
The Systemic Effects of Exercise on the Systemic Effects of Alzheimer’s Disease

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. AD and Exercise
3. Exercise and Peripheral Organs with AD
4. Exercise and the Cardiovascular System with AD
5. Exercise and Gut Microbiome in AD
6. Exercise and Liver with AD
7. Exercise and Gonads with AD
8. Exercise and Kidney in AD
9. The Effects of Exercise on Peripheral Organs in Alzheimer’s Disease
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef]
- Hill, J.M.; Lukiw, W.J. Microbial-generated amyloids and Alzheimer’s disease (AD). Front. Aging Neurosci. 2015, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Tiraboschi, P.; Hansen, L.A.; Thal, L.J.; Corey-Bloom, J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 2004, 62, 1984–1989. [Google Scholar] [CrossRef]
- Pomilio, C.; Gorojod, R.M.; Riudavets, M.; Vinuesa, A.; Presa, J.; Gregosa, A.; Bentivegna, M.; Alaimo, A.; Alcon, S.P.; Sevlever, G.; et al. Microglial autophagy is impaired by prolonged exposure to β-amyloid peptides: Evidence from experimental models and Alzheimer’s disease patients. GeroScience 2020, 42, 613–632. [Google Scholar] [CrossRef]
- Turner, R.S.; Stubbs, T.; Davies, D.A.; Albensi, B.C. Potential new approaches for diagnosis of Alzheimer’s disease and related dementias. Front. Neurol. 2020, 11, 496. [Google Scholar] [CrossRef]
- Serý, O.; Povová, J.; Míšek, I.; Pešák, L.; Janout, V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol. 2013, 51, 1–9. [Google Scholar] [CrossRef]
- Tavares, R.S.; Martins, S.; Almeida-Santos, T.; Sousa, A.P.; Ramalho-Santos, J.; da Cruz, E.S.O.A. Alzheimer’s disease-related amyloid-β(1-42) peptide induces the loss of human sperm function. Cell Tissue Res. 2017, 369, 647–651. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship between exercise and Alzheimer’s disease: A narrative literature review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Doody, R.; Kurz, A.; Mohs, R.C.; Morris, J.C.; Rabins, P.V.; Ritchie, K.; Rossor, M.; Thal, L.; Winblad, B. Current concepts in mild cognitive impairment. Arch. Neurol. 2001, 58, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Amieva, H.; Jacqmin-Gadda, H.; Orgogozo, J.M.; le Carret, N.; Helmer, C.; Letenneur, L.; Barberger-Gateau, P.; Fabrigoule, C.; Dartigues, J.F. The 9 year cognitive decline before dementia of the Alzheimer type: A prospective population-based study. Brain J. Neurol. 2005, 128, 1093–1101. [Google Scholar] [CrossRef]
- Griffith, C.M.; Eid, T.; Rose, G.M.; Patrylo, P.R. Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology 2018, 136, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Tarantini, S.; Fülöp, G.A.; Kiss, T.; Valcarcel-Ares, M.N.; Galvan, V.; Ungvari, Z.; Yabluchanskiy, A. Hypertension impairs neurovascular coupling and promotes microvascular injury: Role in exacerbation of Alzheimer’s disease. GeroScience 2017, 39, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer’s disease a systemic disease? Biochim. Biophys. Acta 2014, 1842, 1340–1349. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Han, P.; Tang, Z.; Yin, J.; Maalouf, M.; Beach, T.G.; Reiman, E.M.; Shi, J. Pituitary adenylate cyclase-activating polypeptide protects against β-amyloid toxicity. Neurobiol. Aging 2014, 35, 2064–2071. [Google Scholar] [CrossRef]
- Lee, E.H.; Seo, S.R. Neuroprotective roles of pituitary adenylate cyclase-activating polypeptide in neurodegenerative diseases. BMB Rep. 2014, 47, 369–375. [Google Scholar] [CrossRef]
- Reglodi, D.; Atlasz, T.; Szabo, E.; Jungling, A.; Tamas, A.; Juhasz, T.; Fulop, B.D.; Bardosi, A. PACAP deficiency as a model of aging. GeroScience 2018, 40, 437–452. [Google Scholar] [CrossRef]
