
Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease


Abstract
1. Introduction
2. Antioxidants and Oxidative Stress
2.1. Antioxidant Classification and Mechanisms of Action
2.1.1. Enzymatic Antioxidants
2.1.2. Non-Enzymatic Antioxidants
Vitamin A
Vitamin D
Vitamin E
Vitamin K
B Vitamins
Vitamin C
Carotenoids
Polyphenols
Minerals
Estrogens
3. Oxidative Stress in Alzheimer’s Disease
4. Current Treatments for Alzheimer’s Disease
5. Conjugate Therapies and the Blood Brain Barrier
6. Clinical Trials
6.1. Vitamins
6.2. Polyphenols
6.3. Minerals
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Halliwell, B. Reactive Oxygen Species and the Central Nervous System. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Mishra, S.K.; Pant, H.C. Oxidative Stress in Neurodegeneration. Adv. Pharmacol. Sci. 2011, 2011, 572634. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Nunomura, A.; Hirai, K.; Zhu, X.; Pérez, M.; Avila, J.; Castellani, R.J.; Atwood, C.S.; Aliev, G.; Sayre, L.M.; et al. Is Oxidative Damage the Fundamental Pathogenic Mechanism of Alzheimer’s and Other Neurodegenerative Diseases? Free Radic. Biol. Med. 2002, 33, 1475–1479. [Google Scholar] [CrossRef]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of Oxidative Stress in Alzheimer’s Disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Frontiers|Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease|Aging Neuroscience. Available online: https://www.frontiersin.org/articles/10.3389/fnagi.2021.617588/full (accessed on 28 December 2021).
- Barber, S.C.; Shaw, P.J. Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Oxidative Stress in ALS: A Mechanism of Neurodegeneration and a Therapeutic Target—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0925443906000524 (accessed on 28 December 2021).
- Castellani, R.; Smith, M.A.; Richey, P.L.; Kalaria, R.; Gambetti, P.; Perry, G. Evidence for Oxidative Stress in Pick Disease and Corticobasal Degeneration. Brain Res. 1995, 696, 268–271. [Google Scholar] [CrossRef]
- Prasad, K.N.; Bondy, S.C. Oxidative and Inflammatory Events in Prion Diseases: Can They Be Therapeutic Targets? Curr. Aging Sci. 2018, 11, 216–225. [Google Scholar] [CrossRef]
- Muchová, J.; Žitňanová, I.; Ďuračková, Z. Oxidative Stress and Down Syndrome. Do Antioxidants Play a Role in Therapy? Physiol. Res. 2014, 63, 535–542. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Sima, A.; Stevens, M. Diabetic Neuropathy and Oxidative Stress. Diabetes Metab. Res. Rev. 2006, 22, 257–273. [Google Scholar] [CrossRef]
- Hosseini, A.; Abdollahi, M. Diabetic Neuropathy and Oxidative Stress: Therapeutic Perspectives. Oxid. Med. Cell. Longev. 2013, 2013, 168039. [Google Scholar] [CrossRef]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The Role of Oxidative Stress in Friedreich’s Ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef]
- Frontiers|Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications|Molecular Neuroscience. Available online: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00068/full (accessed on 28 December 2021).
- Velusamy, T.; Panneerselvam, A.S.; Purushottam, M.; Anusuyadevi, M.; Pal, P.K.; Jain, S.; Essa, M.M.; Guillemin, G.J.; Kandasamy, M. Protective Effect of Antioxidants on Neuronal Dysfunction and Plasticity in Huntington’s Disease. Oxidative Med. Cell. Longev. 2017, 2017, e3279061. [Google Scholar] [CrossRef]
- Túnez, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Tasset-Cuevas, I. Important Role of Oxidative Stress Biomarkers in Huntington’s Disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef] [PubMed]
- Dalfó, E.; Portero-Otín, M.; Ayala, V.; Martínez, A.; Pamplona, R.; Ferrer, I. Evidence of Oxidative Stress in the Neocortex in Incidental Lewy Body Disease. J. Neuropathol. Exp. Neurol. 2005, 64, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative Stress in Multiple Sclerosis: Central and Peripheral Mode of Action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yanjanin, N.M.; Bianconi, S.; Pavan, W.J.; Porter, F.D. Oxidative Stress in Niemann-Pick Disease, Type C. Mol. Genet. Metab. 2010, 101, 214–218. [Google Scholar] [CrossRef]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative Stress: A Pathogenic Mechanism for Niemann-Pick Type C Disease. Oxidative Med. Cell. Longev. 2012, 2012, e205713. [Google Scholar] [CrossRef]
