
Loss and Recovery of Glutaredoxin 5 Is Inducible by Diet in a Murine Model of Diabesity and Mediated by Free Fatty Acids In Vitro

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
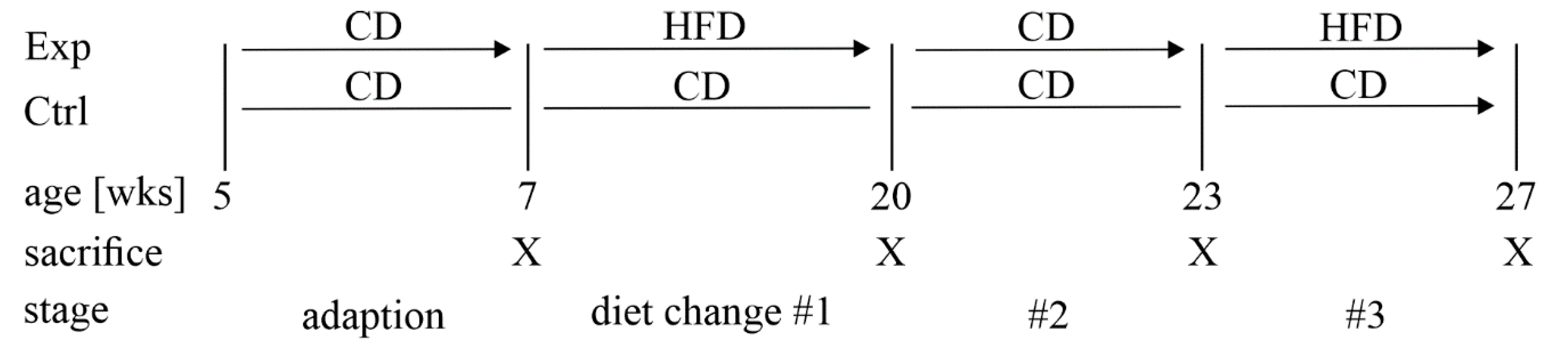
2.1. Research Animals
2.2. Pancreatic Islet Isolation
2.3. Immunohistochemistry
2.4. ROS Production Analysis
2.5. mRNA Expression Analysis
2.6. Glucose Stimulation of Pancreatic Islets
2.7. Cell Culture
2.8. Cell Metabolic Activity
2.9. Immunocytochemistry
2.10. Determination of Cellular ATP Levels
2.11. Mitochondrial Respiration
2.12. ELISA Analysis
2.13. Statistical Analysis
3. Results
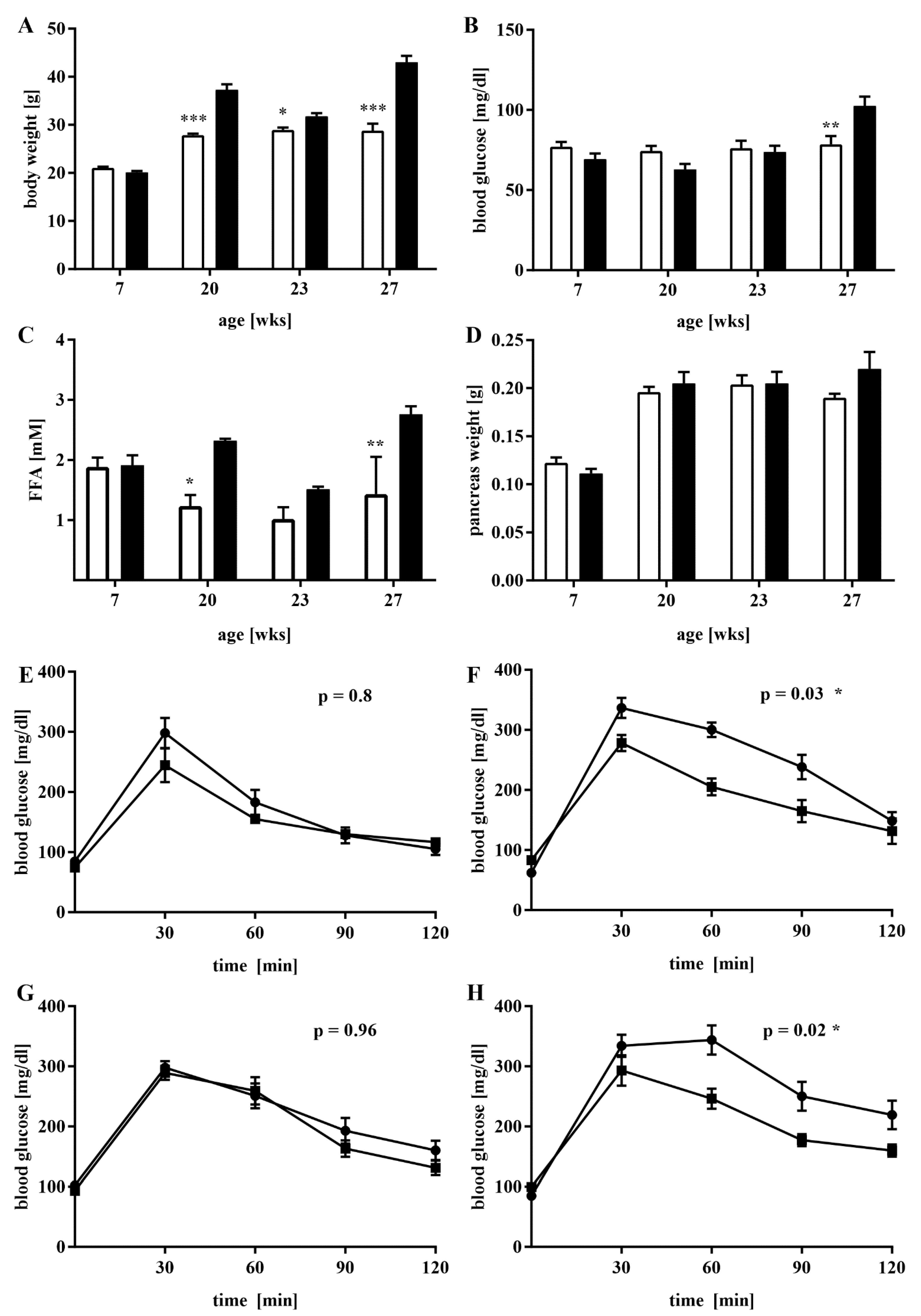
3.1. A Mouse Model of Dietary-Induced Diabesity
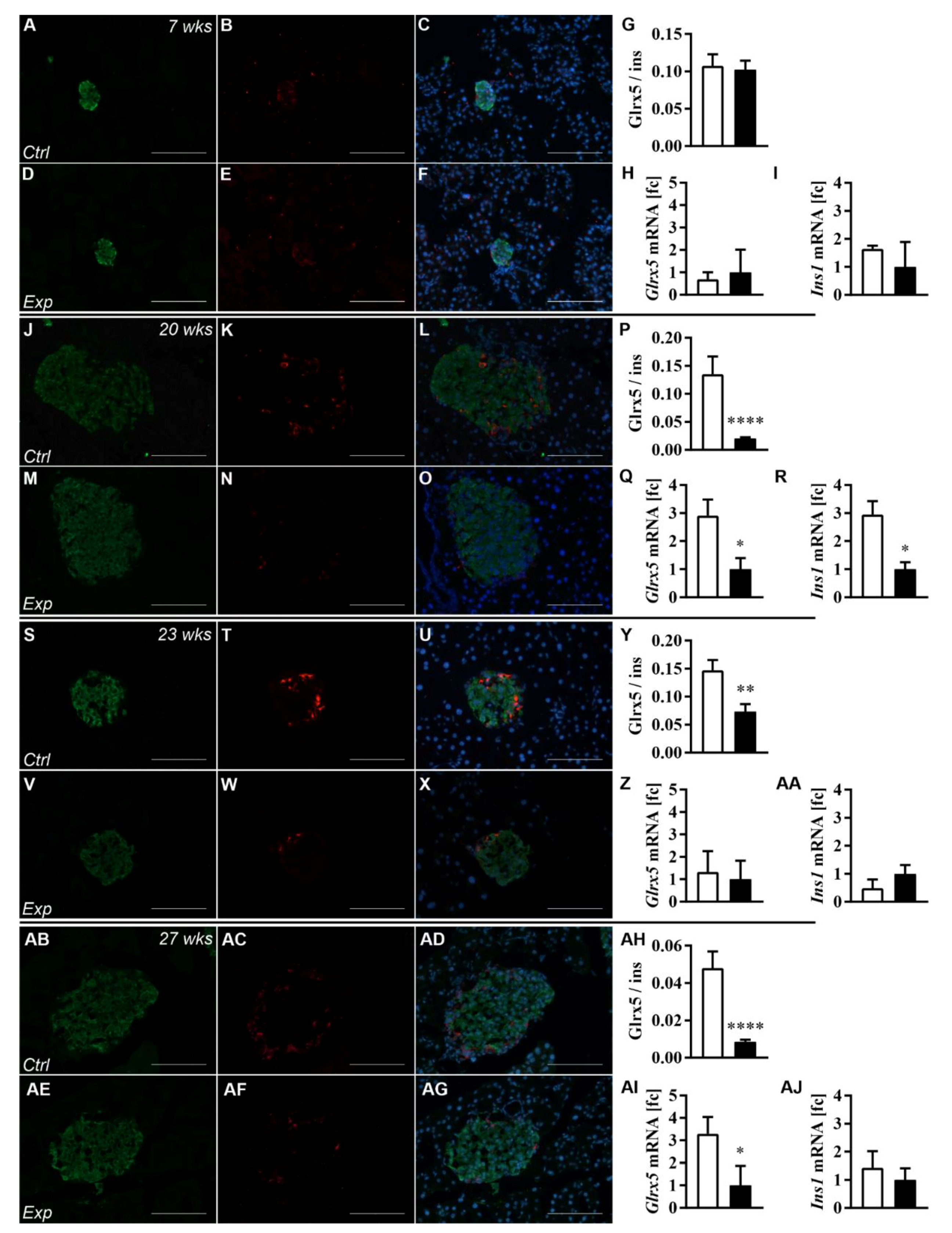
3.2. The Islet Glrx5 Content Is Diet-Dependent
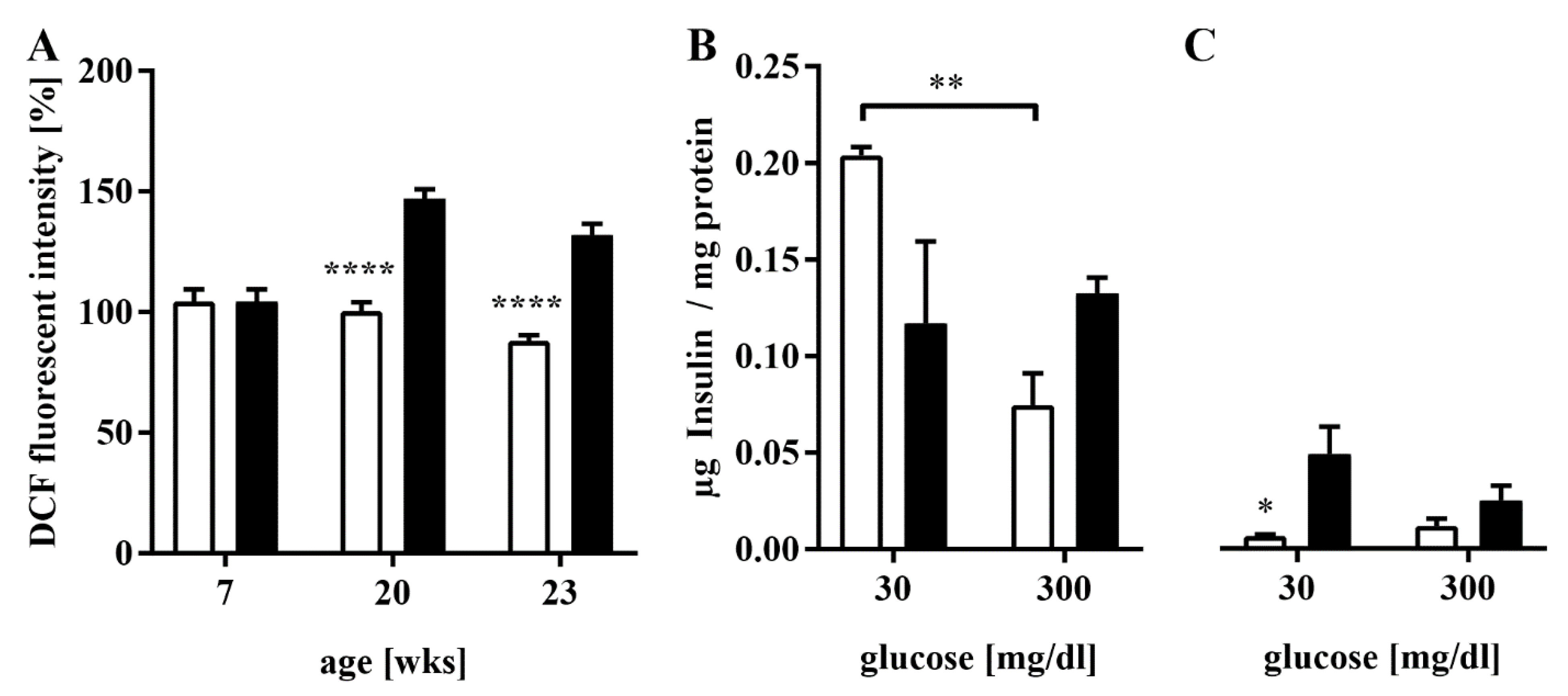
3.3. Elevated ROS Production and Impaired GSIS in HFD-Fed Mice
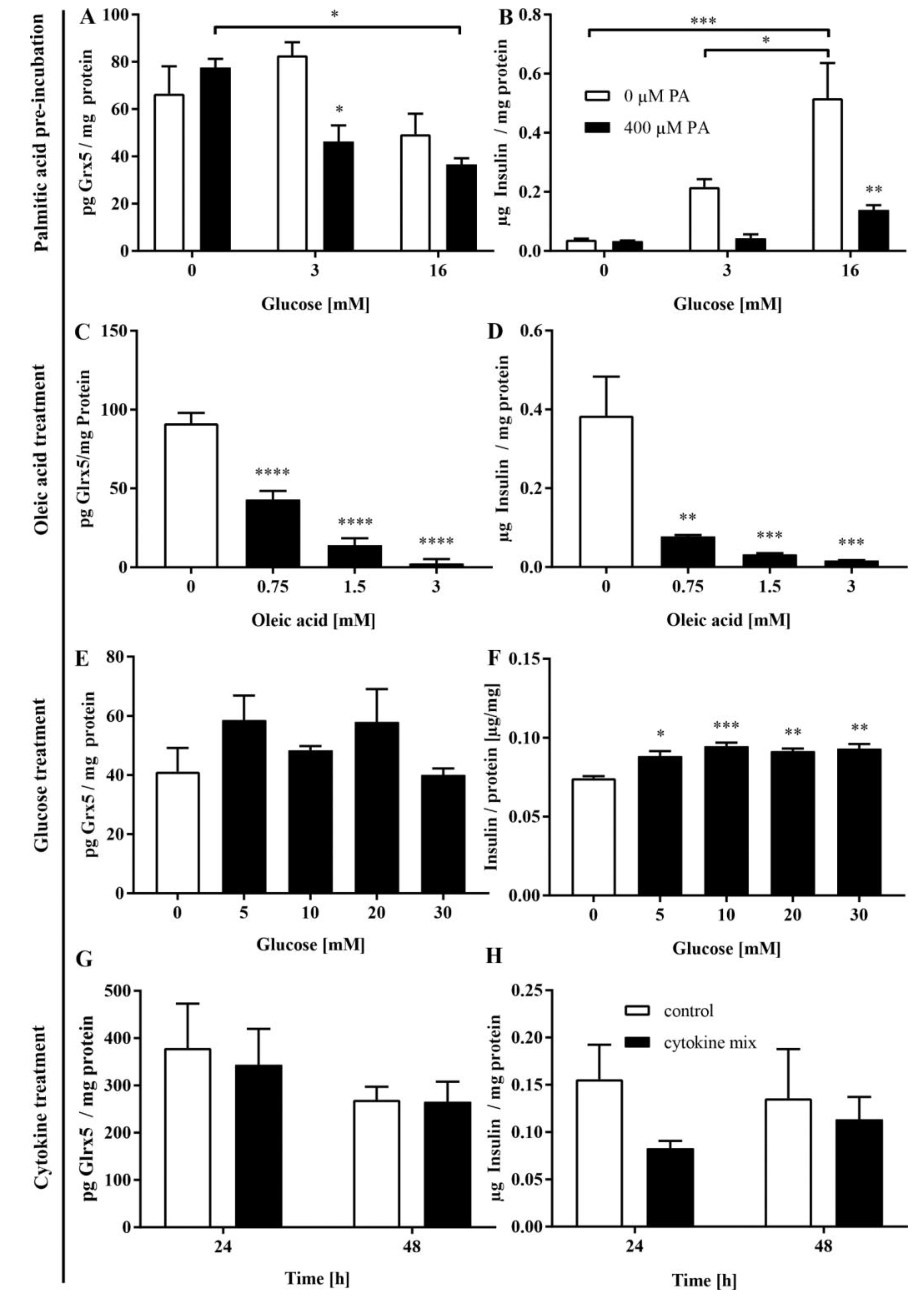
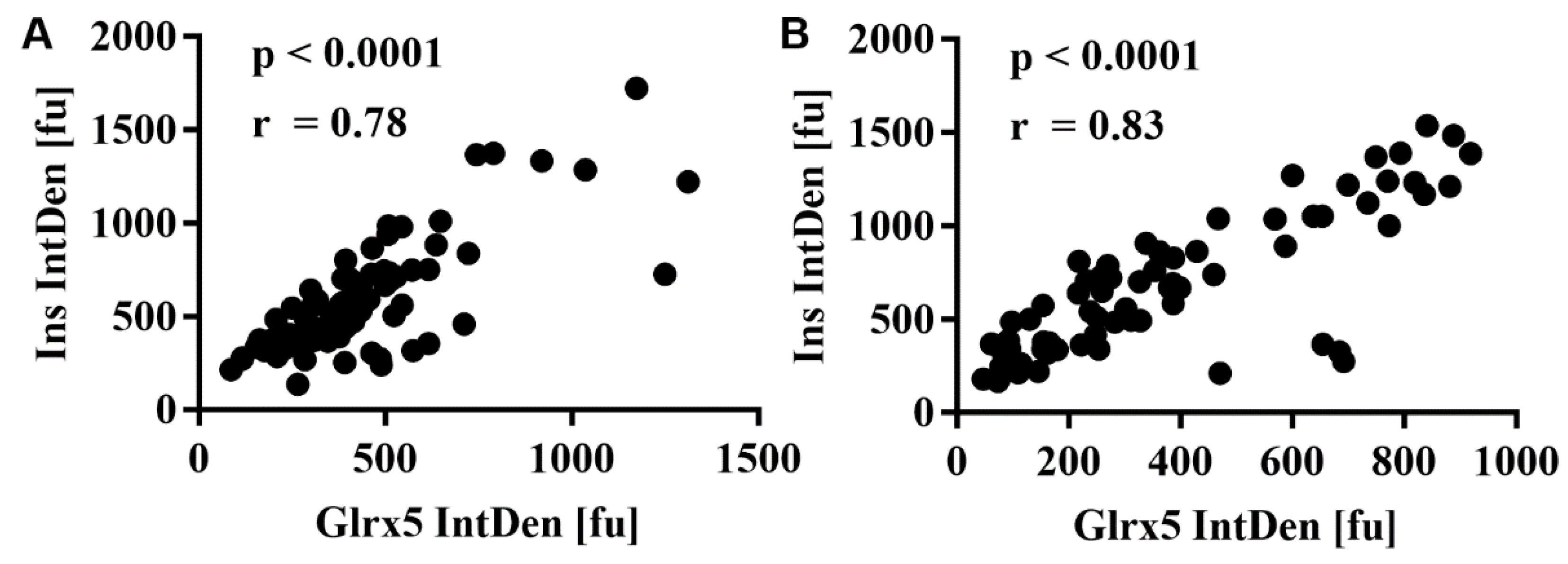
3.4. FFA Mediate the Loss of Glrx5 in MIN6 Cells
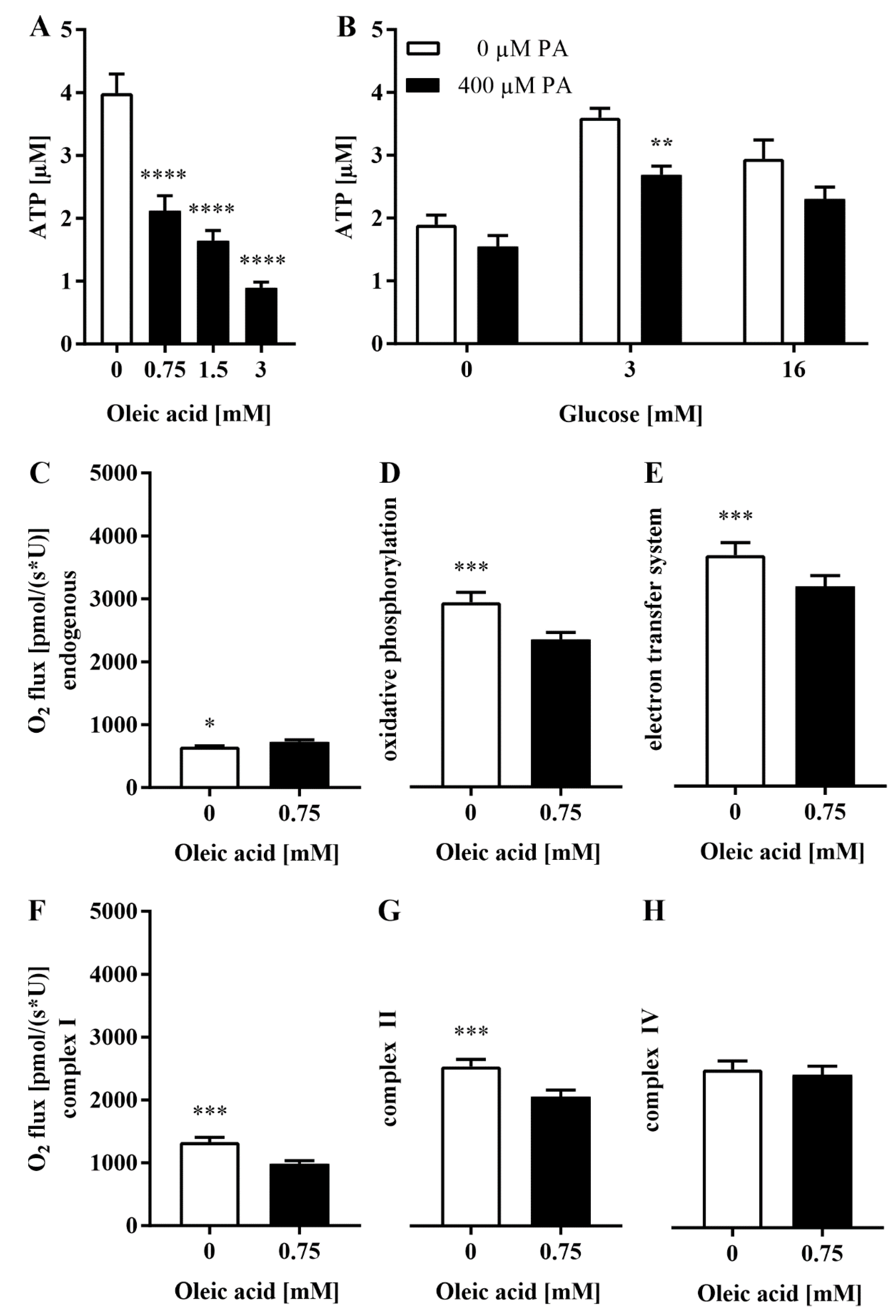
3.5. The Loss of Glrx5 Is Correlated with Mitigated ATP and Impaired O2 Flux in the Respiratory Chain
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barlow, J.; Solomon, T.P.J.; Affourtit, C. Pro-inflammatory cytokines attenuate glucose-stimulated insulin secretion from INS-1E insulinoma cells by restricting mitochondrial pyruvate oxidation capacity—Novel mechanistic insight from real-time analysis of oxidative phosphorylation. PLoS ONE 2018, 13, e0199505. [Google Scholar] [CrossRef] [PubMed]
- Thiébaud, D.; DeFronzo, R.A.; Jacot, E.; Golay, A.; Acheson, K.; Maeder, E.; Jéquier, E.; Felber, J.P. Effect of long chain triglyceride infusion on glucose metabolism in man. Metabolism 1982, 31, 1128–1136. