
Is Nucleoredoxin a Master Regulator of Cellular Redox Homeostasis? Its Implication in Different Pathologies

 , , , , and
, , , , and
Abstract
:1. Introduction
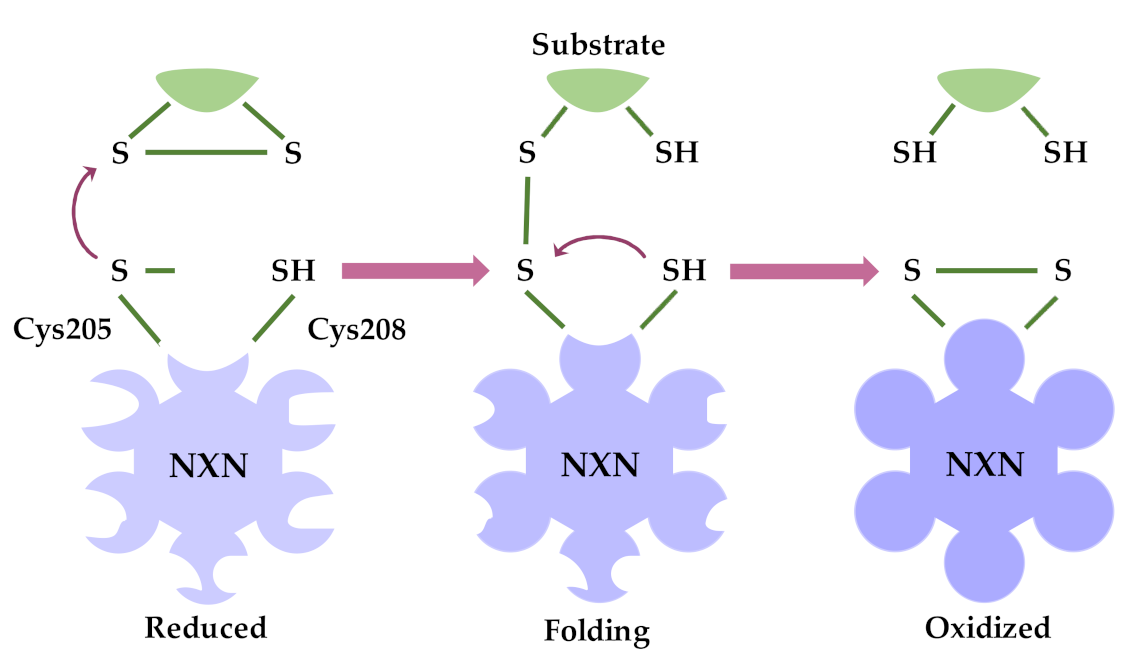
2. Oxidoreductase Activity of NXN
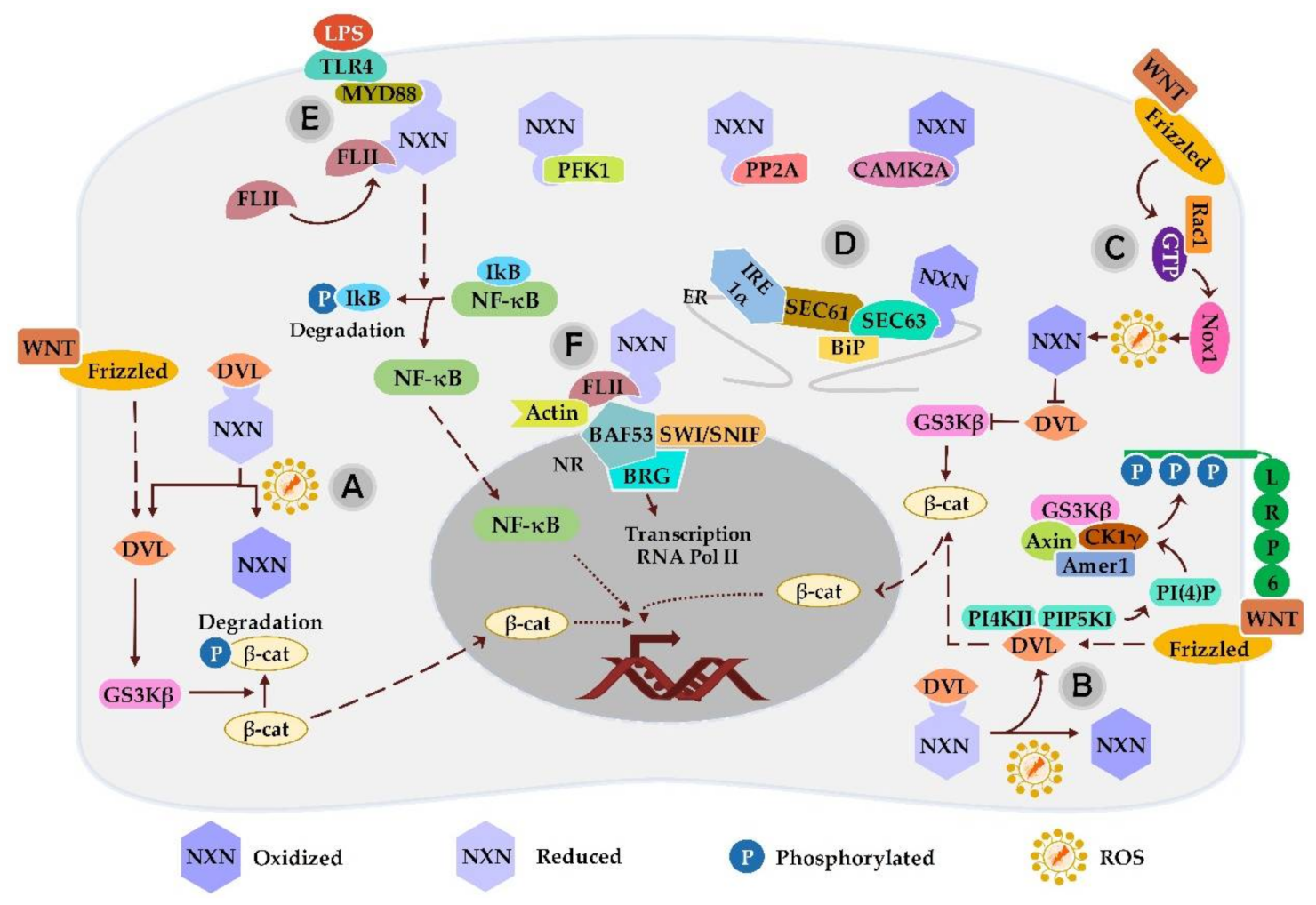
3. Redox-Sensitive Interactions of NXN
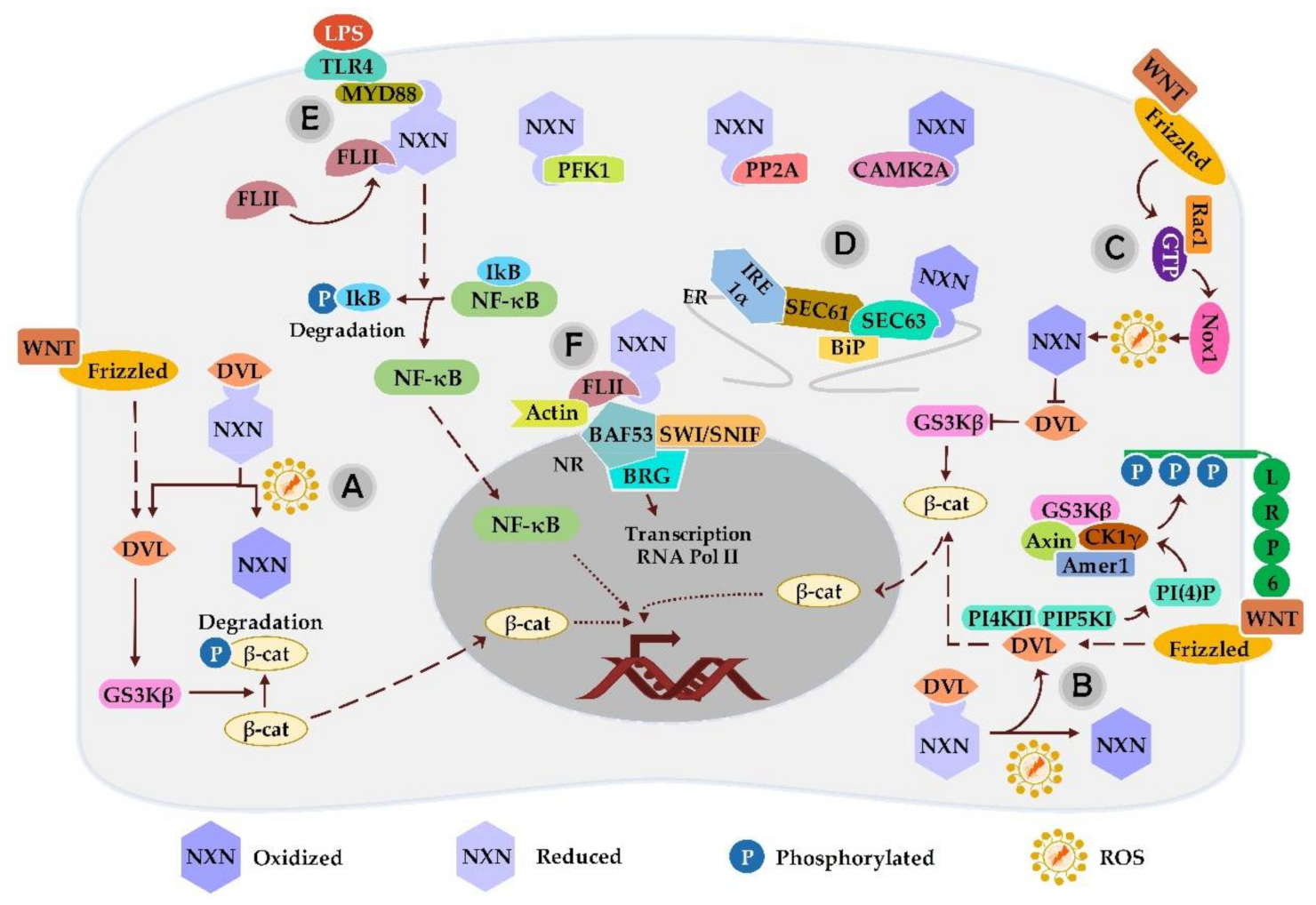
3.1. NXN Interaction with DVL
3.2. NXN Interaction with PFK1
3.3. NXN Interaction with PP2A
3.4. NXN Interaction with MYD88, FLII and Actin
3.5. NXN Interaction with SEC63
3.6. NXN Interaction with CAMK2A
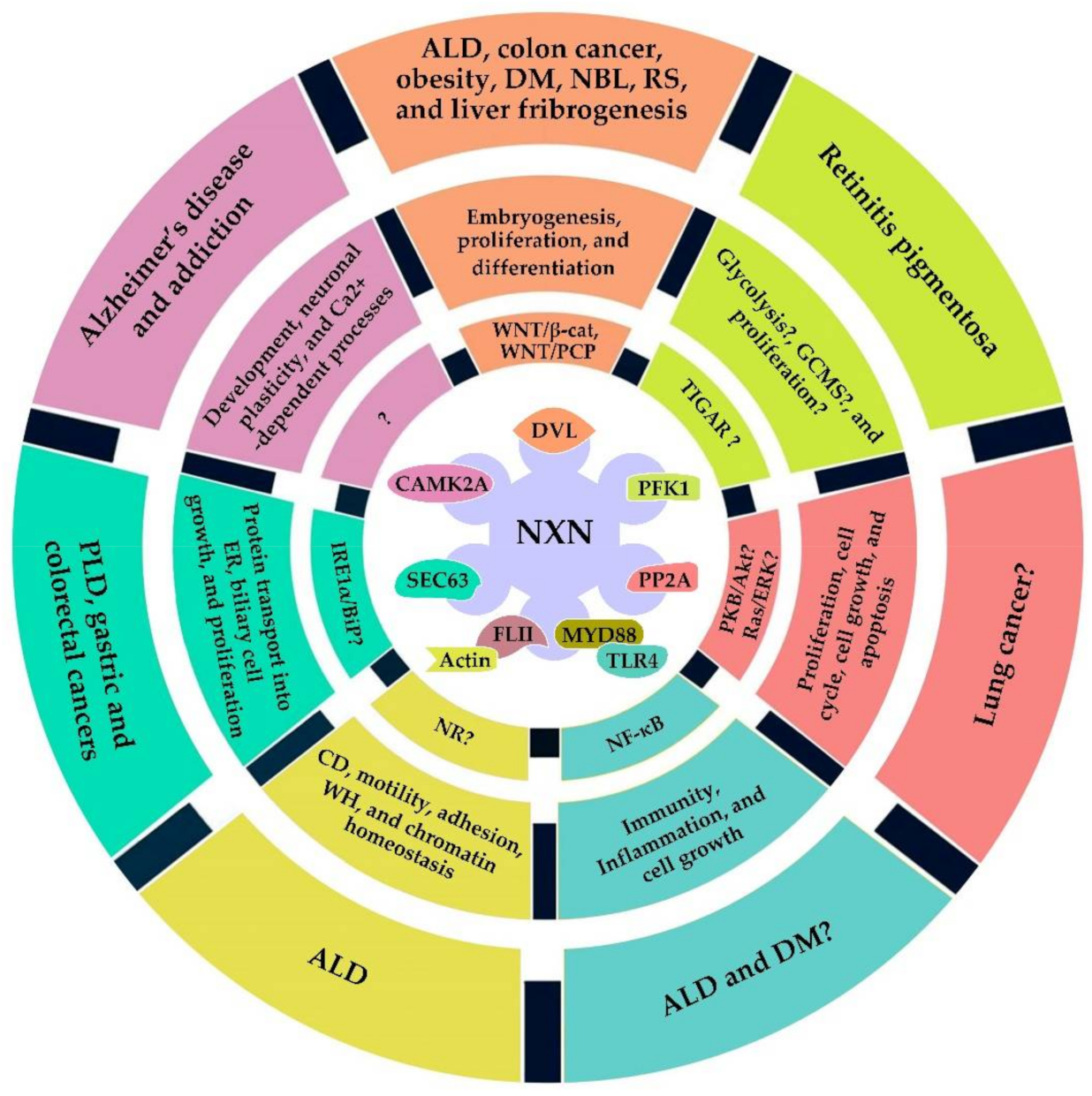
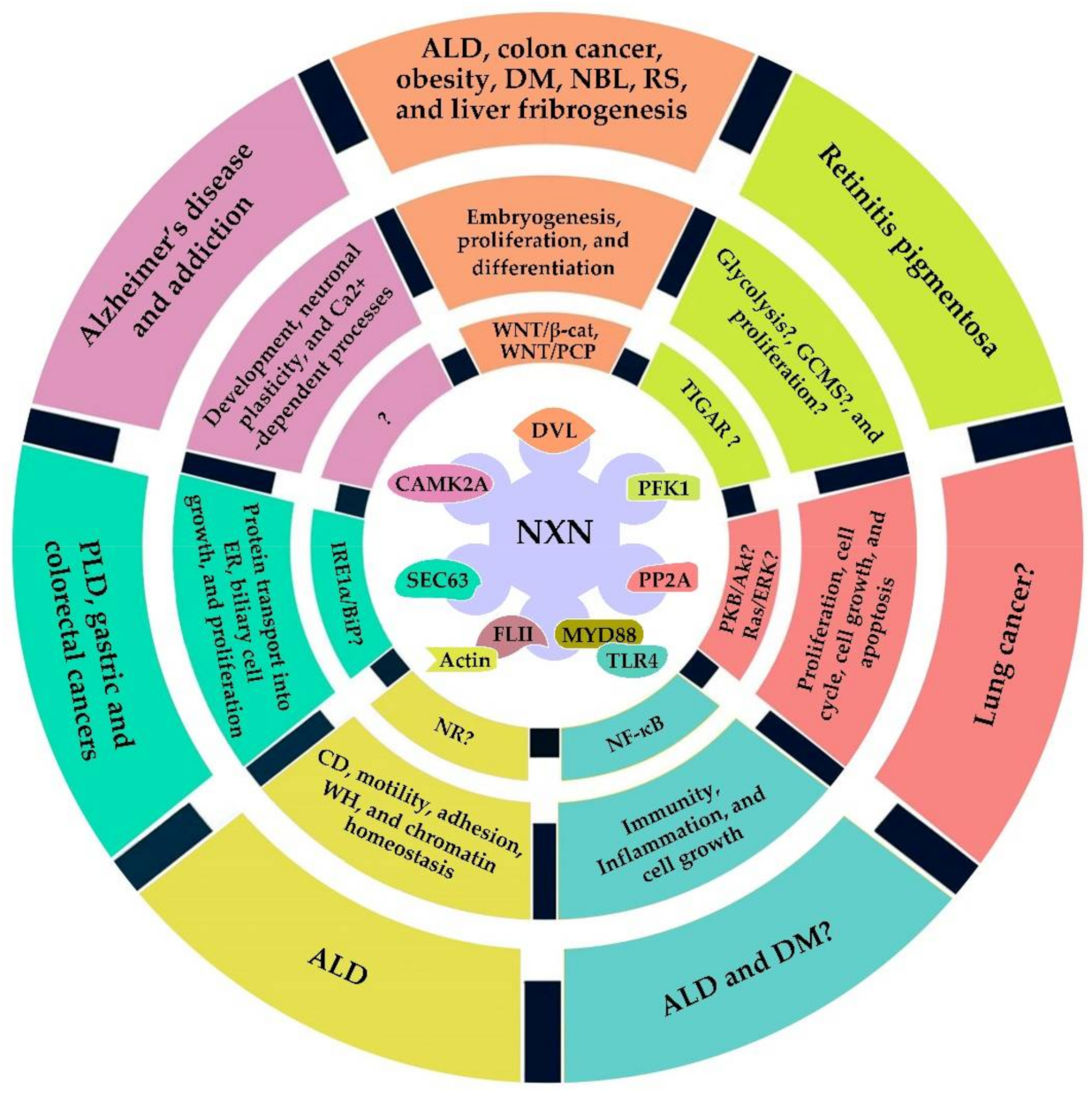
4. NXN Implication in Pathologies
4.1. Diabetes Mellitus
4.2. Obesity
4.3. Brain Diseases
4.4. Hepatic Diseases
4.5. Retinitis Pigmentosa
4.6. Robinow Syndrome
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Léveillard, T.; Aït-Ali, N. Cell Signaling with Extracellular Thioredoxin and Thioredoxin-Like Proteins: Insight into Their Mechanisms of Action. Oxidative Med. Cell. Longev. 2017, 2017, 8475125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, Y.; Siala, W.; Bashandy, T.; Riondet, C.; Vignols, F.; Reichheld, J.P. Glutaredoxins and thioredoxins in plants. Biochim. Biophys. Acta 2008, 1783, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Miki, H. Nucleoredoxin, a Novel Thioredoxin Family Member Involved in Cell Growth and Differentiation. Antioxid. Redox Signal. 2007, 9, 1035–1058. [Google Scholar] [CrossRef]
- Kurooka, H.; Kato, K.; Minoguchi, S.; Takahashi, Y.; Ikeda, J.; Habu, S.; Osawa, N.; Buchberg, A.M.; Moriwaki, K.; Shisa, H.; et al. Cloning and Characterization of the Nucleoredoxin Gene That Encodes a Novel Nuclear Protein Related to Thioredoxin. Genomics 1997, 39, 331–339. [Google Scholar] [CrossRef]
- Funato, Y.; Miki, H. Redox regulation of Wnt signalling via nucleoredoxin. Free Radic. Res. 2010, 44, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Marchal, C.; Delorme-Hinoux, V.; Bariat, L.; Siala, W.; Belin, C.; Saez-Vasquez, J.; Riondet, C.; Reichheld, J.-P. NTR/NRX Define a New Thioredoxin System in the Nucleus of Arabidopsis thaliana Cells. Mol. Plant 2014, 7, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbainsky, C.; Nölker, R.; Imber, M.; Lübken, A.; Mostertz, J.; Hochgräfe, F.; Godoy, J.R.; Hanschmann, E.-M.; Lillig, C.H. Nucleoredoxin-Dependent Targets and Processes in Neuronal Cells. Oxidative Med. Cell. Longev. 2018, 2018, 4829872. [Google Scholar] [CrossRef]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The Thioredoxin-Related Redox-Regulating Protein Nucleoredoxin Inhibits Wnt-Beta-Catenin Signalling through Dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Funato, Y.; Terabayashi, T.; Morinaka, A.; Sakamoto, R.; Ichise, H.; Fukuda, H.; Yoshida, N.; Miki, H. Nucleoredoxin Negatively Regulates Toll-like Receptor 4 Signaling via Recruitment of Flightless-I to Myeloid Differentiation Primary Response Gene (88). J. Biol. Chem. 2010, 285, 18586–18593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechward, K.; Sugajska, E.; De Baere, I.; Goris, J.; Hemmings, B.A.; Zolnierowicz, S. Interaction of nucleoredoxin with protein phosphatase 2A. FEBS Lett. 2006, 580, 3631–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resjö, S.; Göransson, O.; Härndahl, L.; Zolnierowicz, S.; Manganiello, V.; Degerman, E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell. Signal. 2002, 14, 231–238. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, L.; Funato, Y.; Miki, H.; Zimmermann, R. An interaction between human Sec63 and nucleoredoxin may provide the missing link between the SEC63 gene and polycystic liver disease. FEBS Lett. 2011, 585, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, Y.; Hayashi, T.; Irino, Y.; Takenawa, T.; Miki, H. Nucleoredoxin regulates glucose metabolism via phosphofructokinase 1. Biochem. Biophys. Res. Commun. 2013, 440, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Bartrons, R.; Simon-Molas, H.; Rodríguez-García, A.; Castaño, E.; Navarro-Sabaté, À.; Manzano, A.; Martinez-Outschoorn, U.E. Fructose 2,6-Bisphosphate in Cancer Cell Metabolism. Front. Oncol. 2018, 8, 331. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.; Halper, J.P.; Knowles, D.M. Alterations in the activity and isozymic profile of human phosphofructokinase during malignant transformation in vivo and in vitro: Transformation- and progression-linked discriminants of malignancy. Cancer Res. 1985, 45, 2993–3001. [Google Scholar] [PubMed]
