
Mitochondrial Calcium: Effects of Its Imbalance in Disease


 ,
,
Abstract
:1. Introduction
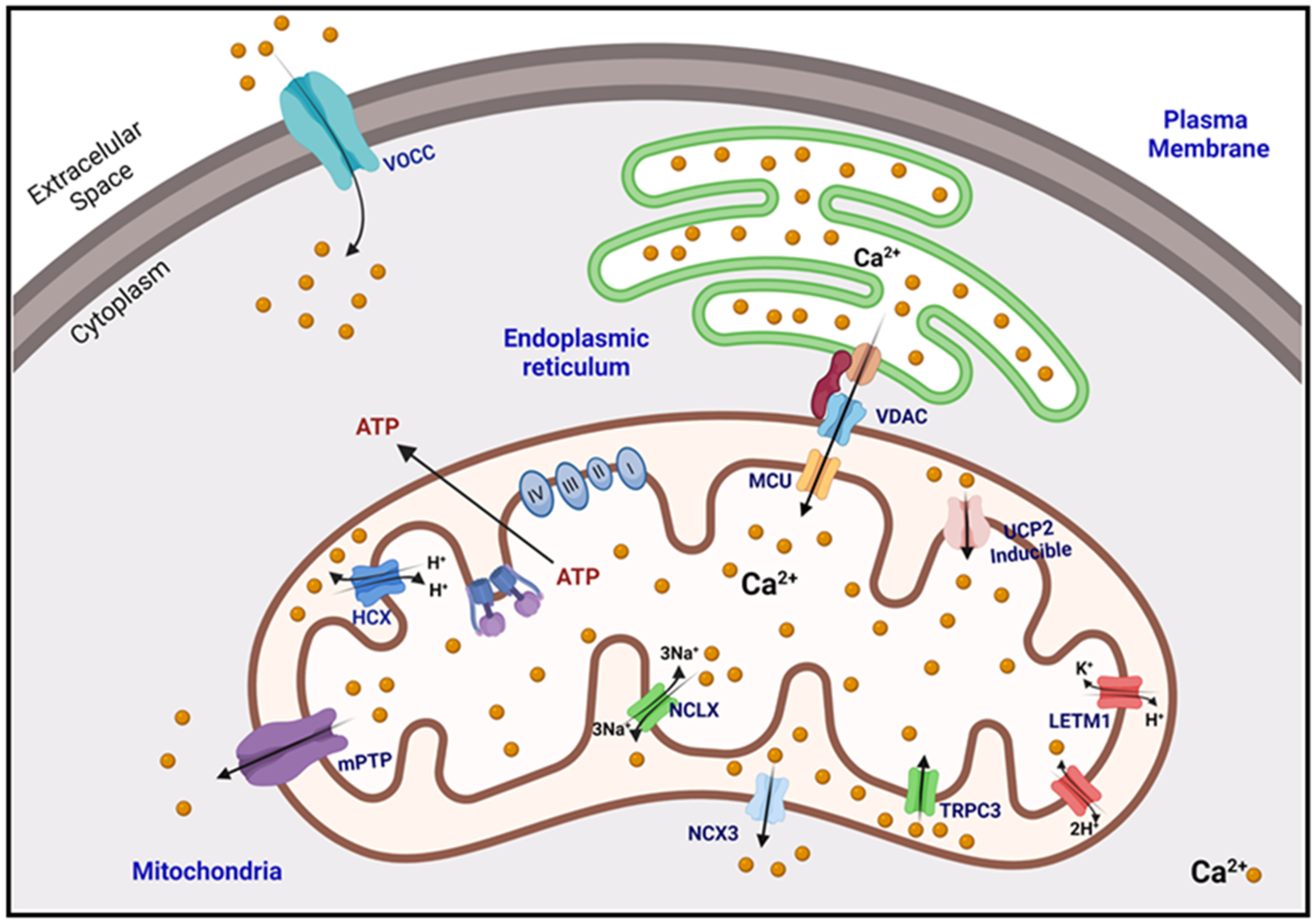
2. Calcium Homeostasis
2.1. Buffering of Matrix Calcium
2.2. Mitochondrial Calcium Uptake
2.3. Mitochondrial Calcium Extrusion
2.4. Microdomains, Connections of the Mitochondria with the ER and the PM
2.4.1. Presenilins
2.4.2. IP3R–SIG1R–GRP75–VDAC
2.4.3. Other Crosstalk Proteins
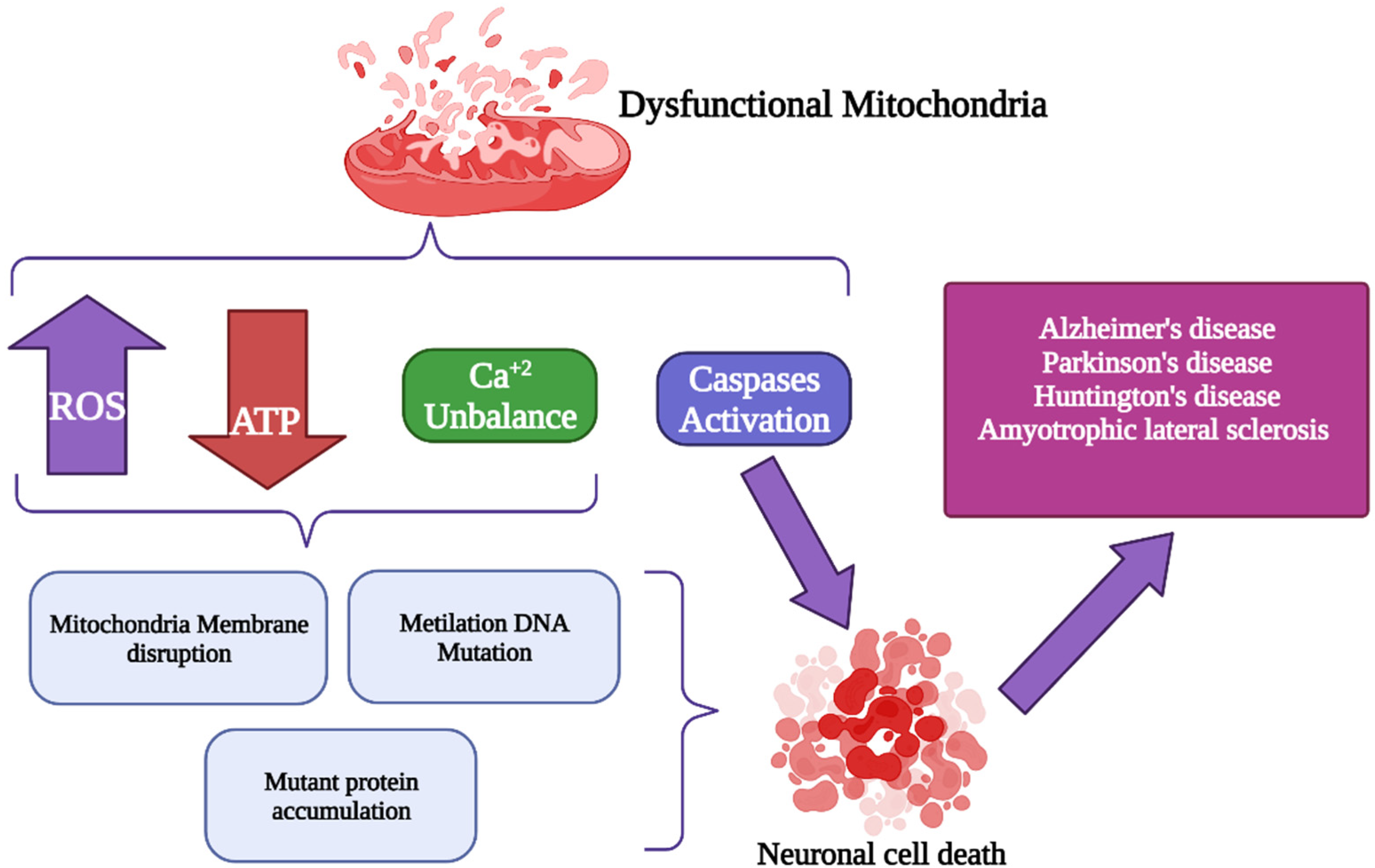
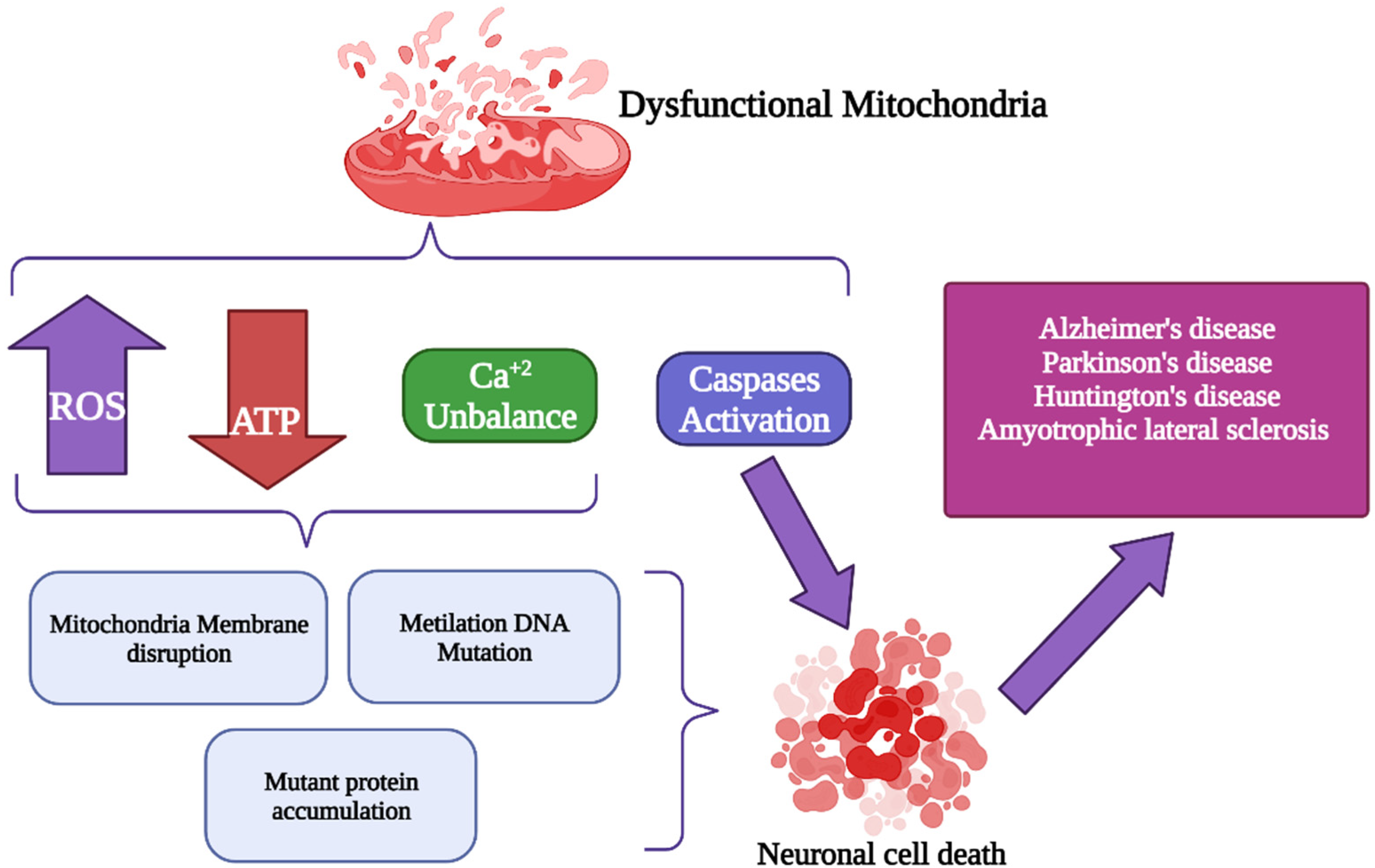
3. Oxidative Stress Generated by Changes in Mitochondrial Calcium Concentration
4. Alterations of Mitochondrial Ca2+ (mCa2+) Signaling in Neurodegenerative Disorders
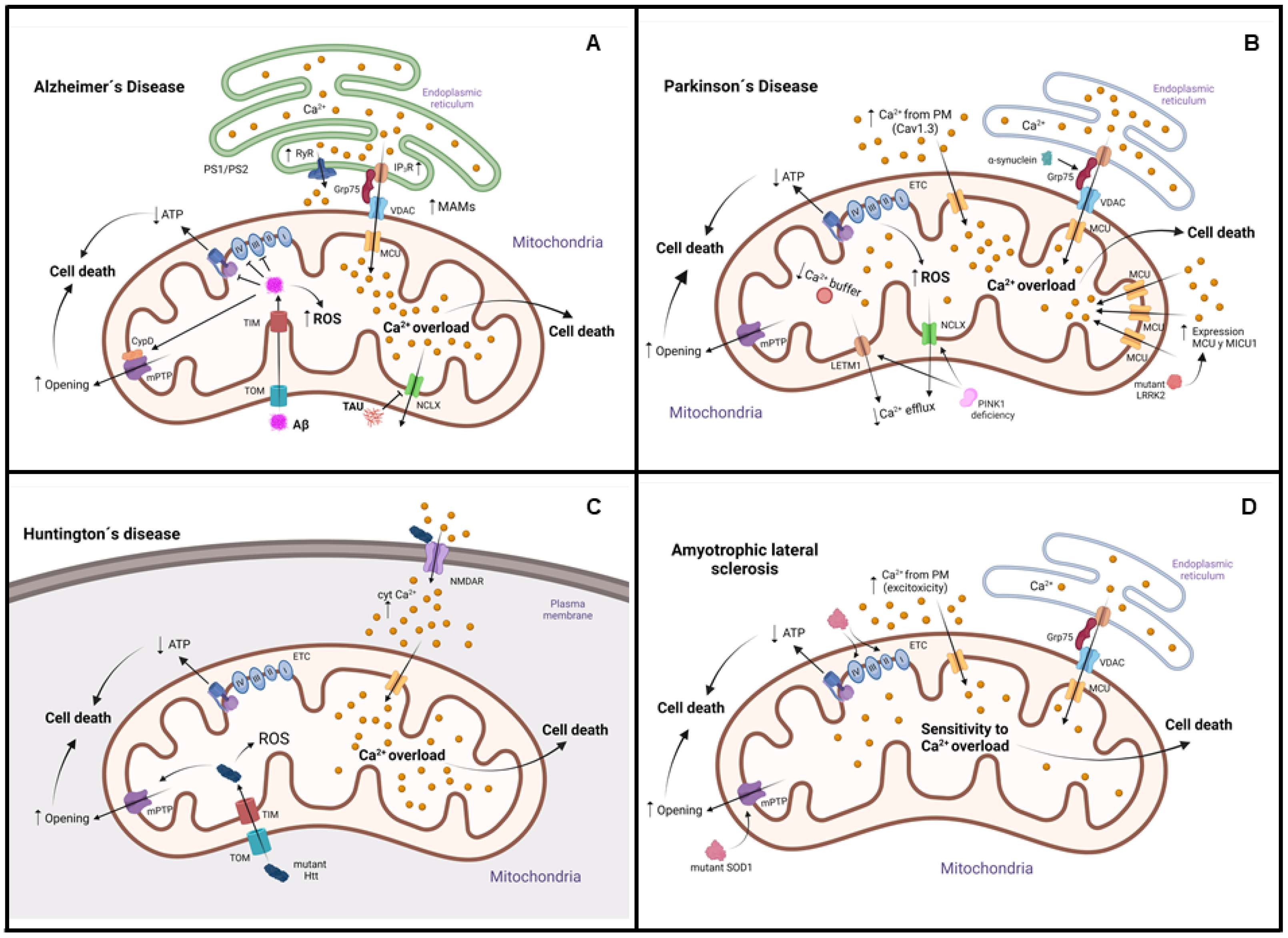
4.1. Alzheimer’s Disease (AD)
4.2. Parkinson’s Disease (PD)
4.3. Huntington’s Disease (HD)
4.4. Amyotrophic Lateral Sclerosis (ALS)
4.5. Other Neurodegenerative Diseases
5. Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dominguez, D.C. Calcium signalling in bacteria. Mol. Microbiol. 2004, 54, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnoczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal Dysfunction in Age-Related Diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. SnapShot: Reactive Oxygen Intermediates (ROI). Cell 2010, 140, 951–951.e2. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Osorio Alves, J.; Matta Pereira, L.; Cabral Coutinho do Rego Monteiro, I.; Pontes Dos Santos, L.H.; Soares Marreiros Ferraz, A.; Carneiro Loureiro, A.C.; Calado Lima, C.; Leal-Cardoso, J.H.; Pires Carvalho, D.; Soares Fortunato, R.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants 2020, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef]
- Blum, T.B.; Hahn, A.; Meier, T.; Davies, K.M.; Kuhlbrandt, W. Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc. Natl. Acad. Sci. USA 2019, 116, 4250–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genova, M.L.; Baracca, A.; Biondi, A.; Casalena, G.; Faccioli, M.; Falasca, A.I.; Formiggini, G.; Sgarbi, G.; Solaini, G.; Lenaz, G. Is supercomplex organization of the respiratory chain required for optimal electron transfer activity? Biochim. Biophys. Acta 2008, 1777, 740–746. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and mechanism of mitochondrial electron transport chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Christakos, S.; Lieben, L.; Masuyama, R.; Carmeliet, G. Vitamin D endocrine system and the intestine. Bonekey Rep. 2014, 3, 496. [Google Scholar] [CrossRef] [Green Version]
- Bronner, F. Extracellular and intracellular regulation of calcium homeostasis. ScientificWorldJournal 2001, 1, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.; Verkhratsky, A. Relations between intracellular Ca2+ stores and store-operated Ca2+ entry in primary cultured human glioblastoma cells. J. Physiol. 1998, 513 Pt 2, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Dey, K.; Hershfinkel, M.; Ohana, E.; Sekler, I. Identification of residues that control Li+ versus Na+ dependent Ca2+ exchange at the transport site of the mitochondrial NCLX. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; Tavakoli, M.; Pfeiffer, C.; Seifert, J.; Mattarei, A.; De Stefani, D.; Zoratti, M.; Nowikovsky, K. LETM1-Mediated K+ and Na+ Homeostasis Regulates Mitochondrial Ca2+ Efflux. Front. Physiol. 2017, 8, 839. [Google Scholar] [CrossRef] [Green Version]
