
3-Hydroxybutyrate Ameliorates the Progression of Diabetic Nephropathy

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
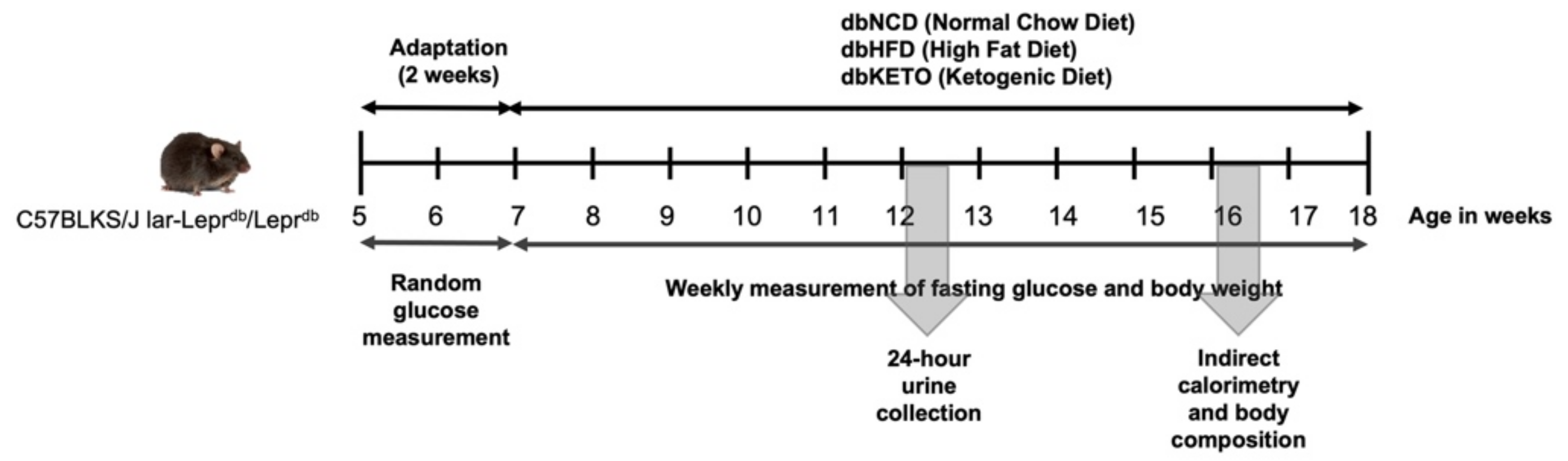
2.1. Animals
2.2. Indirect Calorimetry and Body Composition
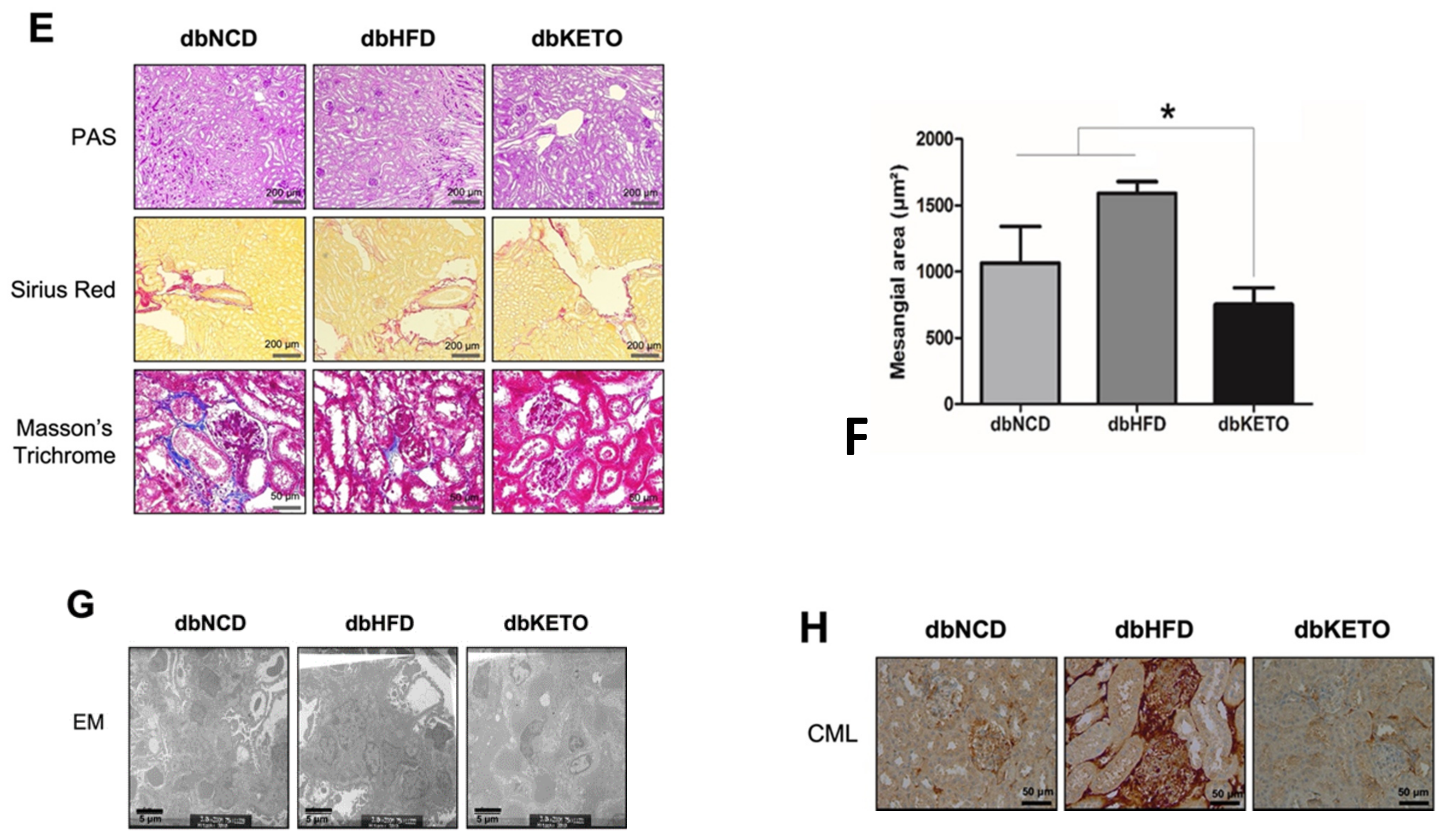
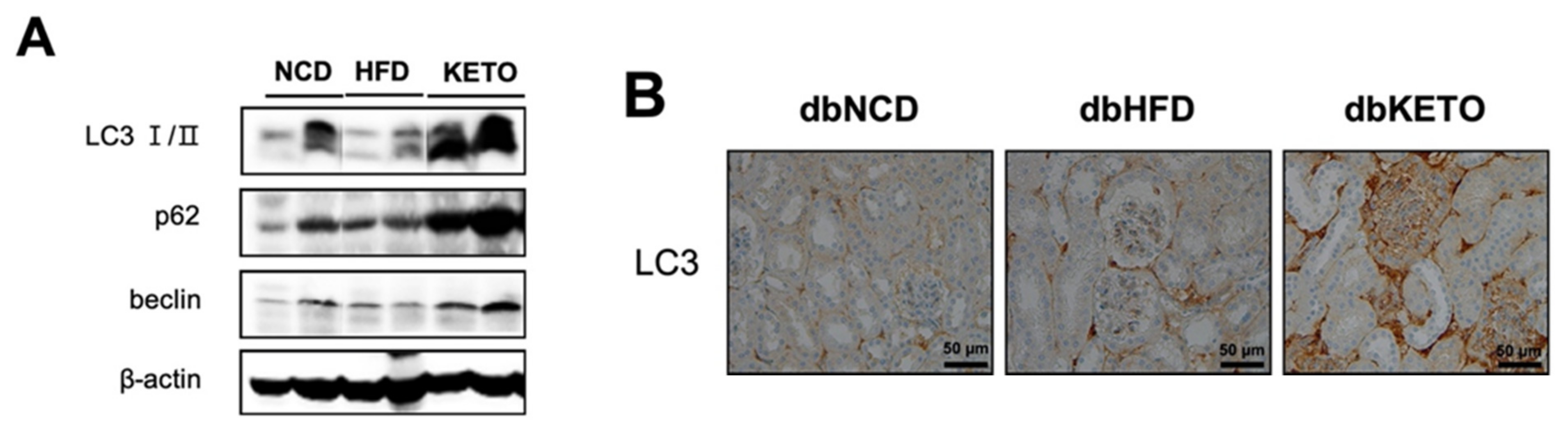
2.3. Immunohistochemistry (IHC) Analyses
2.4. Electron Microscopy (EM) Analyses
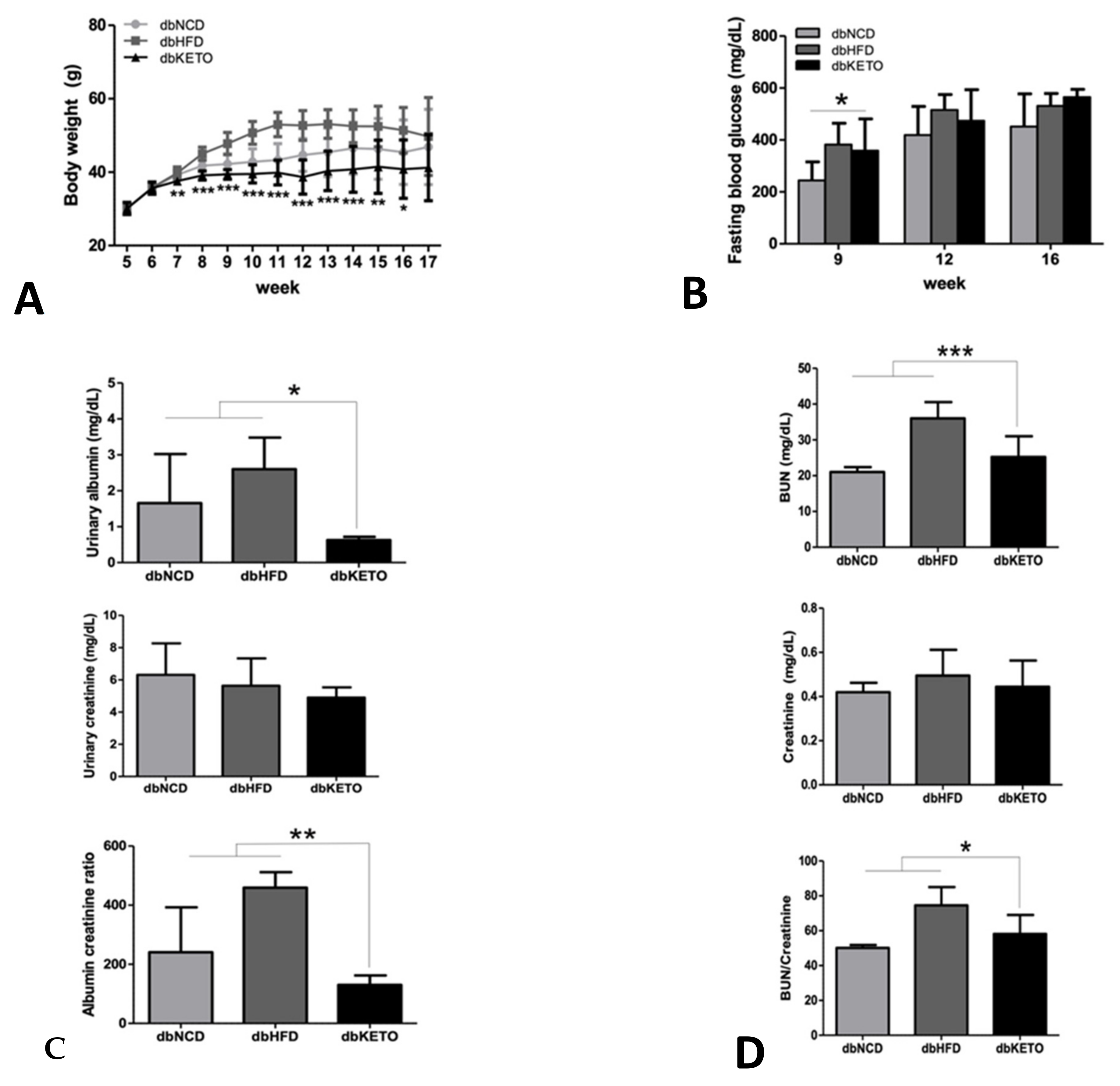
2.5. Blood Chemistry and Urine Analysis
2.6. Cell Culture
2.7. Real-Time Reverse-Transcriptase Polymerase Chain (RT-PCR) Analysis
2.8. Western Blot Analysis
2.9. Immunofluorescence
2.10. Reactive Oxygen Species (ROS)
2.11. Statistical Analysis
3. Results
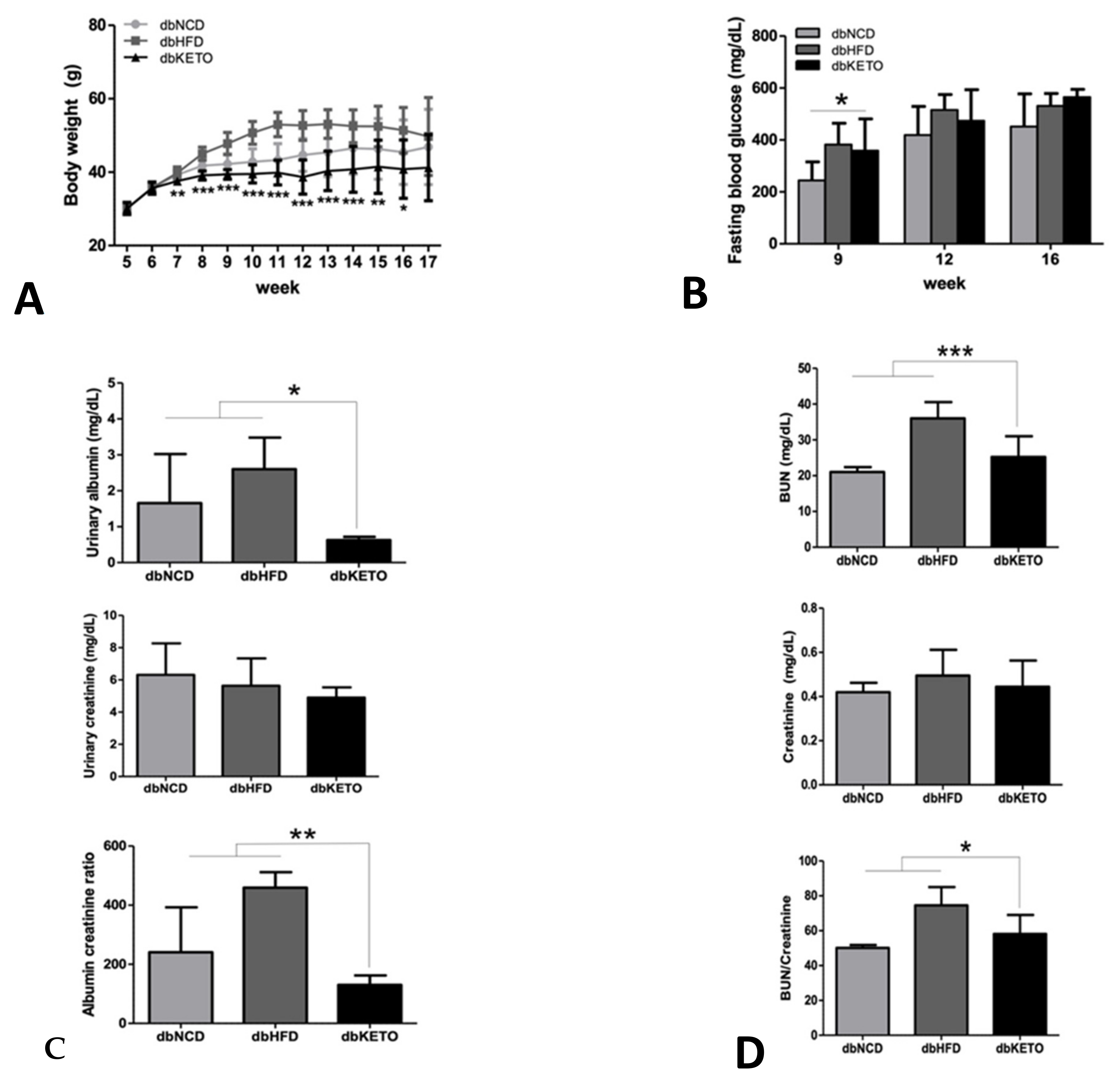
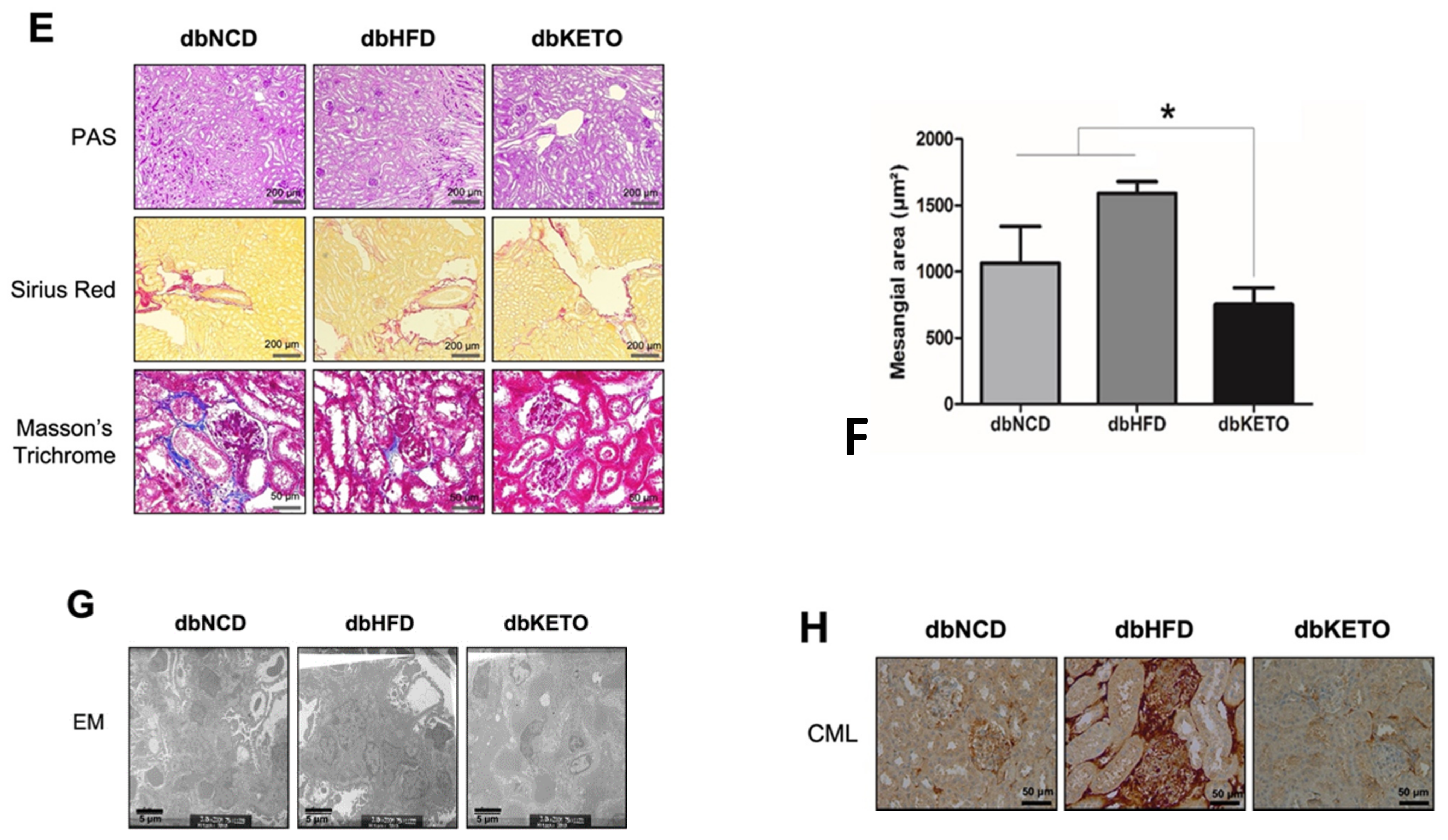
3.1. KD Delays DN-Related Pathological Changes