- Alarcon-Sanchez, R.B.; Guerrero-Escalera, D.; Rosas-Madrigal, S.; Aparicio-Bautista, D.I.; Reyes-Gordillo, K.; Lakshman, M.R.; Ortiz-Fernandez, A.; Quezada, H.; Medina-Contreras, O.; Villa-Trevino, S.; et al. Nucleoredoxin Interaction with Flightless-I/Actin Complex Is Differentially Altered in Alcoholic Liver Disease. Basic Clin. Pharmacol. Toxicol. 2020, 127, 389–404. [Google Scholar] [CrossRef]
- Tran, B.N.; Valek, L.; Wilken-Schmitz, A.; Fuhrmann, D.C.; Namgaladze, D.; Wittig, I.; Tegeder, I. Reduced exploratory behavior in neuronal nucleoredoxin knockout mice. Redox Biol. 2021, 45, 102054. [Google Scholar] [CrossRef]
- Hanschmann, M.E.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Kallis, G.B.; Holmgren, A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J. Biol. Chem. 1980, 255, 10261–10265. [Google Scholar] [CrossRef]
- Dyson, H.J.; Jeng, M.-F.; Tennant, L.L.; Slaby, I.; Lindell, M.; Cui, D.-S.; Kuprin, A.S.; Holmgren, A. Effects of Buried Charged Groups on Cysteine Thiol Ionization and Reactivity in Escherichia coli Thioredoxin: Structural and Functional Characterization of Mutants of Asp 26 and Lys 57. Biochemistry 1997, 36, 2622–2636. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arellanes-Robledo, J.; Reyes-Gordillo, K.; Shah, R.; Dominguez-Rosales, J.A.; Hernandez-Nazara, Z.H.; Ramirez, F.; Rojkind, M.; Lakshman, M.R. Fibrogenic Actions of Acetaldehyde Are Beta-Catenin Dependent but Wingless Independent: A Critical Role of Nucleoredoxin and Reactive Oxygen Species in Human Hepatic Stellate Cells. Free Radic. Biol. Med. 2013, 65, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Nusse, R. Towards an Integrated View of Wnt Signaling in Development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Katoh, M. Wnt Signaling Pathway and Stem Cell Signaling Network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.-C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382, 225–230. [Google Scholar] [CrossRef]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef]
- Semenov, M.V.; Habas, R.; Macdonald, B.T.; He, X. SnapShot: Noncanonical Wnt Signaling Pathways. Cell 2007, 131, 1378.e1–1378.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Chen, Y.-G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Boutros, M.; Paricio, N.; Strutt, D.; Mlodzik, M. Dishevelled Activates JNK and Discriminates between JNK Pathways in Planar Polarity and wingless Signaling. Cell 1998, 94, 109–118. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, T.B.; Tamai, K.; He, X. Wnt/Beta-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilić, J.; Huang, Y.-L.; Davidson, G.; Zimmermann, T.; Cruciat, C.-M.; Bienz, M.; Niehrs, C. Wnt Induces LRP6 Signalosomes and Promotes Dishevelled-Dependent LRP6 Phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Rharass, T.; Lantow, M.; Gbankoto, A.; Weiss, D.G.; Panakova, D.; Lucas, S. Ascorbic Acid Alters Cell Fate Commitment of Human Neural Progenitors in a Wnt/Beta-Catenin/Ros Signaling Dependent Manner. J. Biomed. Sci. 2017, 24, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajla, S.; Mondol, A.S.; Nagasawa, A.; Zhang, Y.; Kato, M.; Matsuno, K.; Yabe-Nishimura, C.; Kamata, T. A Crucial Role for Nox 1 in Redox-Dependent Regulation of Wnt-Beta-Catenin Signaling. FASEB J. 2012, 26, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Michiue, T.; Terabayashi, T.; Yukita, A.; Danno, H.; Asashima, M.; Miki, H. Nucleoredoxin regulates the Wnt/planar cell polarity pathway inXenopus. Genes Cells 2008, 13, 965–975. [Google Scholar] [CrossRef]