- Melchionda, M.; Pittman, J.K.; Mayor, R.; Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 2016, 212, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef]
- Fujimoto, M.; Hayashi, T.; Su, T.P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- AhYoung, A.P.; Jiang, J.; Zhang, J.; Khoi Dang, X.; Loo, J.A.; Zhou, Z.H.; Egea, P.F. Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E3179–E3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, S.; Ponnusamy, M. Calcium Ion in Biological Systems. In Calcium Signaling: From Physiology to Diseases; Springer: Singapore, 2017; pp. 1–14. [Google Scholar]
- Van Rossum, D.B.; Patterson, R.L.; Kiselyov, K.; Boehning, D.; Barrow, R.K.; Gill, D.L.; Snyder, S.H. Agonist-induced Ca2+ entry determined by inositol 1,4,5-trisphosphate recognition. Proc. Natl. Acad. Sci. USA 2004, 101, 2323–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, R.L.; Boehning, D.; Snyder, S.H. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 2004, 73, 437–465. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G. Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr. 1994, 26, 495–508. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Zhu, L.P.; Yu, X.D.; Ling, S.; Brown, R.A.; Kuo, T.H. Mitochondrial Ca2+ homeostasis in the regulation of apoptotic and necrotic cell deaths. Cell Calcium 2000, 28, 107–117. [Google Scholar] [CrossRef]
- Kudla, J.; Becker, D.; Grill, E.; Hedrich, R.; Hippler, M.; Kummer, U.; Parniske, M.; Romeis, T.; Schumacher, K. Advances and current challenges in calcium signaling. New Phytol. 2018, 218, 414–431. [Google Scholar] [CrossRef]
- Kamishima, T.; McCarron, J.G. Regulation of the cytosolic Ca2+ concentration by Ca2+ stores in single smooth muscle cells from rat cerebral arteries. J. Physiol. 1997, 501 Pt 3, 497–508. [Google Scholar] [CrossRef]
- Kamishima, T.; Davies, N.W.; Standen, N.B. Mechanisms that regulate [Ca2+]i following depolarization in rat systemic arterial smooth muscle cells. J. Physiol. 2000, 522 Pt 2, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, J.A. Cytoplasmic Calcium Buffering: An Integrative Crosstalk. Adv. Exp. Med. Biol. 2020, 1131, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, A.; Rapizzi, E.; Tosello, V.; Pinton, P.; de Virgilio, M.; Fogarty, K.E.; Rizzuto, R. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem. J. 2001, 355, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnoczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef]
- Duvvuri, B.; Lood, C. Mitochondrial Calcification. Immunometabolism 2021, 3, e210008. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef] [Green Version]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef]
- Foskett, J.K.; Philipson, B. The mitochondrial Ca2+ uniporter complex. J. Mol. Cell. Cardiol. 2015, 78, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Liu, J.; Holmstrom, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Matsui, A.; Akabane, S.; Tamura, Y.; Hatano, A.; Miyano, Y.; Omote, H.; Kajikawa, M.; Maenaka, K.; Moriyama, Y.; et al. The mitochondrial inner membrane protein LETM1 modulates cristae organization through its LETM domain. Commun Biol. 2020, 3, 99. [Google Scholar] [CrossRef]
