch is mediated by NAD(P)H oxidase generated reactive oxygen species. Biosci. Rep. 2014, 34, e00089. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: A new genetically tractable model for human cancer associated fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).