3.2. KD Attenuates RER and Increases Energy Expenditure
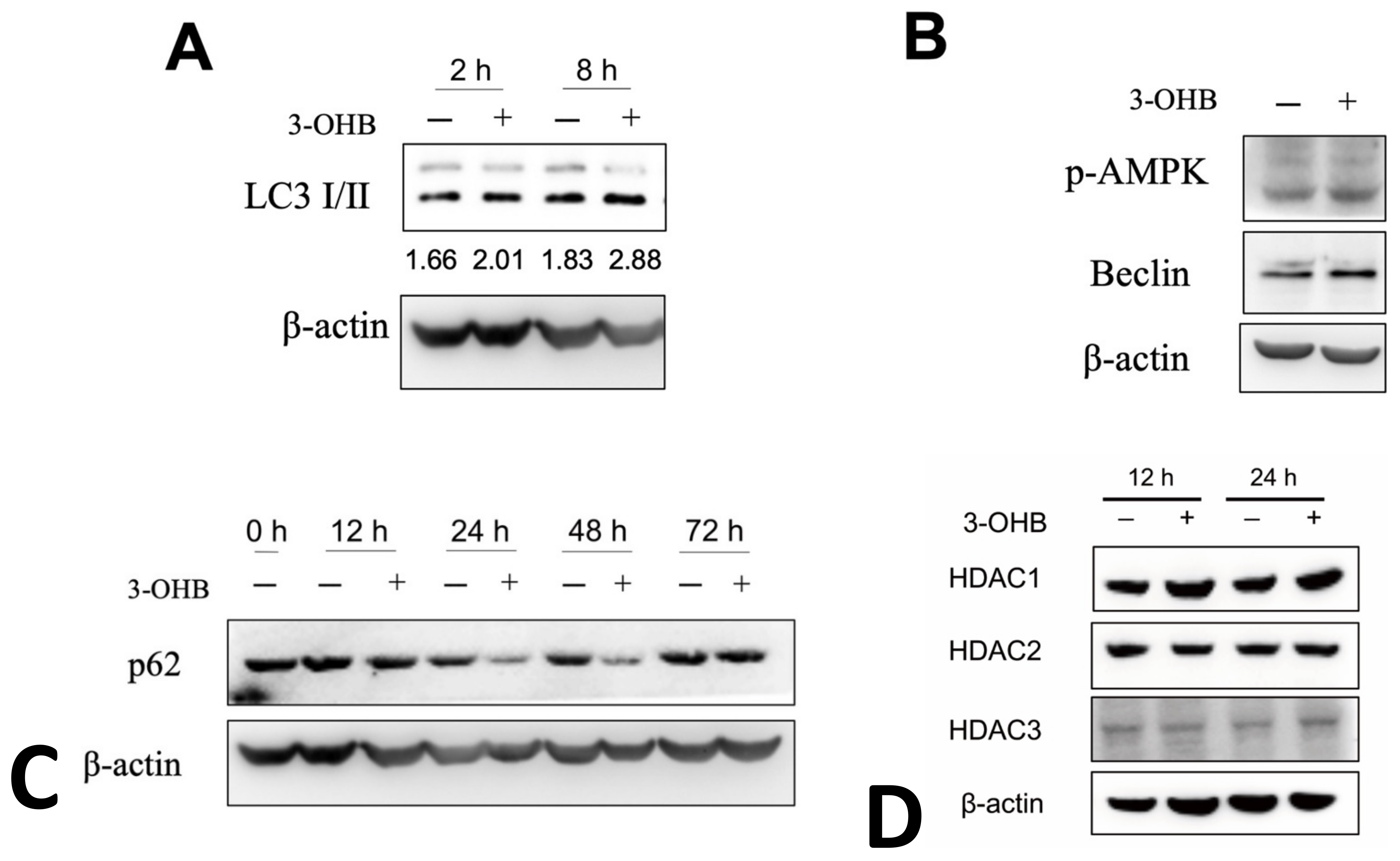
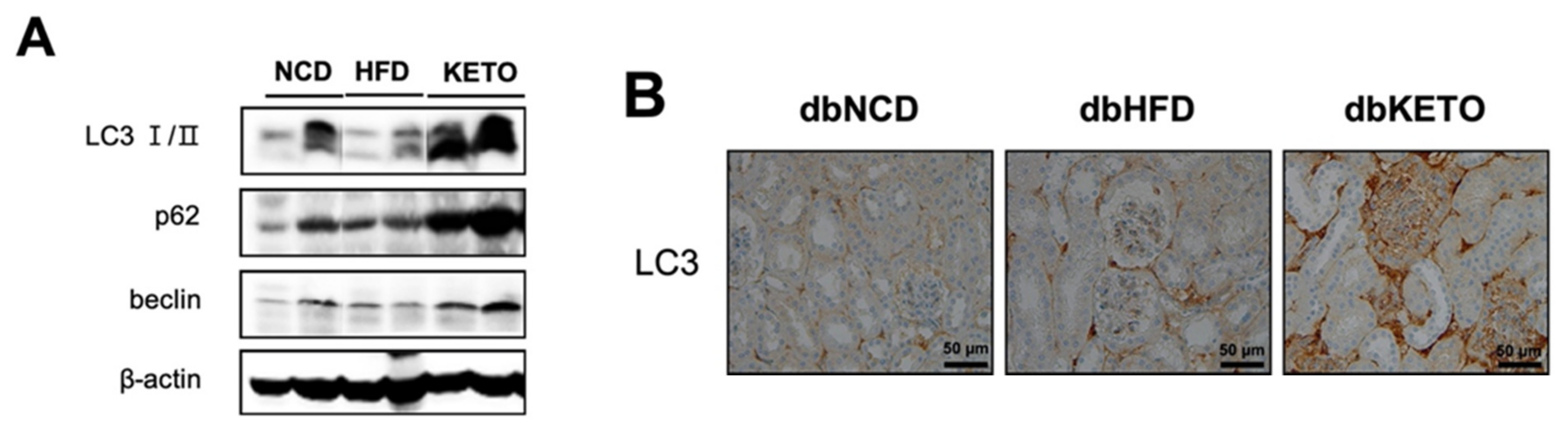
3.3. KD Increases Autophagy in db/db Mice
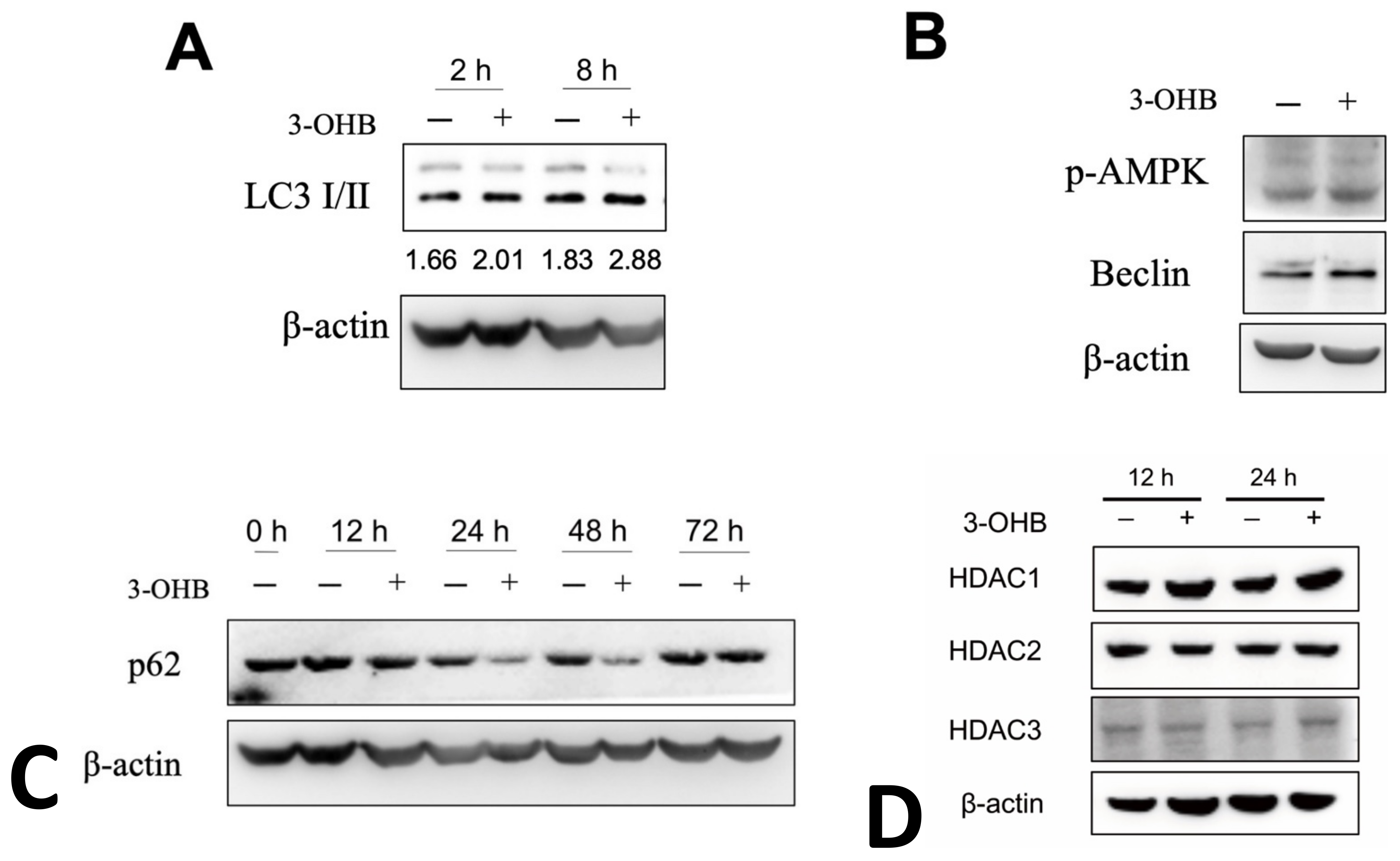
3.4. 3-OHB Increases Autophagy in Renal Proximal Tubular Cells
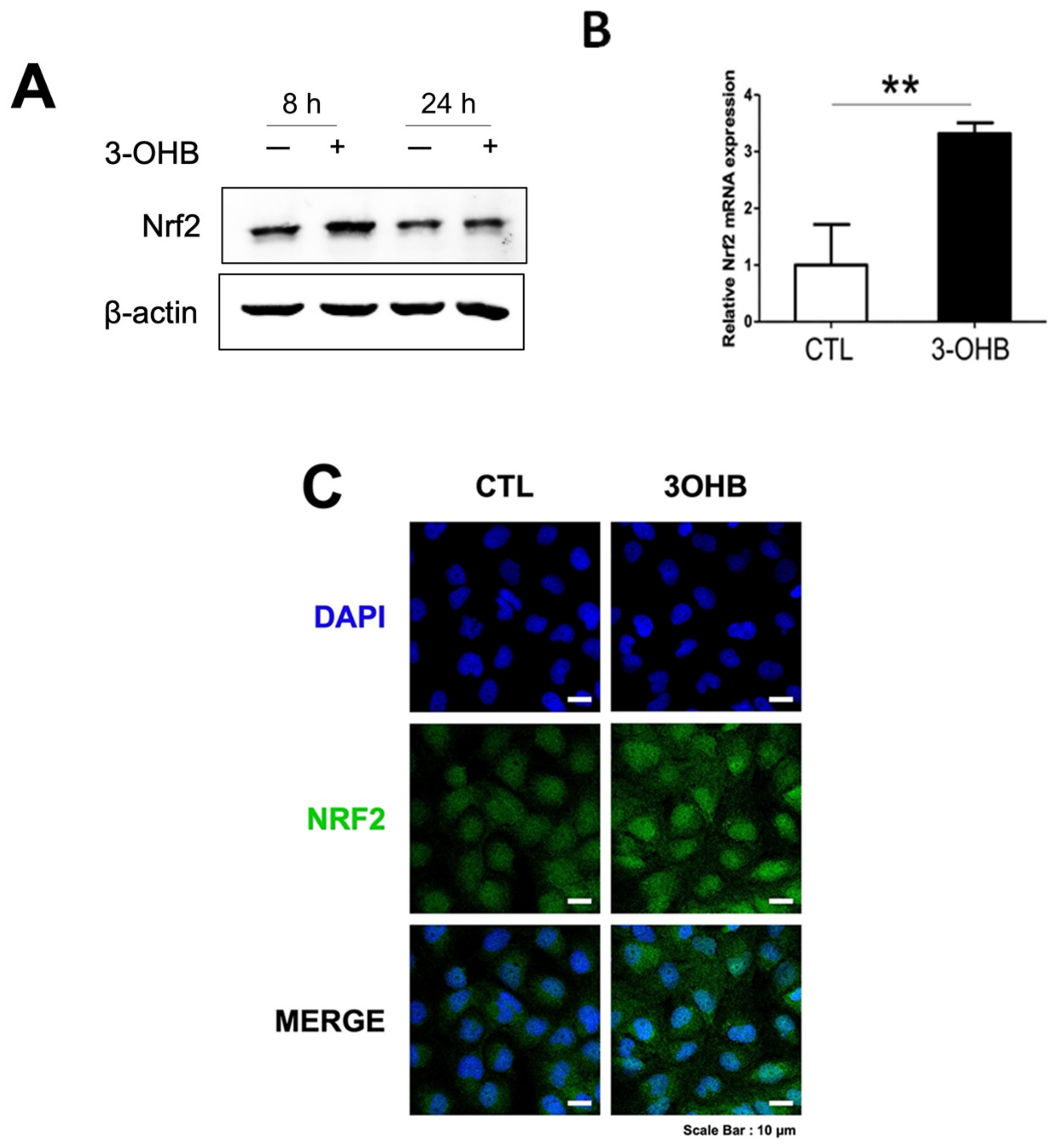
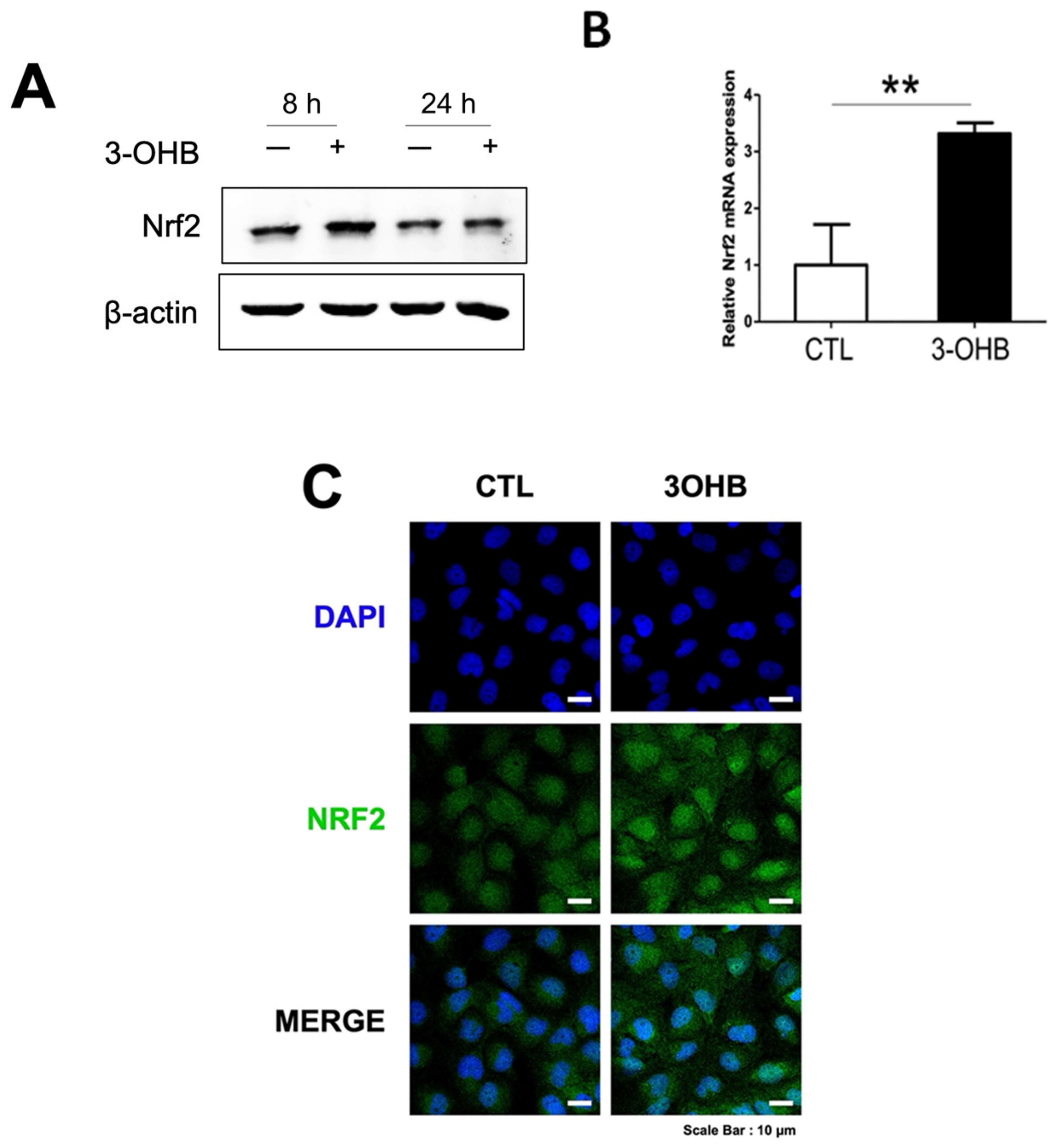
3.5. 3-OHB Increases NRF2 Expression in Renal Proximal Tubular Cells
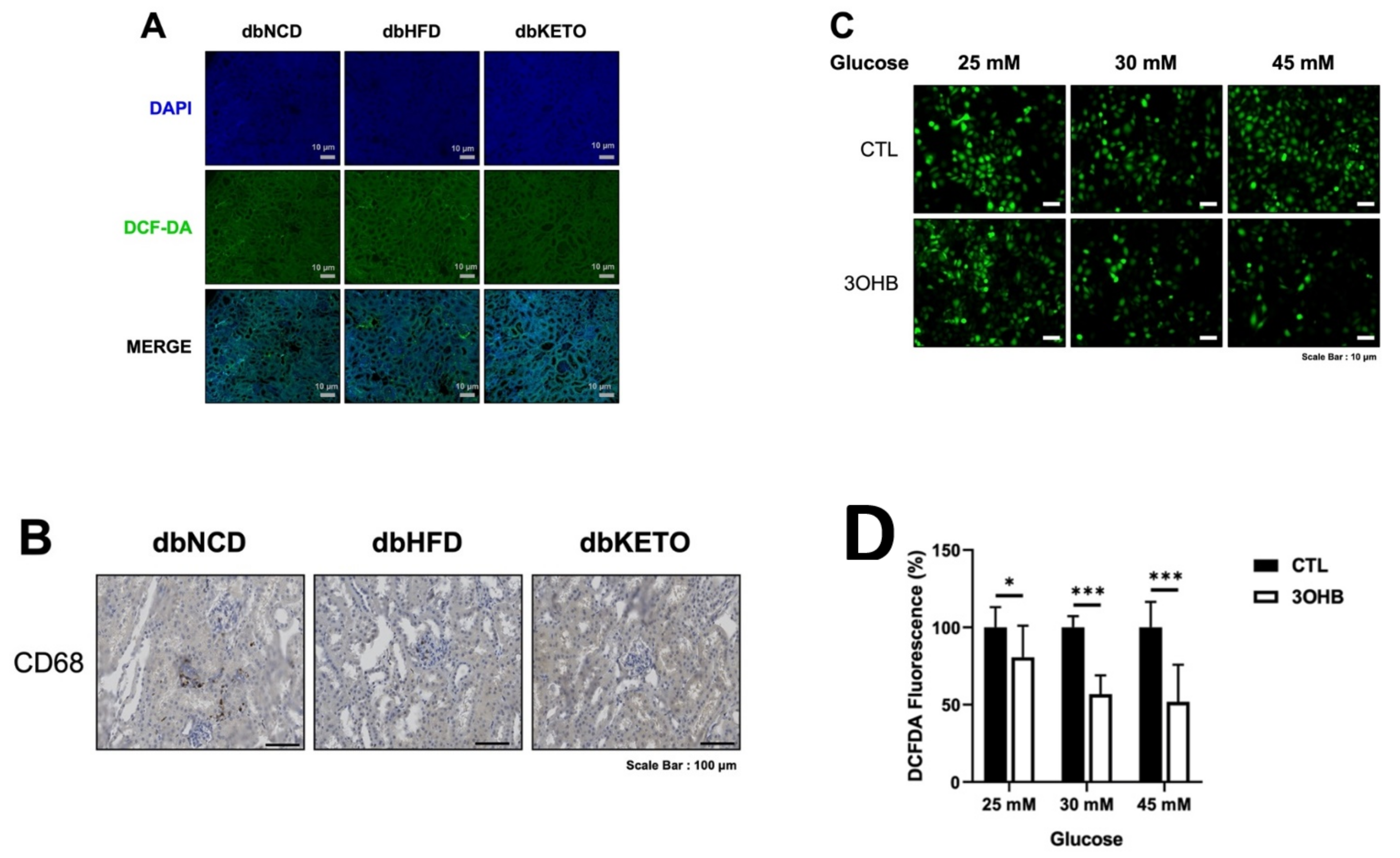
3.6. KD and 3-OHB Decreases ROS Production
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reutens, A.T. Epidemiology of diabetic kidney disease. Med. Clin. N. Am. 2013, 97, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Yu, J.H.; Ko, S.H.; Kwon, H.S.; Kim, D.J.; Kim, J.H.; Kim, C.S.; Song, K.H.; Won, J.C.; Lim, S.; et al. Prevalence and determinants