- Reglodi, D.; Jungling, A.; Longuespée, R.; Kriegsmann, J.; Casadonte, R.; Kriegsmann, M.; Juhasz, T.; Bardosi, S.; Tamas, A.; Fulop, B.D.; et al. Accelerated pre-senile systemic amyloidosis in PACAP knockout mice—A protective role of PACAP in age-related degenerative processes. J. Pathol. 2018, 245, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Liang, W.; Baxter, L.C.; Yin, J.; Tang, Z.; Beach, T.G.; Caselli, R.J.; Reiman, E.M.; Shi, J. Pituitary adenylate cyclase-activating polypeptide is reduced in Alzheimer disease. Neurology 2014, 82, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Kienlen Campard, P.; Crochemore, C.; René, F.; Monnier, D.; Koch, B.; Loeffler, J.P. PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. DNA Cell Biol. 1997, 16, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Juhász, T.; Matta, C.; Katona, É.; Somogyi, C.; Takács, R.; Hajdú, T.; Helgadottir, S.L.; Fodor, J.; Csernoch, L.; Tóth, G.; et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) signalling enhances osteogenesis in UMR-106 cell line. J. Mol. Neurosci. 2014, 54, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Jhanji, N. Natural products as potential anti-Alzheimer agents. Curr. Med. Chem. 2020, 27, 5887–5917. [Google Scholar] [CrossRef]
- Whitehouse, P.J. Ethical issues in early diagnosis and prevention of Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 101–108. [Google Scholar] [CrossRef]
- Raggi, A.; Tasca, D.; Ferri, R. A brief essay on non-pharmacological treatment of Alzheimer’s disease. Rev. Neurosci. 2017, 28, 587–597. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Tanzi, R.E.; George-Hyslop, P.S.; Gusella, J.F. Molecular genetics of Alzheimer disease amyloid. J. Biol. Chem. 1991, 266, 20579–20582. [Google Scholar] [CrossRef]
- Price, D.L.; Borchelt, D.R.; Sisodia, S.S. Alzheimer disease and the prion disorders amyloid beta-protein and prion protein amyloidoses. Proc. Natl. Acad. Sci. USA 1993, 90, 6381–6384. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Hart, N.; Sarga, L.; Koltai, E.; Atalay, M.; Ohno, H.; Boldogh, I. Exercise plays a preventive role against Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Torma, F.; Berkes, I.; Goto, S.; Mimura, T.; Posa, A.; Balogh, L.; Boldogh, I.; Suzuki, K.; Higuchi, M.; et al. Exercise effects on physiological function during aging. Free Radic. Biol. Med. 2019, 132, 33–41. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Mandal, A.; Chanda, D.; Chakraborti, S. Oxidant, antioxidant and physical exercise. Mol. Cell. Biochem. 2003, 253, 307–312. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Martin, B.; Maudsley, S. Anti-inflammatory effects of physical activity in relationship to improved cognitive status in humans and mouse models of Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 86–92. [Google Scholar] [CrossRef]
- Liu, H.L.; Zhao, G.; Zhang, H.; Shi, L.D. Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav. Brain Res. 2013, 256, 261–272. [Google Scholar] [CrossRef]
- Pereira, T.M.C.; Côco, L.Z.; Ton, A.M.M.; Meyrelles, S.S.; Campos-Toimil, M.; Campagnaro, B.P.; Vasquez, E.C. The emerging scenario of the gut-brain axis: The therapeutic actions of the new actor kefir against neurodegenerative diseases. Antioxidants 2021, 10, 1845. [Google Scholar] [CrossRef]
- De Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory cascade in Alzheimer’s disease pathogenesis: A review of experimental findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef] [PubMed]