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wirström, T.; Borg, L.A.H.; Larsson-Nyrén, G.; Hals, I.; Bondo-Hansen, J.; Grill, V.; Björklund, A. Diabetes reduces beta-cell mitochondria and induces distinct morphological abnormalities, which are reproducible by high glucose in vitro with attendant dysfunction. Islets 2012, 4, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208. [Google Scholar] [CrossRef]
- Solinas, G.; Naugler, W.; Galimi, F.; Lee, M.S.; Karin, M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 2006, 103, 16454–16459. [Google Scholar] [CrossRef]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef]
- Lee, K.M.; Seo, Y.J.; Kim, M.K.; Seo, H.A.; Jeong, J.Y.; Choi, H.S.; Lee, I.K.; Park, K.G. Mediation of glucolipotoxicity in INS-1 rat insulinoma cells by small heterodimer partner interacting leucine zipper protein (SMILE). Biochem. Biophys. Res. Commun. 2012, 419, 768–773. [Google Scholar] [CrossRef]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stümpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef]
- Stehling, O.; Wilbrecht, C.; Lill, R. Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 2014, 100, 61–77. [Google Scholar] [CrossRef]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed]
- Linares, G.R.; Xing, W.; Govoni, K.E.; Chen, S.T.; Mohan, S. Glutaredoxin 5 regulates osteoblast apoptosis by protecting against oxidative stress. Bone 2009, 44, 795–804. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodríguez-Manzaneque, M.T.; Ros, J.; Cabiscol, E.; Sorribas, A.; Herrero, E. Grx5 glutaredoxin plays a central role in protection against protein oxidative damage in Saccharomyces cerevisiae. Mol. Cell Biol. 1999, 19, 8180–8190. [Google Scholar] [CrossRef] [PubMed]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmid, B.; et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Investig. 