
Sirtuin 3 Dependent and Independent Effects of NAD+ to Suppress Vascular Inflammation and Improve Endothelial Function in Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals Experiments
2.2. Cell Culture and RNA Interference
2.3. Quantitative RT-PCR Analysis
2.4. Western Blot
2.5. Seahorse Test
2.6. Vascular Reactivity
2.7. MitoSOX Staining
2.8. Histological Staining
2.9. Immunofluorescence Staining
2.10. Serum Chemistry Analysis
2.11. Statistical Analysis
3. Results
3.1. NAD+ Attenuated Inflammatory Response in Endothelial Cells in a SIRT3-Independent Manner
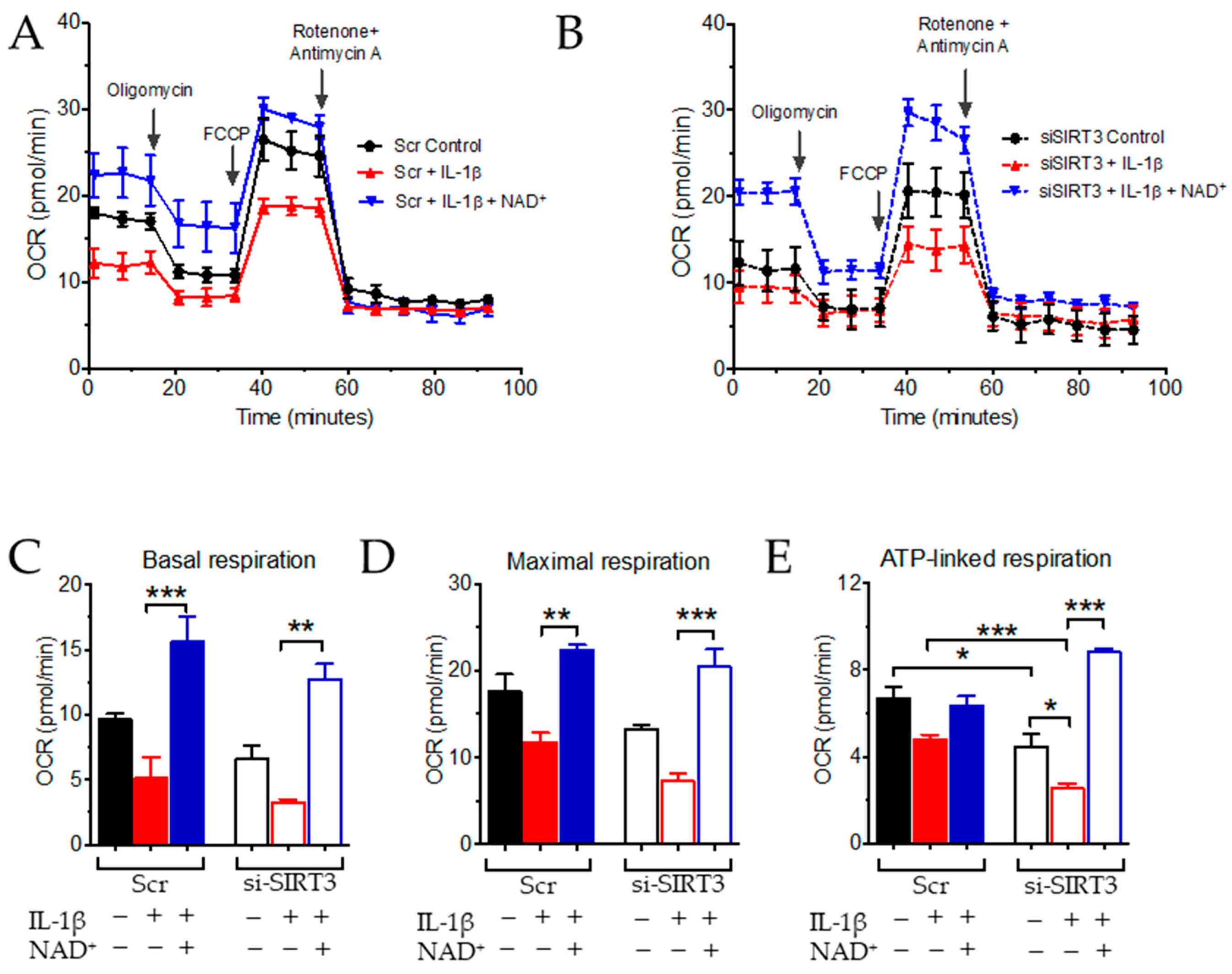
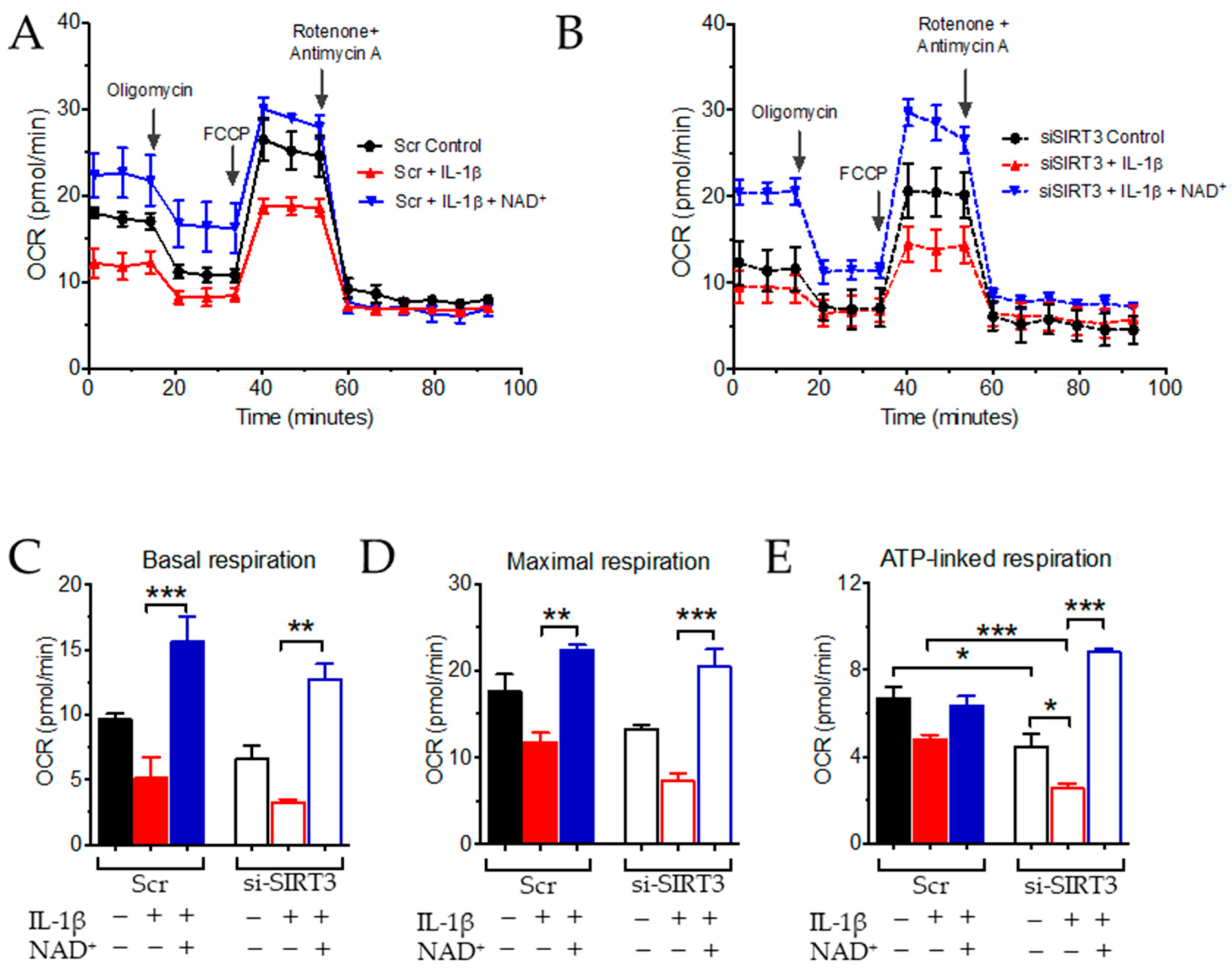
3.2. NAD+ and SIRT3 Independently Improve Mitochondrial Function
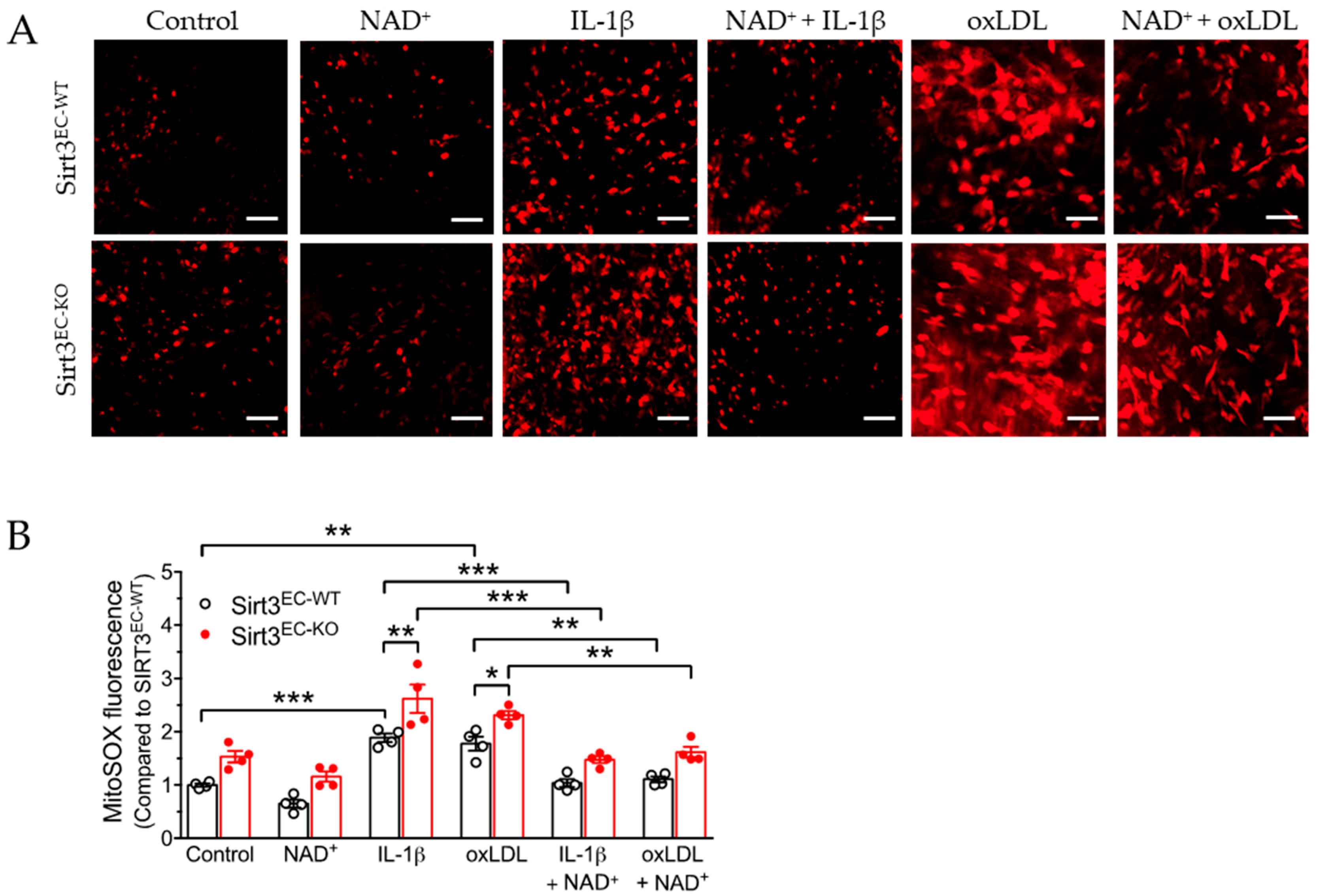
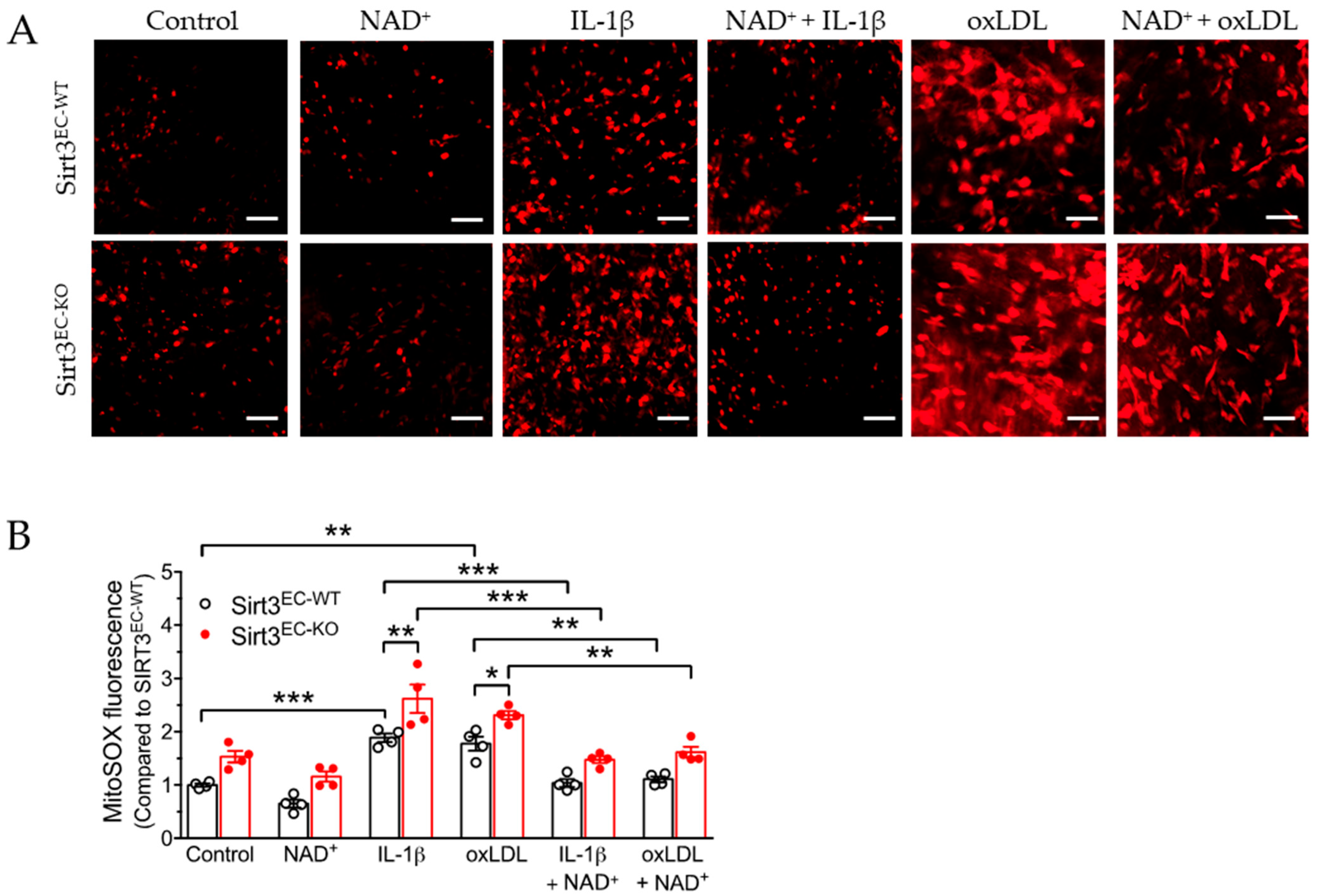
3.3. SIRT3 Deletion Enhanced Oxidative Stress in Endothelial Cells Attenuated by NAD
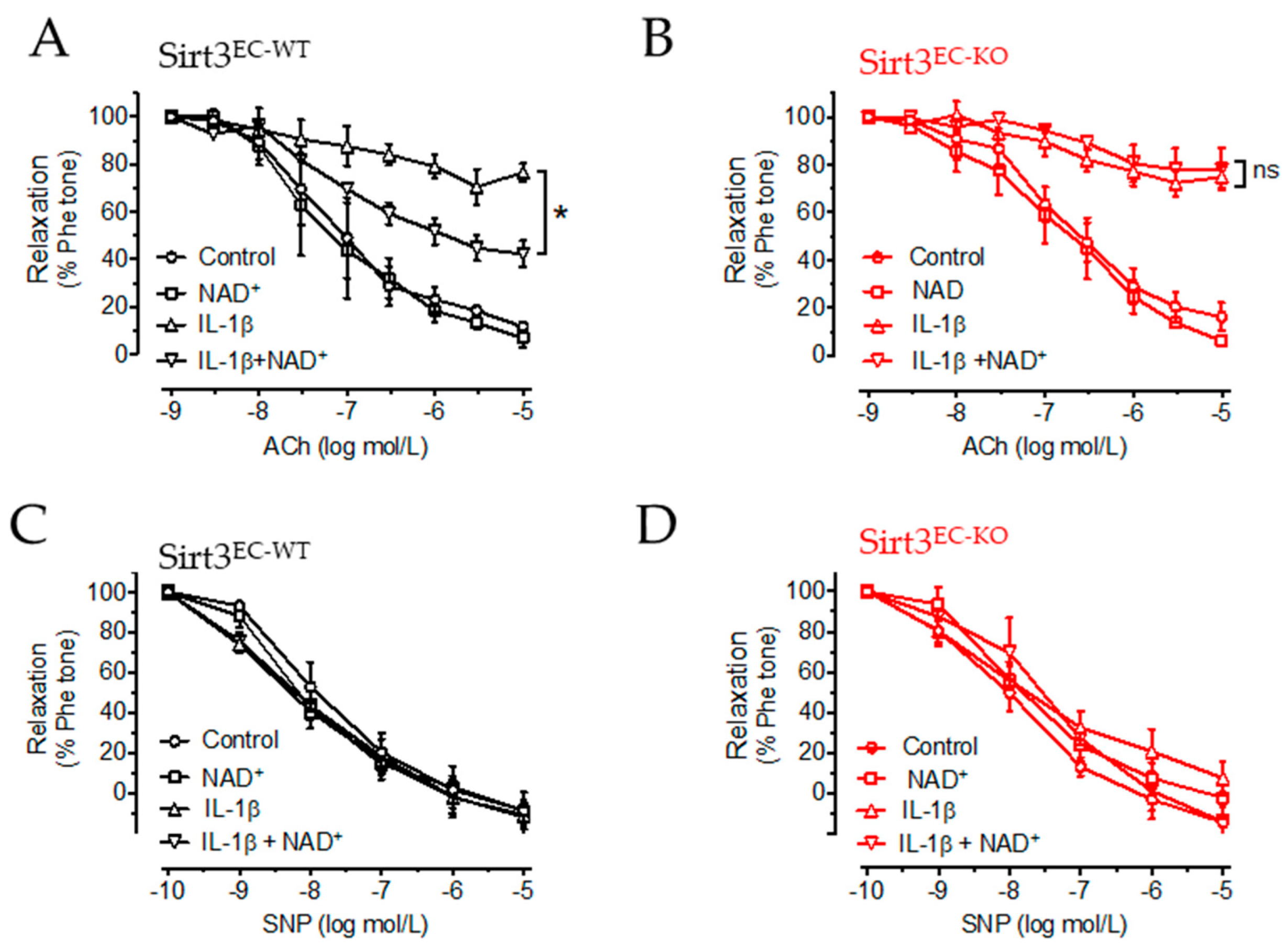
3.4. NAD+ Improved Vascular Function in a SIRT3-Dependent Manner
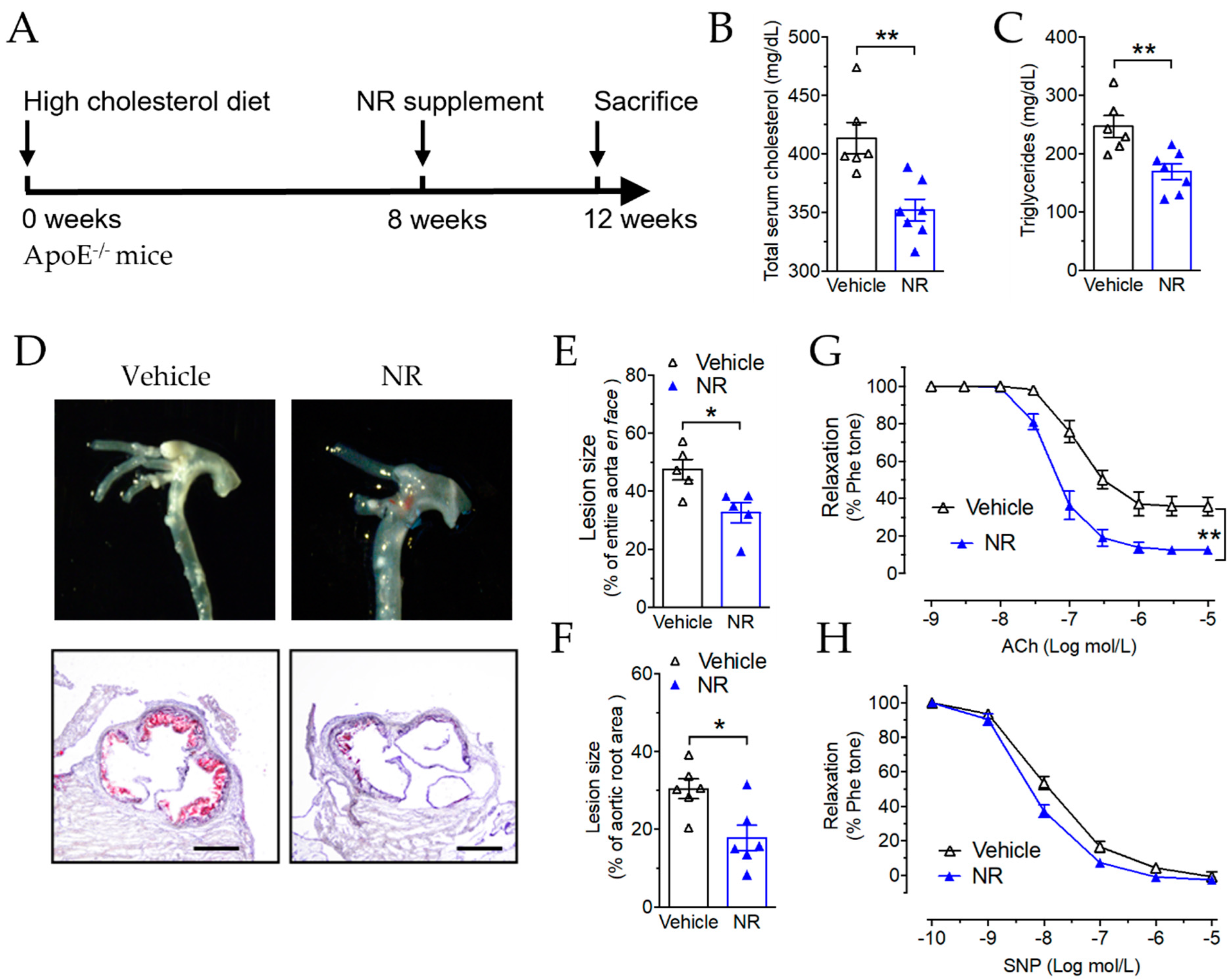
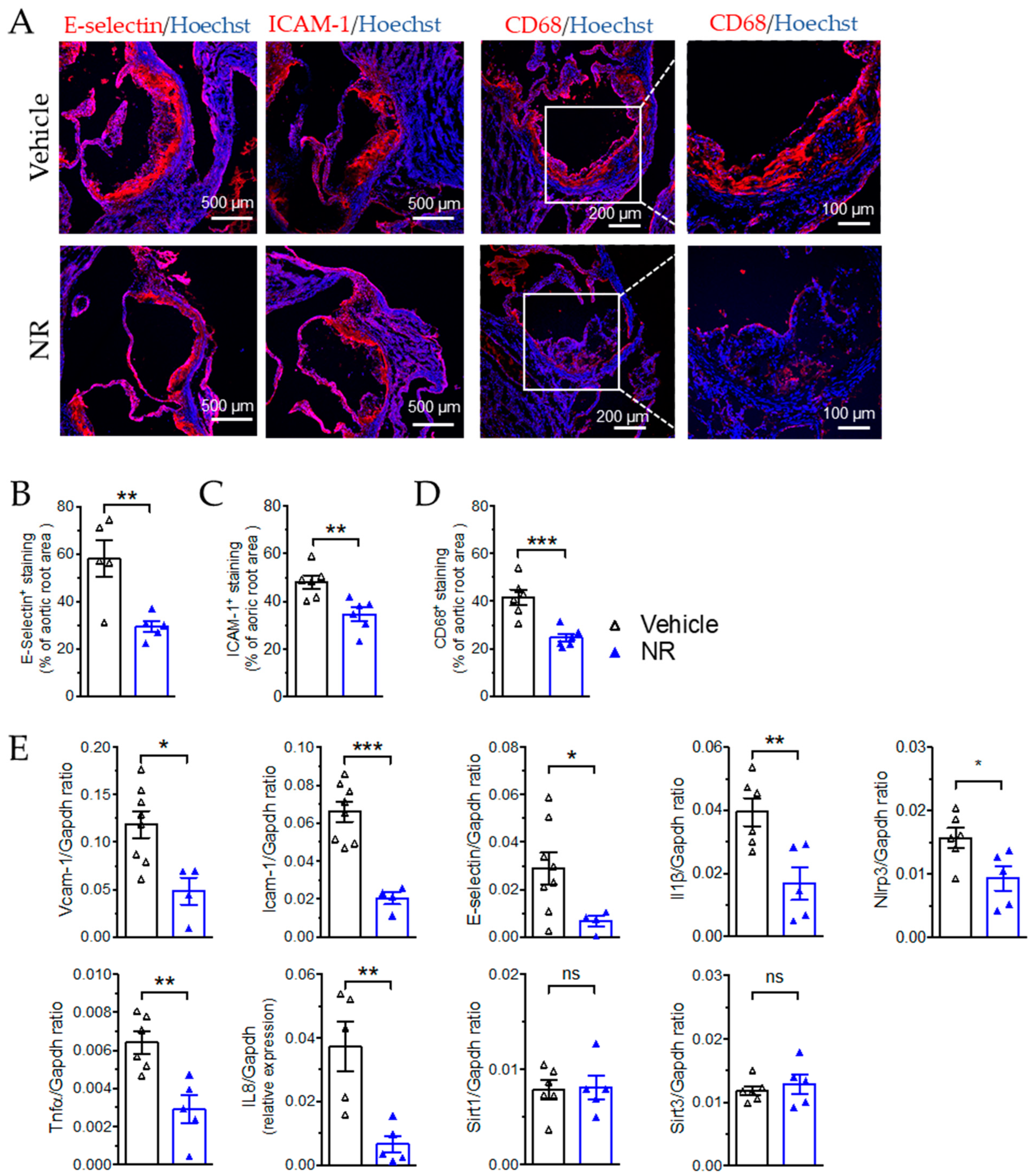
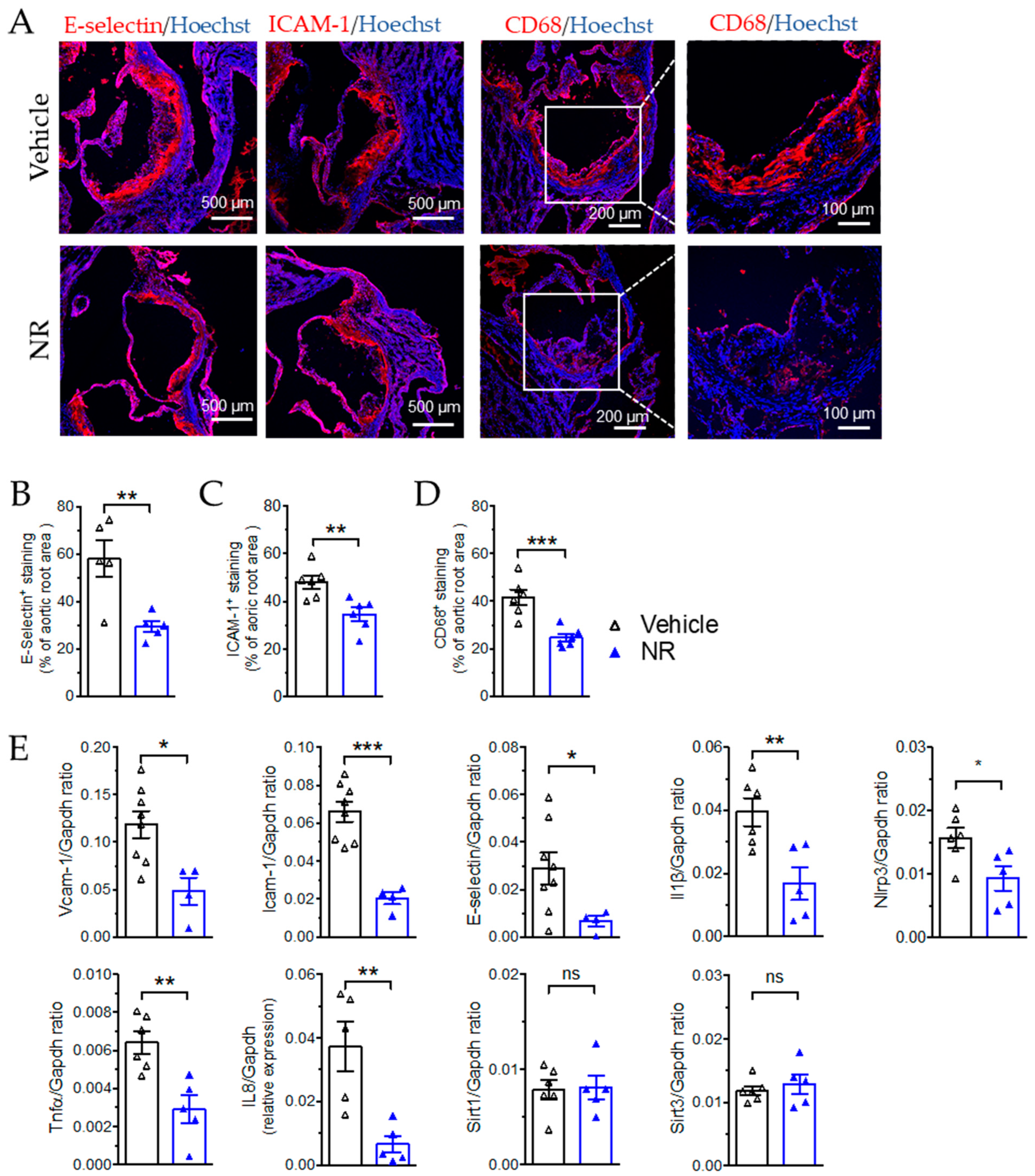
3.5. Nicotinamide Riboside Reduced Atherosclerotic Plaque Formation in ApoE−/− Mice