- Nowikovsky, K.; Pozzan, T.; Rizzuto, R.; Scorrano, L.; Bernardi, P. Perspectives on: SGP symposium on mitochondrial physiology and medicine: The pathophysiology of LETM1. J. Gen. Physiol. 2012, 139, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; Nowikovsky, K. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem. Sci. 2019, 44, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Turner, H.; Fleig, A.; Stokes, A.; Kinet, J.P.; Penner, R. Discrimination of intracellular calcium store subcompartments using TRPV1 (transient receptor potential channel, vanilloid subfamily member 1) release channel activity. Biochem. J. 2003, 371, 341–350. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Ramsey, I.S.; Kotecha, S.; Greka, A.; Clapham, D.E. Rapid vesicular translocation and insertion of TRP channels. Nat. Cell Biol. 2004, 6, 709–720. [Google Scholar] [CrossRef]
- Oancea, E.; Vriens, J.; Brauchi, S.; Jun, J.; Splawski, I.; Clapham, D.E. TRPM1 forms ion channels associated with melanin content in melanocytes. Sci. Signal. 2009, 2, ra21. [Google Scholar] [CrossRef] [Green Version]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci. Signal. 2009, 2, ra23. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockwich, T.; Pant, J.; Makusky, A.; Jankowska-Stephens, E.; Kowalak, J.A.; Markey, S.P.; Ambudkar, I.S. Analysis of TRPC3-interacting proteins by tandem mass spectrometry. J. Proteome Res. 2008, 7, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Poteser, M.; Schleifer, H.; Lichtenegger, M.; Schernthaner, M.; Stockner, T.; Kappe, C.O.; Glasnov, T.N.; Romanin, C.; Groschner, K. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10556–10561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [Green Version]
- Ricquier, D.; Bouillaud, F. Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol. 2000, 529 Pt 1, 3–10. [Google Scholar] [CrossRef]
- Koshenov, Z.; Oflaz, F.E.; Hirtl, M.; Bachkoenig, O.A.; Rost, R.; Osibow, K.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Waldeck-Weiermair, M.; Malli, R.; et al. The contribution of uncoupling protein 2 to mitochondrial Ca2+ homeostasis in health and disease—A short revisit. Mitochondrion 2020, 55, 164–173. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulovic, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhang, D.; He, X.; Huang, Y.; Shao, H. Transport of Calcium Ions into Mitochondria. Curr. Genom. 2016, 17, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [Green Version]
- Bauerova-Hlinkova, V.; Hajduchova, D.; Bauer, J.A. Structure and Function of the Human Ryanodine Receptors and Their Association with Myopathies-Present State, Challenges, and Perspectives. Molecules 2020, 25, 4040. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.W.; Baysal, K.; Brierley, G.P. The sodium-calcium antiport of heart mitochondria is not electroneutral. J. Biol. Chem. 1995, 270, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.K.; Beard, D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008, 586, 3267–3285. [Google Scholar] [CrossRef]
- Gunter, K.K.; Zuscik, M.J.; Gunter, T.E. The Na+-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J. Biol. Chem. 1991, 266, 21640–21648. [Google Scholar] [CrossRef]