2010, 120, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Campanella, A.; De Falco, L.; Boschetto, L.; Merlini, R.; Silvestri, L.; Levi, S.; Iolascon, A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007, 110, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, R.C.; Jones, D.; Gabrielsen, S.; Huang, J.; Simcox, J.A.; Luo, B.; Soesanto, Y.; Rienhoff, H.; Abel, E.D.; McClain, D.A. Dietary iron restriction or iron chelation protects from diabetes and loss of beta-cell function in the obese (ob/ob lep-/-) mouse. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1236–E1243. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Stefanini, M.; De Martino, C.; Zamboni, L. Fixation of ejaculated spermatozoa for electron microscopy. Nature 1967, 216, 173–174. [Google Scholar] [CrossRef]
- Lai, Y.; Schneider, D.; Kidszun, A.; Hauck-Schmalenberger, I.; Breier, G.; Brandhorst, D.; Brandhorst, H.; Iken, M.; Brendel, M.D.; Bretzel, R.G.; et al. Vascular endothelial growth factor increases functional beta-cell mass by improvement of angiogenesis of isolated human and murine pancreatic islets. Transplantation 2005, 79, 1530–1536. [Google Scholar] [CrossRef]
- Petry, S.F.; Sharifpanah, F.; Sauer, H.; Linn, T. Differential expression of islet glutaredoxin 1 and 5 with high reactive oxygen species production in a mouse model of diabesity. PLoS ONE 2017, 12, e0176267. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.; Sharifpanah, F.; Wartenberg, M.; Sauer, H. Omega-3 and Omega-6 polyunsaturated fatty acids stimulate vascular differentiation of mouse embryonic stem cells. J. Cell Physiol. 2020, 235, 7094–7106. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.N.; Hengstler, J.G.; Stewart, J. Analyses of reactive oxygen species. EXCLI J. 2009, 8, 241–245. [Google Scholar]
- Lai, Y.; Brandhorst, H.; Hossain, H.; Bierhaus, A.; Chen, C.; Bretzel, R.G.; Linn, T. Activation of NFkappaB dependent apoptotic pathway in pancreatic islet cells by hypoxia. Islets 2009, 1, 19–25. [Google Scholar] [CrossRef][Green Version]