- Bahn, J.Y.; Lee, K.P.; Lee, S.M.; Choi, J.Y.; Seo, Y.S.; Kwon, K.S. Nucleoredoxin Promotes Adipogenic Differentiation through Regulation of Wnt/Beta-Catenin Signaling. J. Lipid Res. 2015, 56, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Arellanes-Robledo, J.; Reyes-Gordillo, K.; Ibrahim, J.; Leckey, L.; Shah, R.; Lakshman, M.R. Ethanol targets nucleoredoxin/dishevelled interactions and stimulates phosphatidylinositol 4-phosphate production in vivo and in vitro. Biochem. Pharmacol. 2018, 156, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kanai, S.; Shimada, T.; Narita, T.; Okabayashi, K. Phosphofructokinase-1 Subunit Composition and Activity in the Skeletal Muscle, Liver, and Brain of Dogs. J. Vet. Med. Sci. 2019, 81, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zancan, P.; Almeida, F.V.; Faber-Barata, J.; Dellias, J.M.; Sola-Penna, M. Fructose-2,6-bisphosphate counteracts guanidinium chloride-, thermal-, and ATP-induced dissociation of skeletal muscle key glycolytic enzyme 6-phosphofructo-1-kinase: A structural mechanism for PFK allosteric regulation. Arch. Biochem. Biophys. 2007, 467, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Bartrons, R.; Rodríguez-García, A.; Simon-Molas, H.; Castaño, E.; Manzano, A.; Navarro-Sabaté, À. The potential utility of PFKFB3 as a therapeutic target. Expert Opin. Ther. Targets 2018, 22, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Telang, S.; Clem, B.; Chesney, J. Regulation of Glucose Metabolism by 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatases in Cancer. Exp. Mol. Pathol. 2009, 86, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Hennipman, A.; Smits, J.; Van Oirschot, B.; Van Houwelingen, J.; Rijksen, G.; Neyt, J.; Van Unnik, J.; Staal, G. Glycolytic Enzymes in Breast Cancer, Benign Breast Disease and Normal Breast Tissue. Tumor Biol. 1987, 8, 251–263. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodriguez-Enriquez, S.; Marín-Hernández, Á.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, V.W.Y.; Lau, C.P.Y.; Cheung, C.S.F.; Ho, K.; Ng, M.H.L.; Cheng, S.H.; Hong, B.; Tsao, S.-W.; Tsang, C.M.; Lei, K.I.K.; et al. An RNA-directed nucleoside anti-metabolite, 1-(3-C-ethynyl-beta-d-ribo-pentofuranosyl)cytosine (ECyd), elicits antitumor effect via TP53-induced Glycolysis and Apoptosis Regulator (TIGAR) downregulation. Biochem. Pharmacol. 2010, 79, 1772–1780. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., 3rd; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Groves, R.M.; Hanlon, N.; Turowski, P.; Hemmings, B.A.; Barford, D. The Structure of the Protein Phosphatase 2a Pr65/a Subunit Reveals the Conformation of Its 15 Tandemly Repeated Heat Motifs. Cell 1999, 96, 99–110. [Google Scholar] [CrossRef]
- Guo, F.; Stanevich, V.; Wlodarchak, N.; Sengupta, R.; Jiang, L.; Satyshur, K.A.; Xing, Y. Structural Basis of Pp2a Activation by Ptpa, an Atp-Dependent Activation Chaperone. Cell Res. 2014, 24, 190–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Ugi, S.; Imamura, T.; Ricketts, W.; Olefsky, J.M. Protein Phosphatase 2A Forms a Molecular Complex with Shc and Regulates Shc Tyrosine Phosphorylation and Downstream Mitogenic Signaling. Mol. Cell. Biol. 2002, 22, 2375–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, A.J.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/Mek/Erk Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, G.; Wang, W.; Chai, K.; Yang, S.; Li, F.; Jiang, K. Combination Treatment with Triptolide and Hydroxycamptothecin Synergistically Enhances Apoptosis in A549 Lung Adenocarcinoma Cells through Pp2a-Regulated Erk, P38 Mapks and Akt Signaling Pathways. Int. J. Oncol. 2015, 46, 1007–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [Green Version]
- Arriazu, E.; Pippa, R.; Odero, M.D. Protein Phosphatase 2a as a Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 2016, 6, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, S.J.; James, D.E. Uncaging Akt. Sci. Signal 2012, 5, pe20. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, Z.; Renier, N.; Simon, D.J.; Uryu, K.; Park, D.; Greer, P.A.; Tournier, C.; Davis, R.J.; Tessier-Lavigne, M. Pathological Axonal Death through a MAPK Cascade that Triggers a Local Energy Deficit. Cell 2015, 160, 161–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Y.; Lin, K.T.; Chen, S.C.; Gu, D.L.; Chen, C.F.; Tu, P.H.; Jou, Y.S. Overexpressed-Eif3i Interacted and Activated Oncogenic Akt1 Is a Theranostic Target in Human Hepatocellular Carcinoma. Hepatology 2013, 58, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.D.; Coleman, C.S.; Berg, A.; Jefferson, L.S.; Kimball, S.R. REDD1 enhances protein phosphatase 2A–mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci. Signal. 2014, 7, ra68. [Google Scholar] [CrossRef] [Green Version]