- Scorziello, A.; Savoia, C.; Sisalli, M.J.; Adornetto, A.; Secondo, A.; Boscia, F.; Esposito, A.; Polishchuk, E.V.; Polishchuk, R.S.; Molinaro, P.; et al. NCX3 regulates mitochondrial Ca2+ handling through the AKAP121-anchored signaling complex and prevents hypoxia-induced neuronal death. J. Cell Sci. 2013, 126, 5566–5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca2+ transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnoczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P. Mitochondria-plasma membrane interactions and communication. J. Biol. Chem. 2021, 297, 101164. [Google Scholar] [CrossRef]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta 2010, 1797, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Sancho, J. The coupling of plasma membrane calcium entry to calcium uptake by endoplasmic reticulum and mitochondria. J. Physiol. 2014, 592, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012, 464, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Zampese, E.; Fasolato, C.; Pozzan, T.; Pizzo, P. Presenilin-2 modulation of ER-mitochondria interactions: FAD mutations, mechanisms and pathological consequences. Commun. Integr. Biol. 2011, 4, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S265–S279. [Google Scholar] [CrossRef] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Y.B.; Giffard, R.G. ER-Mitochondria Crosstalk during Cerebral Ischemia: Molecular Chaperones and ER-Mitochondrial Calcium Transfer. Int. J. Cell Biol. 2012, 2012, 493934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- De Vos, K.J.; Morotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.A.; Elrod, J.W. Mediating ER-mitochondrial cross-talk. Science 2017, 358, 591–592. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Ahuir, A.; Manzanares-Estreder, S.; Proft, M. Pro- and Antioxidant Functions of the Peroxisome-Mitochondria Connection and Its Impact on Aging and Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9860841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bartosz, G. Reactive oxygen species: Destroyers or messengers? Biochem. Pharmacol. 2009, 77, 1303–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox. Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of reactive oxygen species in cell signalling pathways. Biochem. Soc. Trans. 2001, 29, 345–350. [Google Scholar] [CrossRef]
- Metere, A.; Giacomelli, L. Absorption, metabolism and protective role of fruits and vegetables polyphenols against gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5850–5858. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox. Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox. Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S413–S426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68, 1077–1080. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, P.T.; Chen, C.L.; Green, K.B.; Chen, Y.R. Oxidative modifications of mitochondria complex II. Methods Mol. Biol. 2013, 1005, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Allen, R.G. Relationship between metabolic rate, free radicals, differentiation and aging: A unified theory. Basic Life Sci. 1985, 35, 75–104. [Google Scholar] [CrossRef]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Andriantsitohaina, R.; Duluc, L.; Garcia-Rodriguez, J.C.; Gil-del Valle, L.; Guevara-Garcia, M.; Simard, G.; Soleti, R.; Su, D.F.; Velasquez-Perez, L.; Wilson, J.X.; et al. Systems biology of antioxidants. Clin. Sci. 2012, 123, 173–192. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millare, B.; O’Rourke, B.; Trayanova, N. Hydrogen peroxide diffusion and scavenging shapes mitochondrial network instability and failure by sensitizing ROS-induced ROS release. Sci. Rep. 2020, 10, 15758. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Zandalinas, S.I.; Mittler, R. ROS-induced ROS release in plant and animal cells. Free Radic Biol. Med. 2018, 122, 21–27. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Gozalbo, A.; Rodriguez-Blazquez, C.; Forjaz, M.J.; Martinez-Martin, P. Clinical Characterization of Parkinson’s Disease Patients with Cognitive Impairment. Front. Neurol. 2020, 11, 731. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by beta-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Jin, H.; Huang, Y. Mitochondria-associated membranes (MAMs): A potential therapeutic target for treating Alzheimer’s disease. Clin. Sci. 2021, 135, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.H.; Mei, L.; Mak, D.O.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarasija, S.; Norman, K.R. A gamma-Secretase Independent Role for Presenilin in Calcium Homeostasis Impacts Mitochondrial Function and Morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierer, L.M.; Haroutunian, V.; Gabriel, S.; Knott, P.J.; Carlin, L.S.; Purohit, D.P.; Perl, D.P.; Schmeidler, J.; Kanof, P.; Davis, K.L. Neurochemical correlates of dementia severity in Alzheimer’s disease: Relative importance of the cholinergic deficits. J. Neurochem. 1995, 64, 749–760. [Google Scholar] [CrossRef]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanese, C.; Payan-Gomez, C.; Galvani, M.; Molano Gonzalez, N.; Tresini, M.; Nait Abdellah, S.; van Roon-Mom, W.M.C.; Figini, S.; Marinus, J.; van Hilten, J.J.; et al. Peripheral mitochondrial function correlates with clinical severity in idiopathic Parkinson’s disease. Mov. Disord. 2019, 34, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anne, C.; Gasnier, B. Vesicular neurotransmitter transporters: Mechanistic aspects. Curr. Top. Membr. 2014, 73, 149–174. [Google Scholar] [CrossRef]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, V.S.; Berman, S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: Implications for Parkinson’s disease. Neurobiol. Dis. 2013, 51, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafa, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Schumacker, P.T.; Guzman, J.D.; Ilijic, E.; Yang, B.; Zampese, E. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1013–1019. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- German, D.C.; Manaye, K.F.; Sonsalla, P.K.; Brooks, B.A. Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced parkinsonism: Sparing of calbindin-D28k-containing cells. Ann. N. Y. Acad. Sci. 1992, 648, 42–62. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Kostic, M.; Horne, A.; Gandhi, S.; Sekler, I.; Abramov, A.Y. LRRK2 deficiency induced mitochondrial Ca2+ efflux inhibition can be rescued by Na+/Ca2+/Li+ exchanger upregulation. Cell Death Dis. 2019, 10, 265. [Google Scholar] [CrossRef]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 2008, 28, 12305–12317. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Hudec, R.; Lopreiato, R. Huntington’s disease, calcium, and mitochondria. Biofactors 2011, 37, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Zeron, M.M.; Fernandes, H.B.; Krebs, C.; Shehadeh, J.; Wellington, C.L.; Leavitt, B.R.; Baimbridge, K.G.; Hayden, M.R.; Raymond, L.A. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol. Cell Neurosci. 2004, 25, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Pellman, J.J.; Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Ca2+ handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington’s disease. J. Neurochem. 