- Petry, S.F.; Sun, L.M.; Knapp, A.; Reinl, S.; Linn, T. Distinct Shift in Beta-Cell Glutaredoxin 5 Expression Is Mediated by Hypoxia and Lipotoxicity Both In Vivo and In Vitro. Front. Endocrinol. 2018, 9, 84. [Google Scholar] [CrossRef]
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbé, J.C.; Lorca, T.; Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial pathways and respiratory control: An introduction to OXPHOS analysis, 5th ed. Bioenerg. Commun. 2020, 2020, 2. [Google Scholar]
- Stadlmann, S.; Renner, K.; Pollheimer, J.; Moser, P.L.; Zeimet, A.G.; Offner, F.A.; Gnaiger, E. Preserved coupling of oxidative phosphorylation but decreased mitochondrial respiratory capacity in IL-1beta-treated human peritoneal mesothelial cells. Cell Biochem. Biophys. 2006, 44, 179–186. [Google Scholar] [CrossRef]
- Pohland, M.; Hagl, S.; Pellowska, M.; Wurglics, M.; Schubert-Zsilavecz, M.; Eckert, G.P. MH84: A Novel g-Secretase Modulator/PPARg Agonist–Improves Mitochondrial Dysfunction in a Cellular Model of Alzheimer’s Disease. Neurochem. Res. 2016, 41, 231–242. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of β-cell function by thioredoxin. Redox Biol. 2020, 34, 101570. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53 (Suppl. 1), S119–S124. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Scarcia, P.; Li, N.; Gaudet, P.; Duhamel, D.; Palmieri, F.; Maechler, P. Changes in mitochondrial carriers exhibit stress-specific signatures in INS-1Eb-cells exposed to glucose versus fatty acids. PLoS ONE 2013, 8, e82364. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Fex, M.; Nitert, M.D.; Wierup, N.; Sundler, F.; Ling, C.; Mulder, H. Enhanced mitochondrial metabolism may account for the adaptation to insulin resistance in islets from C57BL/6J mice fed a high-fat diet. Diabetologia 2007, 50, 74–83. [Google Scholar] [CrossRef]