of diabetic nephropathy in Korea: Korea national health and nutrition examination survey. Diabetes Metab. J. 2014, 38, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth-Manikowski, S.; Atta, M.G. Diabetic Kidney Disease: Pathophysiology and Therapeutic Targets. J. Diabetes Res. 2015, 2015, 697010. [Google Scholar] [CrossRef] [Green Version]
- Wada, J.; Makino, H. Inflammation and the pathogenesis of diabetic nephropathy. Clin. Sci. 2013, 124, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Elmarakby, A.A.; Sullivan, J.C. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther. 2012, 30, 49–59. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Fan, T.W.; Lane, A.N.; Weiss, H.L.; Evers, B.M. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017, 24, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.A.H.; Deemer, S.E.; Bergeron, J.M.; Little, J.T.; Warren, J.L.; Fisher, G.; Smith, D.L., Jr.; Fontaine, K.R.; Dickinson, S.L.; Allison, D.B.; et al. Dietary R, S-1,3-butanediol diacetoacetate reduces body weight and adiposity in obese mice fed a high-fat diet. FASEB J. 2019, 33, 2409–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-M.; Ramprasath, T.; Zou, M.-H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Hartman, A.L.; Gasior, M.; Vining, E.P.; Rogawski, M.A. The neuropharmacology of the ketogenic diet. Pediatr. Neurol. 2007, 36, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Goday, A.; Bellido, D.; Sajoux, I.; Crujeiras, A.B.; Burguera, B.; García-Luna, P.P.; Oleaga, A.; Moreno, B.; Casanueva, F.F. Short-term safety, tolerability and efficacy of a very low-calorie-ketogenic diet interventional