- Van Valkenburgh, J.; Meuret, C.; Martinez, A.E.; Kodancha, V.; Solomon, V.; Chen, K.; Yassine, H.N. Understanding the exchange of systemic HDL particles into the brain and vascular cells has diagnostic and therapeutic implications for neurodegenerative diseases. Front. Physiol. 2021, 12, 700847. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef]
- Kheirbakhsh, R.; Haddadi, M.; Muhammadnejad, A.; Abdollahi, A.; Shahi, F.; Amanpour-Gharaei, B.; Abrahim-Habibi, A.; Barati, T.; Amanpour, S. Long-term behavioral, histological, biochemical and hematological evaluations of amyloid beta-induced Alzheimer’s disease in rat. Acta Neurobiol. Exp. 2018, 78, 51–59. [Google Scholar] [CrossRef]
- Arbor, S. Targeting amyloid precursor protein shuttling and processing—Long before amyloid beta formation. Neural Regen. Res. 2017, 12, 207–209. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Téglás, T.; Ábrahám, D.; Jókai, M.; Kondo, S.; Mohammadi, R.; Fehér, J.; Szabó, D.; Wilhelm, M.; Radák, Z. Exercise combined with a probiotics treatment alters the microbiome, but moderately affects signalling pathways in the liver of male APP/PS1 transgenic mice. Biogerontology 2020, 21, 807–815. [Google Scholar] [CrossRef]
- Szegeczki, V.; Horváth, G.; Perényi, H.; Tamás, A.; Radák, Z.; Ábrahám, D.; Zákány, R.; Reglodi, D.; Juhász, T. Alzheimer’s disease mouse as a model of testis degeneration. Int. J. Mol. Sci. 2020, 21, 5726. [Google Scholar] [CrossRef]
- Perényi, H.; Szegeczki, V.; Horváth, G.; Hinnah, B.; Tamás, A.; Radák, Z.; Ábrahám, D.; Zákány, R.; Reglodi, D.; Juhász, T. Physical activity protects the pathological alterations of Alzheimer’s disease kidneys via the activation of PACAP and BMP signaling pathways. Front. Cell. Neurosci. 2020, 14, 243. [Google Scholar] [CrossRef]
- Szegeczki, V.; Perényi, H.; Horváth, G.; Hinnah, B.; Tamás, A.; Radák, Z.; Ábrahám, D.; Zákány, R.; Reglodi, D.; Juhász, T. Physical training inhibits the fibrosis formation in Alzheimer’s disease kidney influencing the TGFβ signaling pathways. J. Alzheimer’s Dis. 2021, 81, 1195–1209. [Google Scholar] [CrossRef]
- Zheng, H.; Cai, A.; Shu, Q.; Niu, Y.; Xu, P.; Li, C.; Lin, L.; Gao, H. Tissue-specific metabolomics analysis identifies the liver as a major organ of metabolic disorders in amyloid precursor protein/presenilin 1 mice of Alzheimer’s disease. J. Proteome Res. 2019, 18, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and neurovascular function in the healthy and diseased brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.P.; Baum, L. Lipoproteins and related molecules in Alzheimer’s disease. Microsc. Res. Tech. 2000, 50, 259–260. [Google Scholar] [CrossRef]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Shi, J.; Yuan, G.; Shou, X.; Chen, T.; Zhu, X.; Yang, Y.; Hu, Y. Causal association between heart failure and Alzheimer’s disease: A two-sample bidirectional mendelian randomization study. Front. Genet. 2021, 12, 772343. [Google Scholar] [CrossRef]
- Haider, A.W.; Larson, M.G.; Benjamin, E.J.; Levy, D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 1998, 32, 1454–1459. [Google Scholar] [CrossRef]
- Haring, B.; Omidpanah, A.; Suchy-Dicey, A.M.; Best, L.G.; Verney, S.P.; Shibata, D.K.; Cole, S.A.; Ali, T.; Howard, B.V.; Buchwald, D.; et al. Left ventricular mass, brain magnetic resonance imaging, and cognitive performance: Results from the strong heart study. Hypertension 2017, 70, 964–971. [Google Scholar] [CrossRef]
- Frenzel, S.; Wittfeld, K.; Bülow, R.; Völzke, H.; Friedrich, N.; Habes, M.; Felix, S.B.; Dörr, M.; Grabe, H.J.; Bahls, M. Cardiac hypertrophy is associated with advanced brain aging in the general population. J. Am. Heart Assoc. 2021, 10, e020994. [Google Scholar] [CrossRef]