- Ferdaoussi, M.; Bergeron, V.; Zarrouki, B.; Kolic, J.; Cantley, J.; Fielitz, J.; Olson, E.N.; Prentki, M.; Biden, T.; MacDonald, P.E.; et al. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 2012, 55, 2682–2692. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Kim, H.S.; Hwang, Y.C.; Koo, S.H.; Park, K.S.; Lee, M.S.; Kim, K.W.; Lee, M.K. PPAR-g activation increases insulin secretion through the up-regulation of the free fatty acid receptor GPR40 in pancreatic b-cells. PLoS ONE 2013, 8, e50128. [Google Scholar] [CrossRef]
- Sako, Y.; Grill, V.E. A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology 1990, 127, 1580–1589. [Google Scholar] [CrossRef]
- Ritz-Laser, B.; Meda, P.; Constant, I.; Klages, N.; Charollais, A.; Morales, A.; Magnan, C.; Ktorza, A.; Philippe, J. Glucose-induced preproinsulin gene expression is inhibited by the free fatty acid palmitate. Endocrinology 1999, 140, 4005–4014. [Google Scholar] [CrossRef]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patané, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link b cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.M.; Goh, T.; Tchipashvili, V.; Sandhu, H.; Gupta, N.; Lewis, G.F.; Giacca, A. Prolonged elevation of plasma free fatty acids desensitizes the insulin secretory response to glucose in vivo in rats. Diabetes 1999, 48, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Gambardella, A.; Amato, L.; Tortoriello, R.; D’Amore, A.; Varricchio, M.; D’Onofrio, F. Opposite effects of short- and long-term fatty acid infusion on insulin secretion in healthy subjects. Diabetologia 1995, 38, 1295–1299. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Tornheim, K.; Leahy, J.L. Fatty acid-induced beta cell hypersensitivity to glucose. Increased phosphofructokinase activity and lowered glucose-6-phosphate content. J. Clin. Investig. 1998, 101, 1870–1875. [Google Scholar] [CrossRef][Green Version]