- Pentón-Rol, G.; Cervantes-Llanos, M.; Martínez-Sánchez, G.; Cabrera-Gómez, J.A.; Valenzuela-Silva, C.M.; Ramírez-Nuñez, O.; Casanova-Orta, M.; Robinson-Agramonte, M.A.; Lopategui-Cabezas, I.; López-Saura, P.A. TNF-α and IL-10 Downregulation and Marked Oxidative Stress in Neuromyelitis Optica. J. Inflamm. 2009, 6, 18. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative Stress and Parkinson’s Disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J. Oxidative Stress and Cellular Pathologies in Parkinson’s Disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsubara, K.; Kobayashi, S. Aging and Oxidative Stress in Progressive Supranuclear Palsy. Eur. J. Neurol. 2006, 13, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Dennis, A.-G.; Almaguer-Mederos, L.E.; Raúl, R.-A.; Roberto, R.-L.; Luis, V.-P.; Dany, C.-A.; Yanetza, G.-Z.; Yaimeé, V.-M.; Annelié, E.-D.; Arnoy, P.-A.; et al. Redox Imbalance Associates with Clinical Worsening in Spinocerebellar Ataxia Type 2. Oxidative Med. Cell. Longev. 2021, 2021, e9875639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Lee, C.-M.; Lee, L.-C.; Tung, L.-C.; Hsieh-Li, H.-M.; Lee-Chen, G.-J.; Su, M.-T. Mitochondrial Dysfunction and Oxidative Stress Contribute to the Pathogenesis of Spinocerebellar Ataxia Type 12 (SCA12). J. Biol. Chem. 2011, 286, 21742–21754. [Google Scholar] [CrossRef]
- Torres-Ramos, Y.; Montoya-Estrada, A.; Cisneros, B.; Tercero-Pérez, K.; León-Reyes, G.; Leyva-García, N.; Hernández-Hernández, O.; Magaña, J.J. Oxidative Stress in Spinocerebellar Ataxia Type 7 Is Associated with Disease Severity. Cerebellum 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Oxidative Stress and Its Role in the Pathogenesis of Ischaemic Stroke. Int. J. Stroke. 2009, 4, 461–470. [Google Scholar] [CrossRef]
- Žitňanová, I.; Šiarnik, P.; Kollár, B.; Chomová, M.; Pazderová, P.; Andrezálová, L.; Ježovičová, M.; Koňariková, K.; Laubertová, L.; Krivošíková, Z.; et al. Oxidative Stress Markers and Their Dynamic Changes in Patients after Acute Ischemic Stroke. Oxidative Med. Cell. Longev. 2016, 2016, e9761697. [Google Scholar] [CrossRef]
- Komsiiska, D. Oxidative Stress and Stroke: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Comp. Clin. Pathol. 2019, 28, 915–926. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic Brain Injury: Oxidative Stress and Neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Mendes Arent, A.; de Souza, L.F.; Walz, R.; Dafre, A.L. Perspectives on Molecular Biomarkers of Oxidative Stress and Antioxidant Strategies in Traumatic Brain Injury. Biomed. Res. Int. 2014, 2014, e723060. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Yankner, B.A. The Aging Stress Response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Yu, B.P. Cellular Defenses against Damage from Reactive Oxygen Species. Physiol. Rev. 1994, 74, 139–162. [Google Scholar] [CrossRef]
- Khajeh Dangolani, S.; Panahi, F.; Tavaf, Z.; Nourisefat, M.; Yousefi, R.; Khalafi-Nezhad, A. Synthesis and Antioxidant Activity Evaluation of Some Novel Aminocarbonitrile Derivatives Incorporating Carbohydrate Moieties. ACS Omega 2018, 3, 10341–10350. [Google Scholar] [CrossRef]
- Mohana, K.N.; Kumar, C.B.P. Synthesis and Antioxidant Activity of 2-Amino-5-Methylthiazol Derivatives Containing 1,3,4-Oxadiazole-2-Thiol Moiety. ISRN Org. Chem. 2013, 2013, e620718. [Google Scholar] [CrossRef]
- Li, A.-N.; Li, S.; Zhang, Y.-J.; Xu, X.-R.; Chen, Y.-M.; Li, H.-B. Resources and Biological Activities of Natural Polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.; Zhou, T.; Zheng, J.; Li, S.; Li, H.-B. Dietary Natural Products for Prevention and Treatment of Liver Cancer. Nutrients 2016, 8, 156. [Google Scholar] [CrossRef]
- Peng, C.; Wang, X.; Chen, J.; Jiao, R.; Wang, L.; Li, Y.M.; Zuo, Y.; Liu, Y.; Lei, L.; Ma, K.Y.; et al. Biology of Ageing and Role of Dietary Antioxidants. Biomed. Res. Int. 2014, 2014, 831841. [Google Scholar] [CrossRef] [PubMed]
- Arulselvan, P.; Fard, M.T.; Tan, W.S.; Gothai, S.; Fakurazi, S.; Norhaizan, M.E.; Kumar, S.S. Role of Antioxidants and Natural Products in Inflammation. Oxid. Med. Cell. Longev. 2016, 2016, 5276130. [Google Scholar] [CrossRef] [PubMed]
- Biotransformation of Waste Biomass into High Value Biochemicals. Available online: https://www.springerprofessional.de/en/biotransformation-of-waste-biomass-into-high-value-biochemicals/1889608 (accessed on 28 December 2021).