- Lei, N.; Peng, B.; Zhang, J.-Y. CIP2A regulates cell proliferation via the AKT signaling pathway in human lung cancer. Oncol. Rep. 2014, 32, 1683–1694. [Google Scholar] [CrossRef] [Green Version]
- Ohama, T.; Brautigan, D.L. Endotoxin Conditioning Induces VCP/p97-mediated and Inducible Nitric-oxide Synthase-dependent Tyr284 Nitration in Protein Phosphatase 2A. J. Biol. Chem. 2010, 285, 8711–8718. [Google Scholar] [CrossRef] [Green Version]
- Dagnell, M.; Pace, P.E.; Cheng, Q.; Frijhoff, J.; Östman, A.; Arnér, E.; Hampton, M.B.; Winterbourn, C.C. Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J. Biol. Chem. 2017, 292, 14371–14380. [Google Scholar] [CrossRef] [Green Version]
- Lord, K.; Hoffman-Liebermann, B.; Liebermann, D. Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene 1990, 5, 1095–1097. [Google Scholar]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of MyD88-Deficient Mice to Endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, Y.; Shen, P. Nf-Kappab and Its Regulation on the Immune System. Cell Mol. Immunol. 2004, 1, 343–350. [Google Scholar] [PubMed]
- Pereira, G.S.; Oakley, F. Nuclear Factor-Kappab1: Regulation and Function. Int. J. Biochem. Cell Biol. 2008, 40, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, M.A.; Basith, S.; Choi, S. Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp. Mol. Med. 2013, 45, e11. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Fitzgerald, K.; Bowie, A. The Toll–IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003, 24, 286–289. [Google Scholar] [CrossRef]
- Bieghs, V.; Trautwein, C. Innate immune signaling and gut-liver interactions in non-alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2014, 3, 377–385. [Google Scholar] [CrossRef]
- Yin, H.; Huang, L.; Ouyang, T.; Chen, L. Baicalein Improves Liver Inflammation in Diabetic Db/Db Mice by Regulating Hmgb1/Tlr4/Nf-Kappab Signaling Pathway. Int. Immunopharmacol. 2018, 55, 55–62. [Google Scholar] [CrossRef]
- Liew, F.Y.; Xu, D.; Brint, E.; O’Neill, L. Negative regulation of Toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005, 5, 446–458. [Google Scholar] [CrossRef]
- Wang, T.; Gu, S.; Ronni, T.; Du, Y.-C.; Chen, X. In Vivo Dual-Tagging Proteomic Approach in Studying Signaling Pathways in Immune Response. J. Proteome Res. 2005, 4, 941–949. [Google Scholar] [CrossRef]
- Ruzehaji, N.; Mills, S.J.; Melville, E.; Arkell, R.; Fitridge, R.; Cowin, A.J. The Influence of Flightless I on Toll-Like-Receptor-Mediated Inflammation in a Murine Model of Diabetic Wound Healing. BioMed Res. Int. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chuang, T.-H.; Ronni, T.; Gu, S.; Du, Y.-C.; Cai, H.; Tsung-Hsien, C.; Yin, H.L.; Chen, X. Flightless I Homolog Negatively Modulates the TLR Pathway. J. Immunol. 2006, 176, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davy, D.; Campbell, H.D.; Fountain, S.; De Jong, D.; Crouch, M.F. The flightless I protein colocalizes with actin- and microtubule-based structures in motile Swiss 3T3 fibroblasts: Evidence for the involvement of PI 3-kinase and Ras-related small GTPases. J. Cell Sci. 2001, 114, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Campbell, H.D.; Stallcup, M.R. Developmentally Essential Protein Flightless I Is a Nuclear Receptor Coactivator with Actin Binding Activity. Mol. Cell. Biol. 2004, 24, 2103–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.; Claudianos, C.; Campbell, H.D. Evolution of the gelsolin family of actin-binding proteins as novel transcriptional coactivators. BioEssays 2005, 27, 388–396. [Google Scholar] [CrossRef]
- Jeong, K.W. Flightless I (Drosophila) Homolog Facilitates Chromatin Accessibility of the Estrogen Receptor Alpha Target Genes in Mcf-7 Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2014, 446, 608–613. [Google Scholar] [CrossRef]