3.6. NR Decreased Macrophage Infiltration and Vascular Inflammation in ApoE−/− Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Nagasawa, K.; Munch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, M.P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016, 167, 985–1000.e1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilopoulos, A.; Pennington, J.D.; Andresson, T.; Rees, D.M.; Bosley, A.D.; Fearnley, I.M.; Ham, A.; Flynn, C.R.; Hill, S.; Rose, K.L.; et al. SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid. Redox Signal. 2014, 21, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Dittenhafer-Reed, K.E.; Richards, A.L.; Fan, J.; Smallegan, M.J.; Fotuhi Siahpirani, A.; Kemmerer, Z.A.; Prolla, T.A.; Roy, S.; Coon, J.J.; Denu, J.M. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 2015, 21, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T.t.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ. Res. 2020, 126, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Benigni, A.; Cassis, P.; Conti, S.; Perico, L.; Corna, D.; Cerullo, D.; Zentilin, L.; Zoja, C.; Perna, A.; Lionetti, V.; et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid. Redox Signal. 2019, 31, 1255–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zeng, H.; He, X.; Chen, J.X. Sirtuin 3 Alleviates Diabetic Cardiomyopathy by Regulating TIGAR and Cardiomyocyte Metabolism. J. Am. Heart Assoc. 2021, 10, e018913. [Google Scholar] [CrossRef] [PubMed]