2015, 134, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.; Pellman, J.J.; Brustovetsky, T.; Harris, R.A.; Brustovetsky, N. Oxidative metabolism and Ca2+ handling in isolated brain mitochondria and striatal neurons from R6/2 mice, a model of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2762–2775. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Barrett, E.F.; Barrett, J.N.; David, G. Dysfunctional mitochondrial Ca2+ handling in mutant SOD1 mouse models of fALS: Integration of findings from motor neuron somata and motor terminals. Front. Cell Neurosci. 2014, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275 Pt 1, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Kubalik, N.; Zinchenko, N.; Ridings, D.M.; Radoff, D.A.; Hemendinger, R.; Brooks, B.R.; Bonkovsky, H.L. Metabolic and functional differences between brain and spinal cord mitochondria underlie different predisposition to pathology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R844–R854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Higgins, C.M.; Xu, Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 535–545. [Google Scholar] [CrossRef]
- Tradewell, M.L.; Cooper, L.A.; Minotti, S.; Durham, H.D. Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: Mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 2011, 42, 265–275. [Google Scholar] [CrossRef]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef]
- Ristow, M.; Pfister, M.F.; Yee, A.J.; Schubert, M.; Michael, L.; Zhang, C.Y.; Ueki, K.; Michael, M.D., 2nd; Lowell, B.B.; Kahn, C.R. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12239–12243. [Google Scholar] [CrossRef] [Green Version]
- Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal. Signal. 2018, 2, NS20180061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMauro, S.; Hirano, M.; Schon, E.A. Approaches to the treatment of mitochondrial diseases. Muscle Nerve 2006, 34, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, S.; Mancuso, M.; Naini, A. Mitochondrial encephalomyopathies: Therapeutic approach. Ann. N. Y. Acad. Sci. 2004, 1011, 232–245. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Mancuso, M. Mitochondrial diseases: Therapeutic approaches. Biosci. Rep. 2007, 27, 125–137. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Flatters, S.J.L. Chapter Five—The Contribution of Mitochondria to Sensory Processing and Pain. In Progress in Molecular Biology and Translational Science; Price, T.J., Dussor, G., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 131, pp. 119–146. [Google Scholar]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated with Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [Green Version]
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H.; et al. Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxid. Redox Signal. 2017, 27, 1048–1066. [Google Scholar] [CrossRef]
- Ghosh, A.; Langley, M.R.; Harischandra, D.S.; Neal, M.L.; Jin, H.; Anantharam, V.; Joseph, J.; Brenza, T.; Narasimhan, B.; Kanthasamy, A.; et al. Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2016, 11, 259–278. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.J.; Nam, Y.; Lee, J.W.; Nguyen, P.T.; Yoo, J.E.; Tran, T.V.; Jeong, J.H.; Jang, C.G.; Oh, Y.J.; Youdim, M.B.H.; et al. N-Methyl, N-propynyl-2-phenylethylamine (MPPE), a Selegiline Analog, Attenuates MPTP-induced Dopaminergic Toxicity with Guaranteed Behavioral Safety: Involvement of Inhibitions of Mitochondrial Oxidative Burdens and p53 Gene-elicited Pro-apoptotic Change. Mol. Neurobiol. 2016, 53, 6251–6269. [Google Scholar] [CrossRef]