- Li, X.; Sun, S.; Appathurai, S.; Sundaram, A.; Plumb, R.; Mariappan, M. A Molecular Mechanism for Turning Off IRE1α Signaling during Endoplasmic Reticulum Stress. Cell Rep. 2020, 33, 108563. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Davila, S.; Furu, L.; Gharavi, A.G.; Tian, X.; Onoe, T.; Qian, Q.; Li, A.; Cai, Y.; Kamath, P.S.; King, B.F.; et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 2004, 36, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schäuble, N.; Jalal, C.; Greiner, M.; Haßdenteufel, S.; Tatzelt, J.; et al. Differential effects of Sec61α-, Sec62- and Sec63-depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef]
- Jermy, A.; Willer, M.; Davis, E.; Wilkinson, B.M.; Stirling, C.J. The Brl Domain in Sec63p Is Required for Assembly of Functional Endoplasmic Reticulum Translocons. J. Biol. Chem. 2006, 281, 7899–7906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, J.B.; Devaraneni, P.K.; Yang, Z.; David, L.L.; Skach, W.R. Cotranslational Stabilization of Sec62/63 within the Er Sec61 Translocon Is Controlled by Distinct Substrate-Driven Translocation Events. Mol. Cell 2015, 58, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.X.; Murray, K.D. Neuronal Excitability and Calcium/Calmodulin-Dependent Protein Kinase Type Ii: Location, Location, Location. Epilepsia 2012, 53, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W. CaMKII: Claiming Center Stage in Postsynaptic Function and Organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Hanson, I.P.; Schulman, H. Neuronal Ca2+/Calmodulin-Dependent Protein Kinases. Annu. Rev. Biochem. 1992, 61, 559–601. [Google Scholar] [CrossRef]
- Erondu, E.N.; Kennedy, M.B. Regional Distribution of Type Ii Ca2+/Calmodulin-Dependent Protein Kinase in Rat Brain. J. Neurosci. 1985, 5, 3270–3277. [Google Scholar] [CrossRef] [Green Version]
- Soderling, R.T.; Chang, B.; Brickey, D. Cellular Signaling through Multifunctional Ca2+/Calmodulin-Dependent Protein Kinase Ii. J. Biol. Chem. 2001, 276, 3719–3722. [Google Scholar] [CrossRef] [Green Version]
- Coultrap, S.J.; Freund, R.K.; O’Leary, H.; Sanderson, J.L.; Roche, K.; Dell’Acqua, M.L.; Bayer, K.U. Autonomous CaMKII Mediates Both LTP and LTD Using a Mechanism for Differential Substrate Site Selection. Cell Rep. 2014, 6, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Giese, P.K.; Fedorov, N.B.; Filipkowski, R.K.; Silva, A.J. Autophosphorylation at Thr286 of the Alpha Calcium-Calmodulin Kinase Ii in Ltp and Learning. Science 1998, 279, 870–873. [Google Scholar] [CrossRef]
- Jaillard, C.; Ouechtati, F.; Clérin, E.; Millet-Puel, G.; Corsi, M.; Aït-Ali, N.; Blond, F.; Chevy, Q.; Gales, L.; Farinelli, M.; et al. The metabolic signaling of the nucleoredoxin-like 2 gene supports brain function. Redox Biol. 2021, 48, 102198. [Google Scholar] [CrossRef] [PubMed]
- Valek, L.; Tegeder, I. Nucleoredoxin Knockdown in SH-SY5Y Cells Promotes Cell Renewal. Antioxidants 2021, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- White, J.J.; Mazzeu, J.; Coban-Akdemir, Z.; Bayram, Y.; Bahrambeigi, V.; Hoischen, A.; van Bon, B.W.; Gezdirici, A.; Gulec, E.Y.; Ramond, F.; et al. WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome. Am. J. Hum. Genet. 2017, 102, 27–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clérin, E.; Marussig, M.; Sahel, J.-A.; Léveillard, T. Metabolic and Redox Signaling of the Nucleoredoxin-Like-1 Gene for the Treatment of Genetic Retinal Diseases. Int. J. Mol. Sci. 2020, 21, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssens, V.; Goris, J. Protein Phosphatase 2a: A Highly Regulated Family of Serine/Threonine Phosphatases Implicated in Cell Growth and Signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Hirota, K.; Matsui, M.; Murata, M.; Takashima, Y.; Cheng, F.S.; Itoh, T.; Fukuda, K.; Yodoi, J. Nucleoredoxin, Glutaredoxin, and Thioredoxin Differentially Regulate Nf-Kappab, Ap-1, and Creb Activation in Hek293 Cells. Biochem. Biophys. Res. Commun. 2000, 274, 177–182. [Google Scholar] [CrossRef]
- Mori, Y.; Sato, F.; Selaru, F.M.; Olaru, A.; Perry, K.; Kimos, M.C.; Tamura, G.; Matsubara, N.; Wang, S.; Xu, Y.; et al. Instabilotyping reveals unique mutational spectra in microsatellite-unstable gastric cancers. Cancer Res. 2002, 62, 3641–3645. [Google Scholar]