- Cho, Y.K.; Lee, J.; Kim, H.S.; Park, J.Y.; Lee, W.J.; Kim, Y.J.; Jung, C.H. The risk of Alzheimer’s disease according to dynamic changes in metabolic health and obesity: A nationwide population-based cohort study. Aging 2021, 13, 16974–16989. [Google Scholar] [CrossRef]
- Singh, S.; Schwarz, K.; Horowitz, J.; Frenneaux, M. Cardiac energetic impairment in heart disease and the potential role of metabolic modulators: A review for clinicians. Circ. Cardiovasc. Genet. 2014, 7, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Le, T.N.V.; Fedorova, J.; Yang, Y.; Krause-Hauch, M.; Davitt, K.; Zoungrana, L.I.; Fatmi, M.K.; Lesnefsky, E.J.; Li, J.; et al. The cardiac dysfunction caused by metabolic alterations in Alzheimer’s disease. Front. Cardiovasc. Med. 2022, 9, 850538. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, V.A.; Fagard, R.H. Effects of endurance training on blood pressure, blood pressure-regulating mechanisms, and cardiovascular risk factors. Hypertension 2005, 46, 667–675. [Google Scholar] [CrossRef]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Koike, M.A.; Green, K.N.; Blurton-Jones, M.; Laferla, F.M. Oligemic hypoperfusion differentially affects tau and amyloid-β. Am. J. Pathol. 2010, 177, 300–310. [Google Scholar] [CrossRef]
- Atochin, D.N.; Huang, P.L. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflug. Archiv.-Eur. J. Physiol. 2010, 460, 965–974. [Google Scholar] [CrossRef]
- Moncada, S. Nitric oxide in the vasculature: Physiology and pathophysiology. Ann. N. Y. Acad. Sci. 1997, 811, 60–69, 60–67; discussion 67–69. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Austin, S.A. Endothelial nitric oxide: Protector of a healthy mind. Eur. Heart J. 2014, 35, 888–894. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Kahouli, I.; Dia, S.Y.; Singh, S.P.; Prakash, S. Microbiome, probiotics and neurodegenerative diseases: Deciphering the gut brain axis. Cell. Mol. Life Sci. 2017, 74, 3769–3787. [Google Scholar] [CrossRef]
- Kim, B.S.; Jeon, Y.S.; Chun, J. Current status and future promise of the human microbiome. Pediatric Gastroenterol. Hepatol. Nutr. 2013, 16, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Clement, C.; Pogue, A.I.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front. Aging Neurosci. 2014, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Mahony, S.M. The microbiome-gut-brain axis: From bowel to behavior. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2011, 23, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. 2013, 7, 153. [Google Scholar] [CrossRef]
- Bell, J.S.; Spencer, J.I.; Yates, R.L.; Yee, S.A.; Jacobs, B.M.; DeLuca, G.C. Invited review: From nose to gut—The role of the microbiome in neurological disease. Neuropathol. Appl. Neurobiol. 2019, 45, 195–215. [Google Scholar] [CrossRef]
- Hu, X.; Wang, T.; Jin, F. Alzheimer’s disease and gut microbiota. Sci. China Life Sci. 2016, 59, 1006–1023. [Google Scholar] [CrossRef]
- Szablewski, L. Human gut microbiota in health and Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 549–560. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis in health and disease. Gastroenterol. Clin. North Am. 2017, 46, 77–89. [Google Scholar] [CrossRef]
- Parker, A.P.; Dean, D.H. Temperate Bacillus bacteriophage SP16 genome is circularly permuted and terminally redundant. J. Bacteriol. 1986, 167, 719–721. [Google Scholar] [CrossRef]