- Dolopikou, C.F.; Kourtzidis, I.A.; Margaritelis, N.V.; Vrabas, I.S.; Koidou, I.; Kyparos, A.; Theodorou, A.A.; Paschalis, V.; Nikolaidis, M.G. Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: A double-blind cross-over study. Eur. J. Nutr. 2020, 59, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance Exercise as a Countermeasure for Aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; He, X.C.; Hou, X.W.; Li, L.F.; Chen, J.X. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am. J. Physiol.-Heart C 2014, 306, H585–H597. [Google Scholar] [CrossRef]
- Sun, Y.; Teng, Z.; Sun, X.; Zhang, L.; Chen, J.; Wang, B.; Lu, F.; Liu, N.; Yu, M.; Peng, S.; et al. Exogenous H2S reduces the acetylation levels of mitochondrial respiratory enzymes via regulating the NAD(+)-SIRT3 pathway in cardiac tissues of db/db mice. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E284–E297. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.K.; Watson, P.A.; Birdsey, N.; Bao, J.J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Liu, X.; Su, C.; Wu, F.; Sun, J.; Zhang, J.; Yang, X.; Zhang, C.; Zhou, Z.; Zhang, X.; et al. Inhibition of Mitochondrial Oxidative Damage Improves Reendothelialization Capacity of Endothelial Progenitor Cells via SIRT3 (Sirtuin 3)-Enhanced SOD2 (Superoxide Dismutase 2) Deacetylation in Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1682–1698. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.F.; Huang, C.C.; Xiao, Y.F.; Guan, X.H.; Wang, X.N.; Cao, Q.; Liu, Y.; Huang, X.; Deng, L.B.; Deng, K.Y.; et al. CD38 Deficiency Protects Heart from High Fat Diet-Induced Oxidative Stress Via Activating Sirt3/FOXO3 Pathway. Cell. Physiol. Biochem. 2018, 48, 2350–2363. [Google Scholar] [CrossRef]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, M.; Zeng, X.; Yang, J.; Deng, H.; Yi, L.; Mi, M.T. Resveratrol regulates mitochondrial reactive oxygen species homeostasis through Sirt3 signaling pathway in human vascular endothelial cells. Cell Death Dis. 2014, 5, e1576. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Liu, J.; Liu, S.; Song, Q.; Liu, L.; Xie, L.; Han, Y.; Ji, Y. Hydrogen sulfide pretreatment improves mitochondrial function in myocardial hypertrophy via a SIRT3-dependent manner. Br. J. Pharmacol. 2018, 175, 1126–1145. [Google Scholar] [CrossRef] [Green Version]
- De Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef] [PubMed]
- Holzhauser, E.; Albrecht, C.; Zhou, Q.X.; Buttler, A.; Preusch, M.R.; Blessing, E.; Katus, H.A.; Bea, F. Nicotinic Acid Has Anti-atherogenic and Anti-inflammatory Properties on Advanced Atherosclerotic Lesions Independent of its Lipid-modifying Capabilities. J. Cardiovasc. Pharm. 2011, 57, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Lara, K.A.; Letelier, N.; Farre, N.; Diarte-Anazco, E.M.G.; Nieto-Nicolau, N.; Rodriguez-Millan, E.; Santos, D.; Pallares, V.; Escola-Gil, J.C.; Vazquez Del Olmo, T.; et al. Nicotinamide