- Casper, M.; Weber, S.N.; Kloor, M.; Müllenbach, R.; Grobholz, R.; Lammert, F.; Zimmer, V. Hepatocellular carcinoma as extracolonic manifestation of Lynch syndrome indicatesSEC63as potential target gene in hepatocarcinogenesis. Scand. J. Gastroenterol. 2012, 48, 344–351. [Google Scholar] [CrossRef]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2015, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Association, American Diabetes. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2014, 37, S81–S90. [Google Scholar] [CrossRef] [Green Version]
- Babaya, N.; Ikegami, H.; Fujisawa, T.; Nojima, K.; Itoi-Babaya, M.; Inoue, K.; Ohno, T.; Shibata, M.; Ogihara, T. Susceptibility to streptozotocin-induced diabetes is mapped to mouse chromosome 11. Biochem. Biophys. Res. Commun. 2005, 328, 158–164. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. Nf-Kappabeta: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chooi, C.Y.; Ding, C.; Magkos, F. The Epidemiology of Obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Flier, J.S. Obesity and the Regulation of Energy Balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Rosen, D.E.; Spiegelman, B.M. Molecular Regulation of Adipogenesis. Annu. Rev. Cell Dev. Biol. 2000, 16, 145–171. [Google Scholar] [CrossRef]
- Moldes, M.; Zuo, Y.; Morrison, R.F.; Silva, D.; Park, B.-H.; Liu, J.; Farmer, S.R. Peroxisome-proliferator-activated receptor γ suppresses Wnt/β-catenin signalling during adipogenesis. Biochem. J. 2003, 376, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of Adipogenesis by Wnt Signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Jocken, J.W.E.; Langin, D.; Smit, E.; Saris, W.H.M.; Valle, C.; Hul, G.B.; Holm, C.; Arner, P.; Blaak, E.E. Adipose Triglyceride Lipase and Hormone-Sensitive Lipase Protein Expression Is Decreased in the Obese Insulin-Resistant State. J. Clin. Endocrinol. Metab. 2007, 92, 2292–2299. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Tordjman, J.; Poitou, C.; Darakhshan, F.; Hugol, D.; Basdevant, A.; Aissat, A.; Guerre-Millo, M.; Clément, K. Human Adipose Tissue Macrophages: M1 and M2 Cell Surface Markers in Subcutaneous and Omental Depots and after Weight Loss. J. Clin. Endocrinol. Metab. 2009, 94, 4619–4623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Duthey, B. Background Paper 6.11: Alzheimer Disease and Other Dementias. Public Health Approach Innov. 2013, 6, 1–74. [Google Scholar]
- Jaillard, C.; Mouret, A.; Niepon, M.; Clérin, E.; Yang, Y.; Lee-Rivera, I.; Aït-Ali, N.; Millet-Puel, G.; Cronin, T.; Sedmak, T. Nxnl2 Splicing Results in Dual Functions in Neuronal Cell Survival and Maintenance of Cell Integrity. Hum. Mol. Genet. 2012, 21, 2298–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.; Concannon, C.G.; Ward, M.W.; Walsh, C.M.; Tirniceriu, A.L.; Tribl, F.; Kögel, D.; Prehn, J.H.M.; Egensperger, R. Modulation of Gene Expression and Cytoskeletal Dynamics by the Amyloid Precursor Protein Intracellular Domain (Aicd). Mol. Biol. Cell 2007, 18, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Maris, M.J.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Lamsa, K.; Irvine, E.E.; Giese, K.P.; Kullmann, D.M. Nmda Receptor-Dependent Long-Term Potentiation in Mouse Hippocampal Interneurons Shows a Unique Dependence on Ca(2+)/Calmodulin-Dependent Kinases. J. Physiol. 2007, 584, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Mijakowska, Z.; Łukasiewicz, K.; Ziółkowska, M.; Lipiński, M.; Trąbczyńska, A.; Matuszek, Ż.; Łęski, S.; Radwanska, K. Autophosphorylation of Alpha Isoform of Calcium/Calmodulin-Dependent Kinase Ii Regulates Alcohol Addiction-Related Behaviors. Addict. Biol. 2017, 22, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S. Neural plasticity: Consequences of stress and actions of antidepressant treatment. Dialog. Clin. Neurosci. 2004, 6, 157–169. [Google Scholar]