- Köhnke, D.; Ludwig, B.; Kadenbach, B. A threshold membrane potential accounts for controversial effects of fatty acids on mitochondrial oxidative phosphorylation. FEBS Lett. 1993, 336, 90–94. [Google Scholar] [CrossRef]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.; Hardie, D.G. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem. J. 1998, 335 Pt 3, 533–539. [Google Scholar] [CrossRef]
- Briaud, I.; Harmon, J.S.; Kelpe, C.L.; Segu, V.B.; Poitout, V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 2001, 50, 315–321. [Google Scholar] [CrossRef]
- Hirota, N.; Otabe, S.; Nakayama, H.; Yuan, X.; Yamada, K. Sequential activation of caspases and synergistic beta-cell cytotoxicity by palmitate and anti-Fas antibodies. Life Sci. 2006, 79, 1312–1316. [Google Scholar] [CrossRef]
- Salgin, B.; Ong, K.K.; Thankamony, A.; Emmett, P.; Wareham, N.J.; Dunger, D.B. Higher fasting plasma free fatty acid levels are associated with lower insulin secretion in children and adults and a higher incidence of type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3302–3309. [Google Scholar] [CrossRef]
- Malmgren, S.; Nicholls, D.G.; Taneera, J.; Bacos, K.; Koeck, T.; Tamaddon, A.; Wibom, R.; Groop, L.; Ling, C.; Mulder, H.; et al. Tight coupling between glucose and mitochondrial metabolism in clonal beta-cells is required for robust insulin secretion. J. Biol. Chem. 2009, 284, 32395–32404. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Yamashita, S.; Takahashi, N.; Eto, K.; Shen, L.M.; Izumi, K.; Daniel, S.; Tsubamoto, Y.; Nemoto, T.; Iino, M.; et al. Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics of betaHC9 cells deficient in mitochondrial DNA transcription. J. Biol. 2002, 277, 41817–41826. [Google Scholar]