Prevents Apolipoprotein B-Containing Lipoprotein Oxidation, Inflammation and Atherosclerosis in Apolipoprotein E-Deficient Mice. Antioxidants 2020, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; da Silva, R.F.; Fraga-Silva, R.A.; Steffens, S.; Fabre, M.; Bauer, I.; Caffa, I.; Magnone, M.; Sociali, G.; Quercioli, A.; et al. Nicotinamide phosphoribosyltransferase inhibition reduces intraplaque CXCL1 production and associated neutrophil infiltration in atherosclerotic mice. Thromb. Haemost. 2014, 111, 308–322. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, Ł.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Xu, S.W.; Bai, P.; Little, P.J.; Liu, P.Q. Poly(ADP-ribose) Polymerase 1 (PARP1) in Atherosclerosis: From Molecular Mechanisms to Therapeutic Implications. Med. Res. Rev. 2014, 34, 644–675. [Google Scholar] [CrossRef]
- Zha, S.Y.; Wang, F.; Li, Z.; Ma, Z.Y.; Yang, L.; Liu, F. PJ34, a PARP1 inhibitor, promotes endothelial repair in a rabbit model of high fat diet-induced atherosclerosis. Cell Cycle 2019, 18, 2099–2109. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an Endothelial NAD+-H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89.e20. [Google Scholar] [CrossRef] [Green Version]
- Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Csipo, T.; Nyúl-Tóth, Á.; Lipecz, A.; Szabo, C.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. Geroscience 2019, 41, 419–439. [Google Scholar] [CrossRef]