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J. Chapter 14—Oxidative Stress and Cell Death in Cardiovascular Disease: A Post-Genomic Appraisal. In Post-Genomic Cardiology, 2nd ed.; Marín-García, J., Ed.; Academic Press: Boston, MA, USA, 2014; pp. 471–498. [Google Scholar] [CrossRef]
- van Lith, R.; Ameer, G.A. Chapter Ten—Antioxidant Polymers as Biomaterial. In Oxidative Stress and Biomaterials; Dziubla, T., Butterfield, D.A., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 251–296. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Dalvi, S.M.; Patil, V.W.; Ramraje, N.N. The Roles of Glutathione, Glutathione Peroxidase, Glutathione Reductase and the Carbonyl Protein in Pulmonary and Extra Pulmonary Tuberculosis. J. Clin. Diagn. Res. 2012, 6, 1462–1465. [Google Scholar] [CrossRef]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione Peroxidase-Catalase Cooperativity Is Required for Resistance to Hydrogen Peroxide by Mature Rat Oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef]
- Mulholland, C.W.; Elwood, P.C.; Davis, A.; Thurnham, D.I.; Kennedy, O.; Coulter, J.; Fehily, A.; Strain, J.J. Antioxidant Enzymes, Inflammatory Indices and Lifestyle Factors in Older Men: A Cohort Analysis. QJM Int. J. Med. 1999, 92, 579–585. [Google Scholar] [CrossRef][Green Version]
- Nimse, S.B.; Pal, D. Free Radicals, Natural Antioxidants, and Their Reaction Mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Shahidi, F.; Zhong, Y. Novel Antioxidants in Food Quality Preservation and Health Promotion. Eur. J. Lipid Sci. Technol. 2010, 112, 930–940. [Google Scholar] [CrossRef]
- Reddy, P.; Jialal, I. Biochemistry, Fat Soluble Vitamins. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Micronutrients, I. Vitamin A; National Academies Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Dowling, J.E. Vitamin A: Its Many Roles-from Vision and Synaptic Plasticity to Infant Mortality. J. Comp. Physiol. A Neuroethol. Sens. Neural. Behav. Physiol. 2020, 206, 389–399. [Google Scholar] [CrossRef]
- Tanumihardjo, S.A. Vitamin A and Bone Health: The Balancing Act. J. Clin. Densitom. 2013, 16, 414–419. [Google Scholar] [CrossRef] [PubMed]
- McGrane, M.M. Vitamin A Regulation of Gene Expression: Molecular Mechanism of a Prototype Gene. J. Nutr. Biochem. 2007, 18, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Pozniakov, S.P. Mechanism of action of vitamin A on cell differentiation and function. Ontogenez 1986, 17, 578–586. [Google Scholar] [PubMed]
- Clagett-Dame, M.; Knutson, D. Vitamin A in Reproduction and Development. Nutrients 2011, 3, 385–428. [Google Scholar] [CrossRef]
- Bar-El Dadon, S.; Reifen, R. Vitamin A and the Epigenome. Crit. Rev. Food Sci. Nutr. 2017, 57, 2404–2411. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Snyder, L.; Arora, J. Vitamin A and Vitamin D Regulate the Microbial Complexity, Barrier Function, and the Mucosal Immune Responses to Ensure Intestinal Homeostasis. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 184–192. [Google Scholar] [CrossRef]
- Holick, M.F. Chapter 4—Photobiology of Vitamin D. In Vitamin D, 4th ed.; Feldman, D., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 45–55. [Google Scholar] [CrossRef]
- Jones, G. Metabolism and Biomarkers of Vitamin D. Scand. J. Clin. Lab. Investig. Suppl. 2012, 243, 7–13. [Google Scholar] [CrossRef]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Cytochrome P450-Mediated Metabolism of Vitamin D. J. Lipid. Res. 2014, 55, 13–31. [Google Scholar] [CrossRef]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-Hydroxylase (CYP24A1): Its Important Role in the Degradation of Vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef]
- Norman, A.W. From Vitamin D to Hormone D: Fundamentals of the Vitamin D Endocrine System Essential for Good Health. Am. J. Clin. Nutr. 2008, 88, 491S–499S. [Google Scholar] [CrossRef]
- Samuel, S.; Sitrin, M.D. Vitamin D’s role in cell proliferation and differentiation. Nutr. Rev. 2008, 66, S116–S124. [Google Scholar] [CrossRef]
- Shaker, J.L.; Deftos, L. Calcium and Phosphate Homeostasis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bowry, V.W.; Ingold, K.U.; Stocker, R. Vitamin E in Human Low-Density Lipoprotein. When and How This Antioxidant Becomes a pro-Oxidant. Biochem. J. 1992, 288 Pt 2, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Jilani, T.; Iqbal, M.P. Does Vitamin E Have a Role in Treatment and Prevention of Anemia? Pak. J. Pharm. Sci. 2011, 24, 237–242. [Google Scholar] [PubMed]