- Miller, A.M.; Horiguchi, N.; Jeong, W.-I.; Radaeva, S.; Gao, B. Molecular Mechanisms of Alcoholic Liver Disease: Innate Immunity and Cytokines. Alcohol. Clin. Exp. Res. 2011, 35, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Taylor, B.; Mohapatra, S.; Irving, H.; Baliunas, D.; Patra, J.; Roerecke, M. Alcohol as a risk factor for liver cirrhosis: A systematic review and meta-analysis. Drug Alcohol Rev. 2010, 29, 437–445. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, G.; Noguchi, T.; Takeda, K.; Ichijo, H. Thioredoxin and protein kinases in redox signaling. Semin. Cancer Biol. 2006, 16, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Arellanes-Robledo, J.; Ibrahim, J.; Reyes-Gordillo, K.; Shah, R.; Leckey, L.; Lakshman, M.R. Flightless-I is a potential biomarker for the early detection of alcoholic liver disease. Biochem. Pharmacol. 2020, 183, 114323. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, S.V.; So, J.-S.; Shrikhande, A.; Lee, S.H.; Gallagher, A.-R.; Barkauskas, C.E.; Somlo, S.; Lee, A.-H. Sec63 and Xbp1 regulate IRE1α activity and polycystic disease severity. J. Clin. Investig. 2015, 125, 1955–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedeles, S.V.; Gallagher, A.-R.; Somlo, S. Polycystin-1: A master regulator of intersecting cystic pathways. Trends Mol. Med. 2014, 20, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Janssen, M.J.; Salomon, J.; Morsche, R.H.M.T.; Drenth, J.P.H. Loss of Heterozygosity Is Present in SEC63 Germline Carriers with Polycystic Liver Disease. PLoS ONE 2012, 7, e50324. [Google Scholar] [CrossRef] [Green Version]
- Dryja, T.P.; McGee, T.L.; Reichel, E.; Hahn, L.B.; Cowley, G.S.; Yandell, D.W.; Sandberg, M.A.; Berson, E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343, 364–366. [Google Scholar] [CrossRef]
- Marlhens, F.; Bareil, C.; Griffoin, J.; Zrenner, E.; Amalric, P.; Eliaou, C.; Liu, S.; Harris, E.; Redmond, T.M.; Arnaud, B. Mutations in Rpe65 Cause Leber’s Congenital Amaurosis. Nat. Genet. 1997, 17, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Redmond, T.M..; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.-X.; Crouch, R.K.; Pfeifer, K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Léveillard, T.; Picaud, S.; Dalkara, D.; Marazova, K.; Safran, A.; Paques, M.; Duebel, J.; Roska, B.; Mohand-Said, S. Functional Rescue of Cone Photoreceptors in Retinitis Pigmentosa. Graefe Arch. Clin. Exp. Ophthalmol. 2013, 251, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Chaffiol, A.; Kole, C.; Yang, Y.; Millet-Puel, G.; Clérin, E.; Aït-Ali, N.; Bennett, J.; Dalkara, D.; Sahel, J. The Thioredoxin Encoded by the Rod-Derived Cone Viability Factor Gene Protects Cone Photoreceptors against Oxidative Stress. Antioxid. Redox Signal. 2016, 24, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.C.; Applebury, M.L.; Farber, D.B. Retinal Degeneration in the Rd Mouse Is Caused by a Defect in the Β Subunit of Rod Cgmp-Phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.-Q.; Hao, Y.; Wong, F. Apoptosis: Final Common Pathway of Photoreceptor Death in Rd, Rds, and Mutant Mice. Neuron 1993, 11, 595–605. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef] [Green Version]
- Portera-Cailliau, C.; Sung, C.H.; Nathans, J.; Adler, R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1994, 91, 974–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronin, T.; Raffelsberger, W.; Lee-Rivera, I.; Jaillard, C.; Niepon, M.-L.; Kinzel, B.; Clérin, E.; Petrosian, A.; Picaud, S.; Poch, O.; et al. The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ. 2010, 17, 1199–1210. [Google Scholar] [CrossRef]
- Warman, M.L.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Lachman, R.; LeMerrer, M.; Mortier, G.; Mundlos, S.; Nishimura, G.; Rimoin, D.L.; et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. Part A 2011, 155, 943–968. [Google Scholar] [CrossRef]
- Patton, M.A.; Afzal, A.R. Robinow Syndrome. J. Med. Genet. 2002, 39, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Van Bokhoven, H.; Celli, J.; Kayserili, H.; Van Beusekom, E.; Balci, S.; Brussel, W.; Skovby, F.; Kerr, B.; Percin, E.F.; Akarsu, N.; et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 2000, 25, 423–426. [Google Scholar] [CrossRef]