- Cartwright, D.M.; Oakey, L.A.; Fletcher, R.S.; Doig, C.L.; Heising, S.; Larner, D.P.; Nasteska, D.; Berry, C.E.; Heaselgrave, S.R.; Ludwig, C.; et al. Nicotinamide riboside has minimal impact on energy metabolism in mouse models of mild obesity. J. Endocrinol. 2021, 251, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Koves, T.R.; Pettway, Y.D.; Draper, J.A.; Slentz, D.H.; Grimsrud, P.A.; Ilkayeva, O.R.; Muoio, D.M. Nicotinamide riboside supplementation confers marginal metabolic benefits in obese mice without remodeling the muscle acetyl-proteome. IScience 2022, 25, 103635. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.W.; Feng, J.; Zhang, R.; Fan, M.M.; Han, D.; Li, X.; Li, C.Y.; Ren, J.; Wang, Y.B.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxidative Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.H.; Yu, B.; Qiao, H. Correlation between endothelial cell apoptosis and SIRT3 gene expression in atherosclerosis rats. Eur. Rev. Med. Pharmacol. 2019, 23, 9033–9040. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD+ metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef] [Green Version]
- Li, R.B.; Xin, T.; Li, D.D.; Wang, C.B.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef]
- Winnik, S.; Gaul, D.S.; Preitner, F.; Lohmann, C.; Weber, J.; Miranda, M.X.; Liu, Y.L.; van Tits, L.J.; Mateos, J.M.; Brokopp, C.E.; et al. Deletion of Sirt3 does not affect atherosclerosis but accelerates weight gain and impairs rapid metabolic adaptation in LDL receptor knockout mice: Implications for cardiovascular risk factor development. Basic Res. Cardiol. 2014, 109, 399. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Geng, H.Z.; Lv, X.Q.; Ma, J.; Liu, F.C.; Lin, P.F.; Yan, C.Z. Idebenone Protects against Atherosclerosis in Apolipoprotein E-Deficient Mice Via Activation of the SIRT3-SOD2-mtROS Pathway. Cardiovasc. Drugs Ther. 2021, 35, 1129–1145. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Traba, J.; Geiger, S.S.; Kwarteng-Siaw, M.; Han, K.; Ra, O.H.; Siegel, R.M.; Gius, D.; Sack, M.N. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J. Biol. Chem. 2017, 292, 12153–12164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxid. Med. Cell. Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | mRNA | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|---|
| NM_011333 | Ccl2 | CATCCACGTGTTGGCTCA | GATCATCTTGCTGGTGAATGAGT |
| NM_011345 | E-selectin | AGTTGTGAGTTCTCCTGCGA | CACTCCATGACGCCATTCTG |
| NM_001289726 | Gapdh | ATGGTGAAGGTCGGTGTGAA | GAGGTCAATGAAGGGGTCGT |
| NM_010493 | Icam1 | AAACCAGACCCTGGAACTGCAC | GCCTGGCATTTCAGAGTCTGCT |
| NM_008361 | IL-1β | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG |
| NM_011339 | IL-8 | GGTGATATTCGAGACCATTTACTG | GCCAACAGTAGCCTTCACCCAT |