- Napolitano, G.; Fasciolo, G.; Di Meo, S.; Venditti, P. Vitamin E Supplementation and Mitochondria in Experimental and Functional Hyperthyroidism: A Mini-Review. Nutrients 2019, 11, 2900. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.K. Vitamin E Regulation of Mitochondrial Superoxide Generation. Biol. Signals Recept. 2001, 10, 112–124. [Google Scholar] [CrossRef]
- Bozaykut, P.; Ekren, R.; Sezerman, O.U.; Gladyshev, V.N.; Ozer, N.K. High-Throughput Profiling Reveals Perturbation of Endoplasmic Reticulum Stress-Related Genes in Atherosclerosis Induced by High-Cholesterol Diet and the Protective Role of Vitamin E. Biofactors 2020, 46, 653–664. [Google Scholar] [CrossRef]
- Office of Dietary Supplements—Vitamin E. Available online: https://ods.od.nih.gov/factsheets/VitaminE-HealthProfessional/ (accessed on 28 December 2021).
- Zingg, J.-M. Vitamin E: A Role in Signal Transduction. Annu. Rev. Nutr. 2015, 35, 135–173. [Google Scholar] [CrossRef]
- Boscoboinik, D.; Szewczyk, A.; Azzi, A. Alpha-Tocopherol (Vitamin E) Regulates Vascular Smooth Muscle Cell Proliferation and Protein Kinase C Activity. Arch. Biochem. Biophys. 1991, 286, 264–269. [Google Scholar] [CrossRef]
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of Cell Proliferation by Alpha-Tocopherol. Role of Protein Kinase C. J. Biol. Chem. 1991, 266, 6188–6194. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 879. [Google Scholar] [CrossRef]
- Berdnikovs, S.; Abdala-Valencia, H.; McCary, C.; Somand, M.; Cole, R.; Garcia, A.; Bryce, P.; Cook-Mills, J.M. Isoforms of Vitamin E Have Opposing Immunoregulatory Functions during Inflammation by Regulating Leukocyte Recruitment. J. Immunol. 2009, 182, 4395–4405. [Google Scholar] [CrossRef]
- Cook-Mills, J.M. Isoforms of Vitamin E Differentially Regulate PKC α and Inflammation: A Review. J. Clin. Cell. Immunol. 2013, 4, 1000137. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M. Vitamins C and E donate single hydrogen atoms in vivo. FEBS Lett. 1991, 284, 147–151. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid Peroxidation Triggers Neurodegeneration: A Redox Proteomics View into the Alzheimer Disease Brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Neelamegam, M.; Looi, I.; Ng, K.S.; Malavade, S.S. Vitamin E Supplementation for Preventing Recurrent Stroke and Other Vascular Events in Patients with Stroke or Transient Ischaemic Attack. Cochrane Database Syst. Rev. 2017, 2017, CD010797. [Google Scholar] [CrossRef]
- Chow, C.K.; Hong, C.B. Dietary Vitamin E and Selenium and Toxicity of Nitrite and Nitrate. Toxicology 2002, 180, 195–207. [Google Scholar] [CrossRef]
- d’Ischia, M.; Novellino, L. Nitric Oxide-Induced Oxidation of Alpha-Tocopherol. Bioorg. Med. Chem. 1996, 4, 1747–1753. [Google Scholar] [CrossRef]
- Booth, S.L. Vitamin K: Food Composition and Dietary Intakes. Food Nutr. Res. 2012, 56, 5505. [Google Scholar] [CrossRef]
- Imbrescia, K.; Moszczynski, Z. Vitamin K. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Tie, J.-K.; Stafford, D.W. Chapter Fourteen—Functional Study of the Vitamin K Cycle Enzymes in Live Cells. In Methods in Enzymology; Gelb, M.H., Ed.; Enzymology at the Membrane Interface: Intramembrane Proteases; Academic Press: Cambridge, MA, USA, 2017; Volume 584, pp. 349–394. [Google Scholar] [CrossRef]
- Tie, J.-K.; Stafford, D.W. Structural and Functional Insights into Enzymes of the Vitamin K Cycle. J. Thromb. Haemost. 2016, 14, 236–247. [Google Scholar] [CrossRef]
- Oldenburg, J.; Bevans, C.G.; Müller, C.R.; Watzka, M. Vitamin K Epoxide Reductase Complex Subunit 1 (VKORC1): The Key Protein of the Vitamin K Cycle. Antioxid. Redox Signal. 2006, 8, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Suttie, J.W. Vitamin K-Dependent Carboxylase. Annu. Rev. Biochem. 1985, 54, 459–477. [Google Scholar] [CrossRef] [PubMed]
- The Vitamin K Cycle—STAFFORD—2005—Journal of Thrombosis and Haemostasis—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1538-7836.2005.01419.x (accessed on 28 December 2021).