- Nho, K.; Kueider-Paisley, A.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; Kastenmüller, G.; et al. Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: Relationship to neuroimaging and CSF biomarkers. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.; Feher, J.; Scuderi, G.L.; Szabo, D.; Dobolyi, A.; Cservenak, M.; Juhasz, J.; Ligeti, B.; Pongor, S.; Gomez-Cabrera, M.C.; et al. Exercise and probiotics attenuate the development of Alzheimer’s disease in transgenic mice: Role of microbiome. Exp. Gerontol. 2019, 115, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.M.; Leonel, A.J.; Melo, M.A.; Santos, R.R.; Cara, D.C.; Cardoso, V.N.; Correia, M.I.; Alvarez-Leite, J.I. Oral supplementation of butyrate reduces mucositis and intestinal permeability associated with 5-Fluorouracil administration. Lipids 2012, 47, 669–678. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Tabat, M.W.; Marques, T.M.; Markgren, M.; Löfvendahl, L.; Brummer, R.J.; Wall, R. Acute effects of butyrate on induced hyperpermeability and tight junction protein expression in human colonic tissues. Biomolecules 2020, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Leblhuber, F.; Steiner, K.; Schuetz, B.; Fuchs, D.; Gostner, J.M. Probiotic supplementation in patients with Alzheimer’s dementia—An explorative intervention study. Curr. Alzheimer Res. 2018, 15, 1106–1113. [Google Scholar] [CrossRef]
- O’Sullivan, O.; Cronin, O.; Clarke, S.F.; Murphy, E.F.; Molloy, M.G.; Shanahan, F.; Cotter, P.D. Exercise and the microbiota. Gut Microbes 2015, 6, 131–136. [Google Scholar] [CrossRef]
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise alters gut microbiota composition and function in lean and obese humans. Med. Sci. Sports Exerc. 2018, 50, 747–757. [Google Scholar] [CrossRef]
- Pan, G.Z.; Chen, S.P.; Mai, C.R. Ranitidine in acute duodenal ulcer. Double blind controlled trial. Chin. Med. J. 1988, 101, 277–279. [Google Scholar]
- Greenhill, C. Gut microbiome influences exercise response. Nat. Rev. Endocrinol. 2020, 16, 68–69. [Google Scholar] [CrossRef]
- Grosicki, G.J.; Durk, R.P.; Bagley, J.R. Rapid gut microbiome changes in a world-class ultramarathon runner. Physiol. Rep. 2019, 7, e14313. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; de Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Quadri, P.; Fragiacomo, C.; Pezzati, R.; Zanda, E.; Forloni, G.; Tettamanti, M.; Lucca, U. Homocysteine, folate, and vitamin B-12 in mild cognitive impairment, Alzheimer disease, and vascular dementia. Am. J. Clin. Nutr. 2004, 80, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.; Puścion-Jakubik, A.; Markiewicz-Żukowska, R.; Socha, K. Vitamins in Alzheimer’s disease—Review of the latest reports. Nutrients 2020, 12, 3458. [Google Scholar] [CrossRef]
- King, B.C.; Vavitsas, K.; Ikram, N.K.; Schrøder, J.; Scharff, L.B.; Bassard, J.; Hamberger, B.; Jensen, P.E.; Simonsen, H.T. Corrigendum: In vivo assembly of DNA-fragments in the moss, Physcomitrella patens. Sci. Rep. 2016, 6, 31261. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Q. Towards understanding brain-gut-microbiome connections in Alzheimer’s disease. BMC Syst. Biol. 2016, 10 (Suppl. 3), 63. [Google Scholar] [CrossRef]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver dysfunction as a novel player in Alzheimer’s progression: Looking outside the brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef]
- Nho, K.; Kueider-Paisley, A.; Ahmad, S.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; et al. Association of altered liver enzymes with Alzheimer disease diagnosis, cognition, neuroimaging measures, and cerebrospinal fluid biomarkers. JAMA Netw. Open 2019, 2, e197978. [Google Scholar] [CrossRef]
- Torma, F.; Koltai, E.; Nagy, E.; Ziaaldini, M.M.; Posa, A.; Koch, L.G.; Britton, S.L.; Boldogh, I.; Radak, Z. Exercise increases markers of spermatogenesis in rats selectively bred for low running capacity. PLoS ONE 2014, 9, e114075. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Prisco, M.; Rosati, L.; Morgillo, E.; Mollica, M.P.; Agnese, M.; Andreuccetti, P.; Valiante, S. Pituitary adenylate cyclase-activating peptide (PACAP) and its receptors in Mus musculus testis. Gen. Comp. Endocrinol. 2020, 286, 113297. [Google Scholar] [CrossRef] [PubMed]