- Izumi, Y.; Sawada, H.; Yamamoto, N.; Kume, T.; Katsuki, H.; Shimohama, S.; Akaike, A. Novel neuroprotective mechanisms of pramipexole, an anti-Parkinson drug, against endogenous dopamine-mediated excitotoxicity. Eur. J. Pharmacol. 2007, 557, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Sathe, A.G.; Tuite, P.; Chen, C.; Ma, Y.; Chen, W.; Cloyd, J.; Low, W.C.; Steer, C.J.; Lee, B.Y.; Zhu, X.H.; et al. Pharmacokinetics, Safety, and Tolerability of Orally Administered Ursodeoxycholic Acid in Patients with Parkinson’s Disease-A Pilot Study. J. Clin. Pharmacol. 2020, 60, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2016, 53, 810–817. [Google Scholar] [CrossRef]
- Hawking, Z.L. Alzheimer’s disease: The role of mitochondrial dysfunction and potential new therapies. Biosci. Horiz. Int. J. Stud. Res. 2016, 9, hzw014. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannan, A.J. Novel therapeutic targets for Huntington’s disease. Expert Opin. Ther. Targets 2005, 9, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Lee, M.C.; Yoon, D.S.; Han, J.; Kim, M.; Hwang, U.K.; Jung, J.H.; Lee, J.S. Effects of bisphenol A and its analogs bisphenol F and S on life parameters, antioxidant system, and response of defensome in the marine rotifer Brachionus koreanus. Aquat. Toxicol. 2018, 199, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Drug | Possible Mechanism of Action | Therapeutic Uses | References |
|---|---|---|---|
| Olesoxime | Scavenges the toxic by-products of lipid peroxidation. It reduced the overactivation of calpains. | Antioxidant and neuroprotective activities in NP, ALS, HD, PD, peripheral neuropathy, and spinal muscular atrophy. | [195] |
| Cholest-4-en-3-one | This drug is bound directly to two components of the mitochondrial permeability transition pore: the voltage-dependent anion channel and peripheral benzodiazepine receptor, suggesting a potential mechanism for its neuroprotective activity. | Effective in treating painful diabetic, chemotherapy-induced neuropathies, and ALS. | [195] |
| Vitamin C | Maintains the integrity of cell membranes and organelles, including mitochondrial membranes. This compound acts as a natural antioxidant. | Antioxidant and neuroprotective activities in NP | [197] |
| N-acetyl-cysteine | Reduce both excitotoxicity and oxidative stress through its actions on glutamate reuptake and antioxidant capacity. | It showed antioxidative properties measured by increased blood and brain glutathione levels after single-time point administration | [198,199] |
| Apocynin | Prevents mitochondrial dysfunction as an NADPH oxidase (NOX) inhibitor. Recent studies highlight its off-target effects. It can function as a scavenger of non-radical oxidant species, which is relevant for its activity against NOX 4 mediated hydrogen peroxide production. | Has been adjusted to target mitochondria (Mito-Apo), with preclinical PD models showing that it could prevent mPTP-induced nigral cell loss, indicating its potential use for mitochondrial dysfunction in PD. Reduce ROS successfully and uses various disorders, such as diabetic complications, neurodegeneration, cardiovascular disorders, lung cancer, hepatocellular cancer, pancreatic cancer, and pheochromocytoma. | [200,201] |