- Lanzkowsky, P. Chapter 7—Megaloblastic Anemia. In Lanzkowsky’s Manual of Pediatric Hematology and Oncology, 6th ed.; Lanzkowsky, P., Lipton, J.M., Fish, J.D., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 84–101. [Google Scholar] [CrossRef]
- Xv, F.; Chen, J.; Duan, L.; Li, S. Research Progress on the Anticancer Effects of Vitamin K2 (Review). Oncol. Lett. 2018, 15, 8926–8934. [Google Scholar] [CrossRef] [PubMed]
- Harshman, S.G.; Shea, M.K. The Role of Vitamin K in Chronic Aging Diseases: Inflammation, Cardiovascular Disease, and Osteoarthritis. Curr. Nutr. Rep. 2016, 5, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ferland, G. Vitamin K and the Nervous System: An Overview of Its Actions. Adv. Nutr. 2012, 3, 204–212. [Google Scholar] [CrossRef]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef]
- Wilson, D.; Branda, N.R. Turning “On” and “Off” a Pyridoxal 5′-Phosphate Mimic Using Light. Angew. Chem. Int. Ed. 2012, 51, 5431–5434. [Google Scholar] [CrossRef]
- Daugherty, M.; Polanuyer, B.; Farrell, M.; Scholle, M.; Lykidis, A.; de Crécy-Lagard, V.; Osterman, A. Complete Reconstitution of the Human Coenzyme A Biosynthetic Pathway via Comparative Genomics. J. Biol. Chem. 2002, 277, 21431–21439. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the Regulation of Metabolism: Mechanisms and Consequences. Curr. Opin. Cell. Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- New Considerations on the Neuromodulatory Role of Thiamine—Abstract—Pharmacology 2012. Volume 89, No. 1–2—Karger Publishers. Available online: https://www.karger.com/Article/Abstract/336339 (accessed on 28 December 2021).
- Dias, C.; Nylandsted, J. Plasma Membrane Integrity in Health and Disease: Significance and Therapeutic Potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef]
- Sharma, A.; Bist, R. Alteration in Cholinesterases, γ-Aminobutyric Acid and Serotonin Level with Respect to Thiamine Deficiency in Swiss Mice. Turk. J. Biochem. 2019, 44, 218–223. [Google Scholar] [CrossRef]
- Zhao, N.; Zhong, C.; Wang, Y.; Zhao, Y.; Gong, N.; Zhou, G.; Xu, T.; Hong, Z. Impaired Hippocampal Neurogenesis Is Involved in Cognitive Dysfunction Induced by Thiamine Deficiency at Early Pre-Pathological Lesion Stage. Neurobiol. Dis. 2008, 29, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Barclay, L.L.; Gibson, G.E.; Blass, J.P. Impairment of Behavior and Acetylcholine Metabolism in Thiamine Deficiency. J. Pharm. Exp. 1981, 217, 537–543. [Google Scholar]
- Pinnell, S.R. Regulation of Collagen Biosynthesis by Ascorbic Acid: A Review. Yale J. Biol. Med. 1985, 58, 553–559. [Google Scholar] [PubMed]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, Alpha-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial Effects from a Mechanistic Perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef]
- Wilson, J.X. Regulation of Vitamin C Transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic Metals, Ascorbate and Free Radicals: Combinations to Avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C—Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Chakraborthy, A.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A. Antioxidant and Pro-Oxidant Activity of Vitamin C in Oral Environment. Indian J. Dent. Res. 2014, 25, 499–504. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental Ascorbate in the Supportive Treatment of Cancer: Prolongation of Survival Times in Terminal Human Cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef]