- Spinedi, E.; Cardinali, D.P. The polycystic ovary syndrome and the metabolic syndrome: A possible chronobiotic-cytoprotective adjuvant therapy. Int. J. Endocrinol. 2018, 2018, 1349868. [Google Scholar] [CrossRef] [PubMed]
- González, F. Inflammation in polycystic ovary syndrome: Underpinning of insulin resistance and ovarian dysfunction. Steroids 2012, 77, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Sarahian, N.; Sarvazad, H.; Sajadi, E.; Rahnejat, N.; Eskandari Roozbahani, N. Investigation of common risk factors between polycystic ovary syndrome and Alzheimer’s disease: A narrative review. Reprod. Health 2021, 18, 156. [Google Scholar] [CrossRef]
- Woodward, A.; Klonizakis, M.; Broom, D. Exercise and polycystic ovary syndrome. Adv. Exp. Med. Biol. 2020, 1228, 123–136. [Google Scholar] [CrossRef]
- Davey, D.A. Alzheimer’s disease, dementia, mild cognitive impairment and the menopause: A ‘window of opportunity’? Women’s Health 2013, 9, 279–290. [Google Scholar] [CrossRef]
- Thomas, A.; Daley, A.J. Women’s views about physical activity as a treatment for vasomotor menopausal symptoms: A qualitative study. BMC Women’s Health 2020, 20, 203. [Google Scholar] [CrossRef]
- Davey, D.A. Prevention of Alzheimer’s disease, cerebrovascular disease and dementia in women: The case for menopause hormone therapy. Neurodegener. Dis. Manag. 2017, 7, 85–94. [Google Scholar] [CrossRef]
- Ghiso, J.; Calero, M.; Matsubara, E.; Governale, S.; Chuba, J.; Beavis, R.; Wisniewski, T.; Frangione, B. Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett. 1997, 408, 105–108. [Google Scholar] [CrossRef]
- Kiuchi, Y.; Isobe, Y.; Fukushima, K. Type IV collagen prevents amyloid beta-protein fibril formation. Life Sci. 2002, 70, 1555–1564. [Google Scholar] [CrossRef]
- Kajdaniuk, D.; Marek, B.; Borgiel-Marek, H.; Kos-Kudła, B. Transforming growth factor β1 (TGFβ1) in physiology and pathology. Endokrynol. Pol. 2013, 64, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, T.; Lu, D.W.; Zhao, H.; Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Zhao, Y.Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018, 101, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Ovase, R. Cerebrovascular transforming growth factor-beta contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 2002, 160, 1583–1587. [Google Scholar] [CrossRef]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Browne, J.A.; Liu, X.; Schnaper, H.W.; Hayashida, T. Serine-204 in the linker region of Smad3 mediates the collagen-I response to TGF-β in a cell phenotype-specific manner. Exp. Cell Res. 2013, 319, 2928–2937. [Google Scholar] [CrossRef]
- Ma, F.Y.; Sachchithananthan, M.; Flanc, R.S.; Nikolic-Paterson, D.J. Mitogen activated protein kinases in renal fibrosis. Front. Biosci. 2009, 1, 171–187. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Deng, D.; Zhang, Q.; Liu, W. Renalase protects against renal fibrosis by inhibiting the activation of the ERK signaling pathways. Int. J. Mol. Sci. 2017, 18, 855. [Google Scholar] [CrossRef]
- Lee, J.; An, J.N.; Hwang, J.H.; Lee, H.; Lee, J.P.; Kim, S.G. p38 MAPK activity is associated with the histological degree of interstitial fibrosis in IgA nephropathy patients. PLoS ONE 2019, 14, e0213981. [Google Scholar] [CrossRef]