| NM_019812 | Sirt1 | GCTGACGACTTCGACGACG | TCGGTCAACAGGAGGTTGTCT |
| NM_022433 | Sirt3 | ATCCCGGACTTCAGATCCCC | CAACATGAAAAAGGGCTTGGG |
| NM_001278601 | Tnfα | CAGCCTCTTCTCATTCCTGC | ATGAGAGGGAGGCCATTTG |
| NM_011693 | Vcam1 | ACAGACAGTCCCCTCAATGG | TCCTCAAAACCCACAGAGCT |
| NM_145827 | Nlrp3 | GTGTGGATCTTTGCTGCGAT | TATCCCAGCAAACCCATCCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Wu, Y.; Hong, H.; Tian, X.Y. Sirtuin 3 Dependent and Independent Effects of NAD+ to Suppress Vascular Inflammation and Improve Endothelial Function in Mice. Antioxidants 2022, 11, 706. https://doi.org/10.3390/antiox11040706
Cao X, Wu Y, Hong H, Tian XY. Sirtuin 3 Dependent and Independent Effects of NAD+ to Suppress Vascular Inflammation and Improve Endothelial Function in Mice. Antioxidants. 2022; 11(4):706. https://doi.org/10.3390/antiox11040706
Chicago/Turabian StyleCao, Xiaoyun, Yalan Wu, Huiling Hong, and Xiao Yu Tian. 2022. "Sirtuin 3 Dependent and Independent Effects of NAD+ to Suppress Vascular Inflammation and Improve Endothelial Function in Mice" Antioxidants 11, no. 4: 706. https://doi.org/10.3390/antiox11040706
APA StyleCao, X., Wu, Y., Hong, H., & Tian, X. Y. (2022). Sirtuin 3 Dependent and Independent Effects of NAD+ to Suppress Vascular Inflammation and Improve Endothelial Function in Mice. Antioxidants, 11(4), 706. https://doi.org/10.3390/antiox11040706