- Pfister, R.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.-T. Plasma Vitamin C Predicts Incident Heart Failure in Men and Women in European Prospective Investigation into Cancer and Nutrition-Norfolk Prospective Study. Am. Heart. J. 2011, 162, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Osganian, S.K.; Stampfer, M.J.; Rimm, E.; Spiegelman, D.; Hu, F.B.; Manson, J.E.; Willett, W.C. Vitamin C and Risk of Coronary Heart Disease in Women. J. Am. Coll. Cardiol. 2003, 42, 246–252. [Google Scholar] [CrossRef]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Preventive and Therapeutic Potential of Ascorbic Acid in Neurodegenerative Diseases. CNS Neurosci. 2017, 23, 921–929. [Google Scholar] [CrossRef]
- Hansen, S.N.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does Vitamin C Deficiency Affect Cognitive Development and Function? Nutrients 2014, 6, 3818–3846. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Bernardo, A.; Walker, M.J.; Kennard, J.A.; Kim, G.Y.; Kessler, E.S.; Harrison, F.E. Vitamin C Deficiency in the Brain Impairs Cognition, Increases Amyloid Accumulation and Deposition, and Oxidative Stress in APP/PSEN1 and Normally-Aging Mice. ACS Chem. Neurosci. 2015, 6, 570–581. [Google Scholar] [CrossRef]
- Harrison, F.E.; Green, R.J.; Dawes, S.M.; May, J.M. Vitamin C Distribution and Retention in the Mouse Brain. Brain Res. 2010, 1348, 181–186. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M.; McDonald, M.P. Vitamin C Deficiency Increases Basal Exploratory Activity but Decreases Scopolamine-Induced Activity in APP/PSEN1 Transgenic Mice. Pharm. Biochem. Behav. 2010, 94, 543–552. [Google Scholar] [CrossRef]
- Ghosh, M.K.; Chattopadhyay, D.J.; Chatterjee, I.B. Vitamin C Prevents Oxidative Damage. Free Radic. Res. 1996, 25, 173–179. [Google Scholar] [CrossRef]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and Monoamine Changes Following Severe Vitamin C Deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C Function in the Brain: Vital Role of the Ascorbate Transporter (SVCT2). Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Landrum, J.T. (Ed.) Carotenoids: Physical, Chemical, and Biological Functions and Properties; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar] [CrossRef]
- Fiedor, J.; Fiedor, L.; Haessner, R.; Scheer, H. Cyclic Endoperoxides of Beta-Carotene, Potential pro-Oxidants, as Products of Chemical Quenching of Singlet Oxygen. Biochim. Biophys. Acta 2005, 1709, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wisniewska, A.; Subczynski, W.K. Effects of Polar Carotenoids on the Shape of the Hydrophobic Barrier of Phospholipid Bilayers. Biochim. Biophys. Acta 1998, 1368, 235–246. [Google Scholar] [CrossRef]
- Accumulation of Macular Xanthophylls in Unsaturated Membrane Domains—Abstract—Europe PMC. Available online: https://europepmc.org/article/med/16678020 (accessed on 28 December 2021).