- Minami, K.; Yano, H.; Miki, T.; Nagashima, K.; Wang, C.Z.; Tanaka, H.; Miyazaki, J.I.; Seino, S. Insulin secretion and differential gene expression in glucose-responsive and -unresponsive MIN6 sublines. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E773–E781. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R.; Smith, P.A.; Ashcroft, F.M. Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic beta-cells. Biochem. J. 1993, 294 Pt 1, 35–42. [Google Scholar] [CrossRef]
- Daunt, M.; Dale, O.; Smith, P.A. Somatostatin inhibits oxidative respiration in pancreatic beta-cells. Endocrinology 2006, 147, 1527–1535. [Google Scholar] [CrossRef]
- Cheng, K.; Delghingaro-Augusto, V.; Nolan, C.J.; Turner, N.; Hallahan, N.; Andrikopoulos, S.; Gunton, J.E. High passage MIN6 cells have impaired insulin secretion with impaired glucose and lipid oxidation. PLoS ONE 2012, 7, e40868. [Google Scholar] [CrossRef]
- Taddeo, E.P.; Stiles, L.; Sereda, S.; Ritou, E.; Wolf, D.M.; Abdullah, M.; Swanson, Z.; Wilhelm, J.; Bellin, M.; McDonald, P.; et al. Individual islet respirometry reveals functional diversity within the islet population of mice and human donors. Mol. Metab. 2018, 16, 150–159. [Google Scholar] [CrossRef]
- Rodríguez-Manzaneque, M.T.; Tamarit, J.; Bellí, G.; Ros, J.; Herrero, E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol. Biol. Cell 2002, 13, 1109–1121. [Google Scholar] [CrossRef]
- Uzarska, M.A.; Dutkiewicz, R.; Freibert, S.A.; Lill, R.; Mühlenhoff, U. The mitochondrial Hsp70 chaperone Ssq1 facilitates Fe/S cluster transfer from Isu1 to Grx5 by complex formation. Mol. Biol. Cell 2013, 24, 1830–1841. [Google Scholar] [CrossRef]
- Corbett, J.A.; Lancaster, J.R.J.; Sweetland, M.A.; McDaniel, M.L. Interleukin-1 beta-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 beta-induced inhibition of insulin secretion. J. Biol. Chem. 1991, 266, 21351–21354. [Google Scholar] [CrossRef]