weight loss program versus hypocaloric diet in patients with type 2 diabetes mellitus. Nutr. Diabetes 2016, 6, e230. [Google Scholar] [CrossRef]
- Verma, S.; Hussain, M.E. Obesity and diabetes: An update. Diabetes Metab. Syndr. 2017, 11, 73–79. [Google Scholar] [CrossRef]
- Poplawski, M.M.; Mastaitis, J.W.; Isoda, F.; Grosjean, F.; Zheng, F.; Mobbs, C.V. Reversal of diabetic nephropathy by a ketogenic diet. PLoS ONE 2011, 6, e18604. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D'Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Park, C.W. Diabetic kidney disease: From epidemiology to clinical perspectives. Diabetes Metab. J. 2014, 38, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Papademetriou, V.; Lovato, L.; Doumas, M.; Nylen, E.; Mottl, A.; Cohen, R.M.; Applegate, W.B.; Puntakee, Z.; Yale, J.F.; Cushman, W.C. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int. 2015, 87, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Ismail-Beigi, F.; Craven, T.; Banerji, M.A.; Basile, J.; Calles, J.; Cohen, R.M.; Cuddihy, R.; Cushman, W.C.; Genuth, S.; Grimm, R.H., Jr.; et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: An analysis of the ACCORD randomised trial. Lancet 2010, 376, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H., Jr.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Tarabra, E.; Toledo, M.; Garcia-Macia, M.; Sahu, S.; Coletto, L.; Batista-Gonzalez, A.; Barzilai, N.; Pessin, J.E.; Schwartz, G.J.; et al. System-wide Benefits of Intermeal Fasting by Autophagy. Cell Metab. 2017, 26, 856–871. [Google Scholar] [CrossRef] [Green Version]