- Gu, S.; Yuan, B.; Campbell, I.; Beck, C.R.; Carvalho, C.M.; Nagamani, S.C.; Erez, A.; Patel, A.; Bacino, C.A.; Shaw, C.A.; et al. Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum. Mol. Genet. 2015, 24, 4061–4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, K.M.; Wilkinson, B.M.; Wilming, L.G.; Liu, B.; Probst, F.J.; Harrow, J.; Grafham, D.; Hentges, K.E.; Woodward, L.P.; Maxwell, A.; et al. Discovery of Candidate Disease Genes in Enu-Induced Mouse Mutants by Large-Scale Sequencing, Including a Splice-Site Mutation in Nucleoredoxin. PLoS Genet. 2009, 5, e1000759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, Y.; Terabayashi, T.; Sakamoto, R.; Okuzaki, D.; Ichise, H.; Nojima, H.; Yoshida, N.; Miki, H. Nucleoredoxin Sustains Wnt/Beta-Catenin Signaling by Retaining a Pool of Inactive Dishevelled Protein. Curr. Biol. 2010, 20, 1945–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneeshaw, S.; Keyani, R.; Delorme-Hinoux, V.; Imrie, L.; Loake, G.J.; Le Bihan, T.; Reichheld, J.-P.; Spoel, S.H. Nucleoredoxin guards against oxidative stress by protecting antioxidant enzymes. Proc. Natl. Acad. Sci. USA 2017, 114, 8414–8419. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NXN Interaction | Signaling Pathway | Cellular Process | Disease | Experimental Model | References |
|---|---|---|---|---|---|
| Disheveled (DVL) | WNT/β-catenin, phosphatidylinositol 4- phosphate [PI (4) P] production | Embryogenesis and organogenesis Cell proliferation and differentiation Liver fibrogenesis | Alcoholic liver disease (ALD) Hepatocellular carcinoma (HCC) Obesity Neuroblastoma (NBL) Robinow syndrome (RS) | C57BL/6J, 129/SvJ mice, human embryonic kidney (HEK)293, murine embryonic fibroblast cell line (NIH3T3), hepatic stellate cell (HSC) and SY-SY5Y cells | [26,38,39,102,103] |
| Phosphofructo kinase-1 (PFK1) | TP53-induced glycolysis and apoptosis regulator (TIGAR) | Glycolysis Global cell metabolic state (GCMS) Cell proliferation | Retinitis pigmentosa | C57BL/6J and BALB/c mice, mouse embryonic fibroblast (MEF), HEK293 | [14,44,104] |
| Protein phosphatase 2A (PP2A) | Protein kinase B (PKB) or Akt | Cell cycle progression Apoptosis Cell growth | Lung cancer | Lung cancer cell lines (NCI-H838, NCI-H1299 and NCI-H460) | [10,12,64,105] |
| Myeloid differentiation primary response gene-88/Flightless-1 (MYD88/FLII) | Nuclear factor kappa beta (NF-κB) | Inflammation Immunity Cell growth | ALD | MEFs and COS7 cells, C57BL/6J mice HEK293,NIH3T3 | [9,80,106] |
| FLII/ACTIN | Nuclear receptors (NR) | Cytoskeletal dynamics Motility Contraction Adhesion Wound healing (WH) Chromatin homeostasis | ALD | C57BL/6J mice, HSC, VL17A cells | [17] |
| Translocation protein SEC63 homolog (SEC63) | IRE1α/BIP | Transport of proteins into ER Biliary cell growth Proliferation | Gastric and colorectal cancers Polycystic liver disease (PLD) | Mouse, C57BL/6J and DBA/2J | [13,107,108] |
| Calcium/calmodulin-dependent protein kinase II type alpha (CAMK2A) | None | Neuronal plasticity Development Ca2+-dependent processes | AD Autism Schizophrenia Addiction | European Conditional Mouse Mutagenesis Program (EUCOMM), Yeast-2-hybrid | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idelfonso-García, O.G.; Alarcón-Sánchez, B.R.; Vásquez-Garzón, V.R.; Baltiérrez-Hoyos, R.; Villa-Treviño, S.; Muriel, P.; Serrano, H.; Pérez-Carreón, J.I.; Arellanes-Robledo, J. Is Nucleoredoxin a Master Regulator of Cellular Redox Homeostasis? Its Implication in Different Pathologies. Antioxidants 2022, 11, 670. https://doi.org/10.3390/antiox11040670
Idelfonso-García OG, Alarcón-Sánchez BR, Vásquez-Garzón VR, Baltiérrez-Hoyos R, Villa-Treviño S, Muriel P, Serrano H, Pérez-Carreón JI, Arellanes-Robledo J. Is Nucleoredoxin a Master Regulator of Cellular Redox Homeostasis? Its Implication in Different Pathologies. Antioxidants. 2022; 11(4):670. https://doi.org/10.3390/antiox11040670
Chicago/Turabian StyleIdelfonso-García, Osiris Germán, Brisa Rodope Alarcón-Sánchez, Verónica Rocío Vásquez-Garzón, Rafael Baltiérrez-Hoyos, Saúl Villa-Treviño, Pablo Muriel, Héctor Serrano, Julio Isael Pérez-Carreón, and Jaime Arellanes-Robledo. 2022. "Is Nucleoredoxin a Master Regulator of Cellular Redox Homeostasis? Its Implication in Different Pathologies" Antioxidants 11, no. 4: 670. https://doi.org/10.3390/antiox11040670
APA StyleIdelfonso-García, O. G., Alarcón-Sánchez, B. R., Vásquez-Garzón, V. R., Baltiérrez-Hoyos, R., Villa-Treviño, S., Muriel, P., Serrano, H., Pérez-Carreón, J. I., & Arellanes-Robledo, J. (2022). Is Nucleoredoxin a Master Regulator of Cellular Redox Homeostasis? Its Implication in Different Pathologies. Antioxidants, 11(4), 670. https://doi.org/10.3390/antiox11040670