- Corbett, J.A.; Sweetland, M.A.; Wang, J.L.; Lancaster, J.R.J.; McDaniel, M.L. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc. Natl. Acad. Sci. USA 1993, 90, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Scarim, A.L.; Heitmeier, M.R.; Corbett, J.A. Irreversible inhibition of metabolic function and islet destruction after a 36-h exposure to interleukin-1beta. Endocrinology 1997, 138, 5301–5307. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.C.F.D.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 regulates in-sulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat. Commun. 2020, 11, 296. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petry, S.F.; Römer, A.; Rawat, D.; Brunner, L.; Lerch, N.; Zhou, M.; Grewal, R.; Sharifpanah, F.; Sauer, H.; Eckert, G.P.; et al. Loss and Recovery of Glutaredoxin 5 Is Inducible by Diet in a Murine Model of Diabesity and Mediated by Free Fatty Acids In Vitro. Antioxidants 2022, 11, 788. https://doi.org/10.3390/antiox11040788
Petry SF, Römer A, Rawat D, Brunner L, Lerch N, Zhou M, Grewal R, Sharifpanah F, Sauer H, Eckert GP, et al. Loss and Recovery of Glutaredoxin 5 Is Inducible by Diet in a Murine Model of Diabesity and Mediated by Free Fatty Acids In Vitro. Antioxidants. 2022; 11(4):788. https://doi.org/10.3390/antiox11040788
Chicago/Turabian StylePetry, Sebastian Friedrich, Axel Römer, Divya Rawat, Lara Brunner, Nina Lerch, Mengmeng Zhou, Rekha Grewal, Fatemeh Sharifpanah, Heinrich Sauer, Gunter Peter Eckert, and et al. 2022. "Loss and Recovery of Glutaredoxin 5 Is Inducible by Diet in a Murine Model of Diabesity and Mediated by Free Fatty Acids In Vitro" Antioxidants 11, no. 4: 788. https://doi.org/10.3390/antiox11040788
APA StylePetry, S. F., Römer, A., Rawat, D., Brunner, L., Lerch, N., Zhou, M., Grewal, R., Sharifpanah, F., Sauer, H., Eckert, G. P., & Linn, T. (2022). Loss and Recovery of Glutaredoxin 5 Is Inducible by Diet in a Murine Model of Diabesity and Mediated by Free Fatty Acids In Vitro. Antioxidants, 11(4), 788. https://doi.org/10.3390/antiox11040788