- Stambe, C.; Atkins, R.C.; Tesch, G.H.; Masaki, T.; Schreiner, G.F.; Nikolic-Paterson, D.J. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J. Am. Soc. Nephrol. 2004, 15, 370–379. [Google Scholar] [CrossRef]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.H.; Nikolic-Paterson, D.J. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 2007, 18, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Models Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Zhao, H.; Dong, Y.; Tian, X.; Tan, T.K.; Liu, Z.; Zhao, Y.; Zhang, Y.; Harris, D.; Zheng, G. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases. World J. Nephrol. 2013, 2, 84–89. [Google Scholar] [CrossRef]
- Liu, Y.; Shang, D. Transforming growth factor-β1 enhances proliferative and metastatic potential by up-regulating lymphoid enhancer-binding factor 1/integrin αMβ2 in human renal cell carcinoma. Mol. Cell. Biochem. 2020, 465, 165–174. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef]
- Park, G.; Nhan, H.S.; Tyan, S.H.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase activation and caspase-mediated cleavage of APP is associated with amyloid β-protein-induced synapse loss in Alzheimer’s disease. Cell Rep. 2020, 31, 107839. [Google Scholar] [CrossRef]
- Horvath, G.; Opper, B.; Reglodi, D. The neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) is protective in inflammation and oxidative stress-induced damage in the kidney. Int. J. Mol. Sci. 2019, 20, 4944. [Google Scholar] [CrossRef]
- Li, M.; Maderdrut, J.L.; Lertora, J.J.; Arimura, A.; Batuman, V. Renoprotection by pituitary adenylate cyclase-activating polypeptide in multiple myeloma and other kidney diseases. Regul. Pept. 2008, 145, 24–32. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Pavelock, K.A.; Girard, B.M.; Schutz, K.C.; Braas, K.M.; May, V. Bone morphogenetic protein down-regulation of neuronal pituitary adenylate cyclase-activating polypeptide and reciprocal effects on vasoactive intestinal peptide expression. J. Neurochem. 2007, 100, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Yadav, P.S.; Prashar, P. BMP signaling in development and diseases: A pharmacological perspective. Biochem. Pharmacol. 2013, 85, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Józsa, G.; Fülöp, B.D.; Kovács, L.; Czibere, B.; Szegeczki, V.; Kiss, T.; Hajdú, T.; Tamás, A.; Helyes, Z.; Zákány, R.; et al. Lack of pituitary adenylate cyclase-activating polypeptide (PACAP) disturbs callus formation. J. Mol. Neurosci. 2021, 71, 1543–1555. [Google Scholar] [CrossRef]
- Laszlo, E.; Juhasz, T.; Varga, A.; Czibere, B.; Kovacs, K.; Degrell, P.; Horvath, G.; Jancso, G.; Szakaly, P.; Tamas, A.; et al. Protective effect of PACAP on ischemia/reperfusion-induced kidney injury of male and female rats: Gender differences. J. Mol. Neurosci. 2019, 68, 408–419. [Google Scholar] [CrossRef]
- Józsa, G.; Szegeczki, V.; Pálfi, A.; Kiss, T.; Helyes, Z.; Fülöp, B.; Cserháti, C.; Daróczi, L.; Tamás, A.; Zákány, R.; et al. Signalling alterations in bones of pituitary adenylate cyclase activating polypeptide (PACAP) gene deficient mice. Int. J. Mol. Sci. 2018, 19, 2538. [Google Scholar] [CrossRef]
- Von Bubnoff, A.; Cho, K.W. Intracellular BMP signaling regulation in vertebrates: Pathway or network? Dev. Biol. 2001, 239, 1–14. [Google Scholar] [CrossRef]
- Matsubara, T.; Araki, M.; Abe, H.; Ueda, O.; Jishage, K.; Mima, A.; Goto, C.; Tominaga, T.; Kinosaki, M.; Kishi, S.; et al. Bone morphogenetic protein 4 and Smad1 mediate extracellular matrix production in the development of diabetic nephropathy. Diabetes 2015, 64, 2978–2990. [Google Scholar] [CrossRef][Green Version]
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef]