| N-Methyl, N-propynyl-2-phenylethylamine (MPPE) | Maintains the integrity of cell membranes and organelles, including mitochondrial membranes. MPPE is a propargylamine-based monoamine oxidase B (MAO-B) inhibitor. | MPPE serves as an MAO-B inhibitor that prevents MPTP-induced nigral cell loss, upregulates mitochondrial superoxide dismutase to alleviate oxidative stress, and improves complex I (CI) function | [202] |
| S(-) enantiomer of pramipexole | Balance the redox state levels of the cell and is a dopamine agonist. | Using dopamine agonists in routine clinical care has also had antioxidative properties. Used in the symptomatic treatment of PD. | [203] |
| Ursodeoxycholic acid | Shown to prevent mitochondrial membrane depolarization and stabilizes cytochrome c in the mitochondrial membrane. | A drug often used in chronic inflammatory liver disease with an extensive safety profile. | [204,205] |
| Vitamin E | Maintains the integrity of cell membranes and organelles, including mitochondrial membranes, by inhibiting peroxidation of the lipids that make up said membranes. | Antioxidant properties in AD and PD. | [206,207] |
| α-Lipoic acid | Acts as a potent antioxidant and decreased a marker of oxidative stress. | Antioxidant properties in AD. | [206,207] |
| Olanzapine | It has affinity for dopamine D1 and D2 receptors, as well as serotonin 5-HT2A receptors. | Antipsychotic agent for the treatment of schizophrenia and other disorders. | [208] |
| Mitoquinone | Produces direct antioxidant action by scavenging peroxyl, peroxynitrite and superoxide. Is a mitochondria-targeted antioxidant that reduces mitochondrial overproduction of ROS. | Antioxidant properties in PD and NDDs. | [209] |
| Melatonin | Direct scavenger of many ROS species such as free radicals, peroxylnitrites, hydroxyls, peroxyls, and other nitrous oxides under normal conditions. | It is mainly used as a dietary supplement for sleep regulation and re-synchronization of disrupted circadian rhythms and antioxidant properties in PD. | [210] |
| Riluzole | Reduces ROS generation via induction of glutathione production. Inhibits the release of glutamic acid from cultured neurons, from brain slices, and from corticostriatal neurons in vivo. | Antioxidant properties in ALS and as neuroprotective, anticonvulsant, and sedative properties. | [179] |
| Dichloroacetate | Dichloroacetate activates the pyruvate dehydrogenase complex and lowers cerebral lactate amounts. | Neuroprotective activity in HD and treatment of mitochondrial genetic diseases. | [208] |
| Z-FA-MK | Anti-apoptotic in function, it also inhibits effector caspases. | Effective therapeutic target in MS. | [211] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. https://doi.org/10.3390/antiox11050801
Matuz-Mares D, González-Andrade M, Araiza-Villanueva MG, Vilchis-Landeros MM, Vázquez-Meza H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants. 2022; 11(5):801. https://doi.org/10.3390/antiox11050801
Chicago/Turabian StyleMatuz-Mares, Deyamira, Martin González-Andrade, Minerva Georgina Araiza-Villanueva, María Magdalena Vilchis-Landeros, and Héctor Vázquez-Meza. 2022. "Mitochondrial Calcium: Effects of Its Imbalance in Disease" Antioxidants 11, no. 5: 801. https://doi.org/10.3390/antiox11050801
APA StyleMatuz-Mares, D., González-Andrade, M., Araiza-Villanueva, M. G., Vilchis-Landeros, M. M., & Vázquez-Meza, H. (2022). Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants, 11(5), 801. https://doi.org/10.3390/antiox11050801