- Bertram, J.S. Cancer Prevention by Carotenoids. Mechanistic Studies in Cultured Cells. Ann. N. Y. Acad. Sci. 1993, 691, 177–191. [Google Scholar] [CrossRef]
- Krinsky, N.I. Micronutrients and Their Influence on Mutagenicity and Malignant Transformation. Ann. N. Y. Acad. Sci. 1993, 686, 229–242. [Google Scholar] [CrossRef]
- Walk, A.M.; Khan, N.A.; Barnett, S.M.; Raine, L.B.; Kramer, A.F.; Cohen, N.J.; Moulton, C.J.; Renzi-Hammond, L.M.; Hammond, B.R.; Hillman, C.H. From Neuro-Pigments to Neural Efficiency: The Relationship between Retinal Carotenoids and Behavioral and Neuroelectric Indices of Cognitive Control in Childhood. Int. J. Psychophysiol. 2017, 118, 1–8. [Google Scholar] [CrossRef]
- Grodstein, F.; Kang, J.H.; Glynn, R.J.; Cook, N.R.; Gaziano, J.M. A Randomized Trial of Beta Carotene Supplementation and Cognitive Function in Men: The Physicians’ Health Study II. Arch. Intern. Med. 2007, 167, 2184–2190. [Google Scholar] [CrossRef]
- Johnson, E.J.; McDonald, K.; Caldarella, S.M.; Chung, H.-Y.; Troen, A.M.; Snodderly, D.M. Cognitive Findings of an Exploratory Trial of Docosahexaenoic Acid and Lutein Supplementation in Older Women. Nutr. Neurosci. 2008, 11, 75–83. [Google Scholar] [CrossRef]
- Rowles, J.L.; Erdman, J.W. Carotenoids and Their Role in Cancer Prevention. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158613. [Google Scholar] [CrossRef]
- Jonasson, L.; Wikby, A.; Olsson, A.G. Low Serum Beta-Carotene Reflects Immune Activation in Patients with Coronary Artery Disease. Nutr. Metab. Cardiovasc. Dis. 2003, 13, 120–125. [Google Scholar] [CrossRef]
- Adewoyin, M.; Ibrahim, M.; Roszaman, R.; Md Isa, M.L.; Mat Alewi, N.A.; Abdul Rafa, A.A.; Anuar, M.N.N. Male Infertility: The Effect of Natural Antioxidants and Phytocompounds on Seminal Oxidative Stress. Diseases 2017, 5, 9. [Google Scholar] [CrossRef]
- Pike, T.W.; Blount, J.D.; Lindström, J.; Metcalfe, N.B. Dietary Carotenoid Availability, Sexual Signalling and Functional Fertility in Sticklebacks. Biol. Lett. 2010, 6, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Sharoni, Y.; Linnewiel-Hermoni, K.; Zango, G.; Khanin, M.; Salman, H.; Veprik, A.; Danilenko, M.; Levy, J. The Role of Lycopene and Its Derivatives in the Regulation of Transcription Systems: Implications for Cancer Prevention. Am. J. Clin. Nutr. 2012, 96, 1173S–1178S. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Goel, N. Phenolic Acids: Natural Versatile Molecules with Promising Therapeutic Applications. Biotechnol. Rep. Amst. 2019, 24, e00370. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.; Pérez-Gregorio, R.; Soares, S.; Mateus, N.; de Freitas, V. Wine Flavonoids in Health and Disease Prevention. Molecules 2017, 22, 292. [Google Scholar] [CrossRef] [PubMed]
- Musial, C.; Kuban-Jankowska, A.; Gorska-Ponikowska, M. Beneficial Properties of Green Tea Catechins. Int. J. Mol. Sci. 2020, 21, 1744. [Google Scholar] [CrossRef]
- Sultana, B.; Anwar, F. Flavonols (Kaempeferol, Quercetin, Myricetin) Contents of Selected Fruits, Vegetables and Medicinal Plants. Food Chem. 2008, 108, 879–884. [Google Scholar] [CrossRef]
- Reinisalo, M.; Kårlund, A.; Koskela, A.; Kaarniranta, K.; Karjalainen, R.O. Polyphenol Stilbenes: Molecular Mechanisms of Defence against Oxidative Stress and Aging-Related Diseases. Oxidative Med. Cell. Longev. 2015, 2015, 340520. [Google Scholar] [CrossRef]
- Al-Suhaimi, E.A.; Al-Riziza, N.A.; Al-Essa, R.A. Physiological and Therapeutical Roles of Ginger and Turmeric on Endocrine Functions. Am. J. Chin. Med. 2011, 39, 215–231. [Google Scholar] [CrossRef]
- Eren, D.; Betul, Y.M. Revealing the Effect of 6-Gingerol, 6-Shogaol and Curcumin on MPGES-1, GSK-3β and β-Catenin Pathway in A549 Cell Line. Chem. Biol. Interact. 2016, 258, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zheng, J.; Li, Y.; Xu, D.-P.; Li, S.; Chen, Y.-M.; Li, H.-B. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients 2016, 8, E515. [Google Scholar] [CrossRef]