- Qian, W.; Yin, X.; Hu, W.; Shi, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X.; Liu, F. Activation of protein phosphatase 2B and hyperphosphorylation of tau in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 23, 617–627. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Henriques, A.G.; Martins, F.; Rebelo, S.; da Cruz e Silva, O.A. Amyloid-β modulates both AβPP and tau phosphorylation. J. Alzheimer’s Dis. 2015, 45, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Leong, W.; Xu, W.; Wang, B.; Gao, S.; Zhai, X.; Wang, C.; Gilson, E.; Ye, J.; Lu, Y. PP2A subunit PPP2R2C is downregulated in the brains of Alzheimer’s transgenic mice. Aging 2020, 12, 6880–6890. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.G.; Coffee, R.L., Jr.; Zhang, H.; Pelech, S.; Strack, S.; Wadzinski, B.E. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J. Biol. Chem. 2005, 280, 42644–42654. [Google Scholar] [CrossRef]
- Molkentin, J.D. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef]


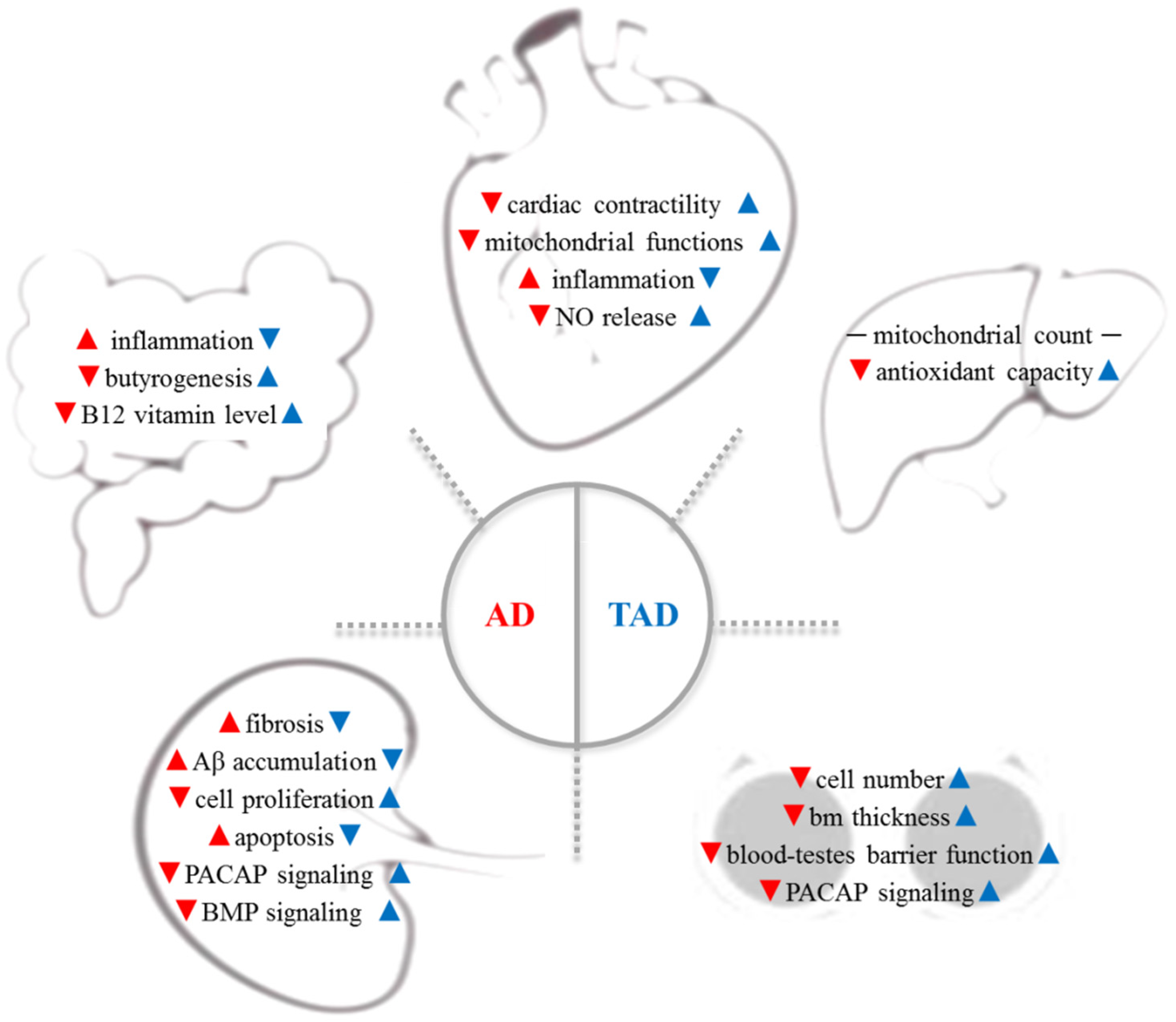{kind=link}
| Changes | AD Mice | TAD Mice | Decisive Study | |
|---|---|---|---|---|
| Cardivascular system | Cardiac contractility | ↓ | ↑ | |
| Mitochondrial functions | ↓ | ↑ | ||
| Inflammation | ↑ | ↓ | ||
| NO release | ↓ | ↑ | ||
| Gut Microbiome | Inflammation | ↑ | ↓ | Abraham et al. [82] |
| Butyrogenesis | ↓ | ↑ | ||
| B12 vitamin level | ↓ | ↑ | ||
| Bacteroides | ↓ | ↑ | ||
| Lactobacillus | ↓ | ↑ | ||
| Prevotella | ↓ | ↑ | ||
| Liver | Mitochondrial antioxidant capacity | ↓ | ↑ | Téglás et al. [47] |
| NRF-2 | ↓ | ↑ | ||
| SOD2 | ↓ | ↑ | ||
| Gonades (testes) | Cell numbers | Szegeczki et al. [48] | ||
| Spermatogonia count | ↓ | ↑ | ||
| Spermatocytes count | ↓ | ↑ | ||
| Leydig cells count | ↓ | ↑ | ||
| Collagen type IV | ↓ | ↑ | ||
| Basement membrane thickness | ↓ | ↑ | ||
| Blood-testes barrier function | ↓ | ↑ | ||
| PACAP signaling | ||||
| PAC1R | ↓ | ↑ | ||
| VPAC1R | - | ↑ | ||
| VPAC2R | - | ↓ | ||
| cAMP | ↓ | - | ||
| PKA | ↓ | ↑ | ||
| P-PKA | ↓ | ↑ | ||
| PP2A | ↓ | ↑ | ||
| Kidney | Aβ accumulation | ↑ | ↓ | Perényi et al. [49] |
| Basement membrane formation | ||||
| Collagen type IV | ↓ | ↑ | ||
| Fibrosis | ||||
| Collagen type I | ↑ | ↓ | ||
| TGFβ1 | - | ↑ | ||
| TGFβRI | ↓ | ↑ | ||
| TGFβRII | ↑ | ↓ | ||
| ERK1/2 | ↓ | ↑ | ||
| Phospho ERK1/2 | ↑ | ↓ | ||
| p38 | ↑ | ↓ | ||
| phospho 38 | ↓ | ↑ | ||
| JNK | ↓ | ↑ | ||
| MMP9 | ↑ | ↑↑ | ||
| Cell proliferation | ||||
| CDKN1/p21 | ↑ | ↓ | ||
| PCNA | ↓ | ↑ | ||
| Apoptosis Cleaved caspase 3 | ↑ | ↓ | ||
| PACAP signaling | ||||
| PAC1R | ↓↓ | ↑↑ | ||
| VPAC1R | ↓↓ | ↑↑ | ||
| VPAC2R | ↓ | ↑ | ||
| PKA | ↓ | ↑ | ||
| CREB | ↓ | - | ||
| Phospho CBEB | ↓↓ | ↑ | ||
| BMP1R | ↓ | ↑ | ||
| BMP4 | ↓ | ↑ | ||
| Smad1 | ↓ | ↑ | ||
| Smad2 | ↑ | ↓ | ||
| Smad3 | ↓ | ↑ | ||
| PP2A | ↓ | ↑ | ||
| PP2B | ↑ | ↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aczel, D.; Gyorgy, B.; Bakonyi, P.; BukhAri, R.; Pinho, R.; Boldogh, I.; Yaodong, G.; Radak, Z. The Systemic Effects of Exercise on the Systemic Effects of Alzheimer’s Disease. Antioxidants 2022, 11, 1028. https://doi.org/10.3390/antiox11051028
Aczel D, Gyorgy B, Bakonyi P, BukhAri R, Pinho R, Boldogh I, Yaodong G, Radak Z. The Systemic Effects of Exercise on the Systemic Effects of Alzheimer’s Disease. Antioxidants. 2022; 11(5):1028. https://doi.org/10.3390/antiox11051028
Chicago/Turabian StyleAczel, Dora, Bernadett Gyorgy, Peter Bakonyi, RehAn BukhAri, Ricardo Pinho, Istvan Boldogh, Gu Yaodong, and Zsolt Radak. 2022. "The Systemic Effects of Exercise on the Systemic Effects of Alzheimer’s Disease" Antioxidants 11, no. 5: 1028. https://doi.org/10.3390/antiox11051028
APA StyleAczel, D., Gyorgy, B., Bakonyi, P., BukhAri, R., Pinho, R., Boldogh, I., Yaodong, G., & Radak, Z. (2022). The Systemic Effects of Exercise on the Systemic Effects of Alzheimer’s Disease. Antioxidants, 11(5), 1028. https://doi.org/10.3390/antiox11051028