- Hostetter, T.H. Diabetic nephropathy. Metabolic versus hemodynamic considerations. Diabetes Care 1992, 15, 1205–1215. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol. 2012, 74, 351–375. [Google Scholar] [CrossRef] [Green Version]
- Slyne, J.; Slattery, C.; McMorrow, T.; Ryan, M.P. New developments concerning the proximal tubule in diabetic nephropathy: In vitro models and mechanisms. Nephrol. Dial. Transplant. 2015, 30 (Suppl. S4), iv60–iv67. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Ma, Z.; Li, L.; Livingston, M.J.; Zhang, D.; Mi, Q.; Zhang, M.; Ding, H.F.; Huo, Y.; Mei, C.; Dong, Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Investig. 2020, 130, 5011–5026. [Google Scholar] [CrossRef]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.; Park, W.Y.; Kim, Y.J.; Kim, M.; Choe, M.; Jin, K.; Seo, J.H.; Ha, E. 3-Hydroxybutyrate Ameliorates the Progression of Diabetic Nephropathy. Antioxidants 2022, 11, 381. https://doi.org/10.3390/antiox11020381
Jung J, Park WY, Kim YJ, Kim M, Choe M, Jin K, Seo JH, Ha E. 3-Hydroxybutyrate Ameliorates the Progression of Diabetic Nephropathy. Antioxidants. 2022; 11(2):381. https://doi.org/10.3390/antiox11020381
Chicago/Turabian StyleJung, Jeeyoun, Woo Yeong Park, Yun Jin Kim, Mikyung Kim, Misun Choe, Kyubok Jin, Ji Hae Seo, and Eunyoung Ha. 2022. "3-Hydroxybutyrate Ameliorates the Progression of Diabetic Nephropathy" Antioxidants 11, no. 2: 381. https://doi.org/10.3390/antiox11020381
APA StyleJung, J., Park, W. Y., Kim, Y. J., Kim, M., Choe, M., Jin, K., Seo, J. H., & Ha, E. (2022). 3-Hydroxybutyrate Ameliorates the Progression of Diabetic Nephropathy. Antioxidants, 11(2), 381. https://doi.org/10.3390/antiox11020381