- Fujiki, H.; Sueoka, E.; Watanabe, T.; Suganuma, M. Primary Cancer Prevention by Green Tea, and Tertiary Cancer Prevention by the Combination of Green Tea Catechins and Anticancer Compounds. J. Cancer Prev. 2015, 20, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sirerol, J.A.; Rodríguez, M.L.; Mena, S.; Asensi, M.A.; Estrela, J.M.; Ortega, A.L. Role of Natural Stilbenes in the Prevention of Cancer. Oxidative Med. Cell. Longev. 2015, 2016, e3128951. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Li, W.; Liu, B.; Li, S.; Zhang, B.; Xu, Y. Resveratrol Protects Vascular Smooth Muscle Cells against High Glucose-Induced Oxidative Stress and Cell Proliferation in Vitro. Med. Sci. Monit. Basic Res. 2014, 20, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Tagami, M.; Yamori, Y. Dietary Polyphenols Regulate Endothelial Function and Prevent Cardiovascular Disease. Nutrition 2015, 31, 28–37. [Google Scholar] [CrossRef]
- Xiao, J.B.; Högger, P. Dietary Polyphenols and Type 2 Diabetes: Current Insights and Future Perspectives. Curr. Med. Chem. 2015, 22, 23–38. [Google Scholar] [CrossRef]
- Zhang, H.; Tsao, R. Dietary Polyphenols, Oxidative Stress and Antioxidant and Anti-Inflammatory Effects. Curr. Opin. Food Sci. 2016, 8, 33–42. [Google Scholar] [CrossRef]
- Rossi, L.; Mazzitelli, S.; Arciello, M.; Capo, C.R.; Rotilio, G. Benefits from Dietary Polyphenols for Brain Aging and Alzheimer’s Disease. Neurochem. Res. 2008, 33, 2390–2400. [Google Scholar] [CrossRef]
- Noguchi-Shinohara, M.; Yuki, S.; Dohmoto, C.; Ikeda, Y.; Samuraki, M.; Iwasa, K.; Yokogawa, M.; Asai, K.; Komai, K.; Nakamura, H.; et al. Consumption of Green Tea, but Not Black Tea or Coffee, Is Associated with Reduced Risk of Cognitive Decline. PLoS ONE 2014, 9, e96013. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of Nitric Oxide Synthases in Parkinson’s Disease: A Review on the Antioxidant and Anti-Inflammatory Activity of Polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- Bao, J.; Liu, W.; Zhou, H.-Y.; Gui, Y.-R.; Yang, Y.-H.; Wu, M.-J.; Xiao, Y.-F.; Shang, J.-T.; Long, G.-F.; Shu, X.-J. Epigallocatechin-3-Gallate Alleviates Cognitive Deficits in APP/PS1 Mice. Curr. Med. Sci. 2020, 40, 18–27. [Google Scholar] [CrossRef]
- Broderick, T.L.; Rasool, S.; Li, R.; Zhang, Y.; Anderson, M.; Al-Nakkash, L.; Plochocki, J.H.; Geetha, T.; Babu, J.R. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7337. [Google Scholar] [CrossRef]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The Curry Spice Curcumin Reduces Oxidative Damage and Amyloid Pathology in an Alzheimer Transgenic Mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.C.G.E.I.; Puerta, E.; Suárez-Santiago, J.E.; Santos-Magalhães, N.S.; Ramirez, M.J.; Irache, J.M. Effect of the oral administration of nanoencapsulated quercetin on a mouse model of Alzheimer’s disease. Int. J. Pharm. 2017, 517, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Napolitano, M.; Tedesco, I.; Moccia, S.; Milito, A.; Russo, G.L. Neuroprotective Role of Natural Polyphenols. Curr. Top. Med. Chem. 2016, 16, 1943–1950. [Google Scholar] [CrossRef]
- Waggoner, D.J.; Bartnikas, T.B.; Gitlin, J.D. The Role of Copper in Neurodegenerative Disease. Neurobiol. Dis. 1999, 6, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, D.; Uzun, H. Copper Intoxication; Antioxidant Defenses and Oxidative Damage in Rat Brain. Biol. Trace Elem. Res. 2009, 127, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, K.; Yasutake, A.; Inoue, M. Free Radicals and Trace Elements. Prog. Clin. Biol. Res. 1993, 380, 257–268. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxidative Med. Cell. Longev. 2018, 2018, e7580707. [Google Scholar] [CrossRef]
- Coskun, M.; Kayis, T.; Gulsu, E.; Alp, E. Effects of Selenium and Vitamin E on Enzymatic, Biochemical, and Immunological Biomarkers in Galleria mellonella L. Sci. Rep. 2020, 10, 9953. [Google Scholar] [CrossRef]
- Jerome-Morais, A.; Bera, S.; Rachidi, W.; Gann, P.H.; Diamond, A.M. The Effects of Selenium and the GPx-1 Selenoprotein on the Phosphorylation of H2AX. Biochim. Biophys. Acta 2013, 1830, 3399–3406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prasad, A.S.; Miale, A.; Farid, Z.; Sandstead, H.H.; Schulert, A.R. Zinc Metabolism in Patients with the Syndrome of Iron Deficiency Anemia, Hepatosplenomegaly, Dwarfism, and Hypognadism. J. Lab. Clin. Med. 1963, 61, 537–549. [Google Scholar] [PubMed]
- Bao, B.; Ahmad, A.; Azmi, A.; Li, Y.; Prasad, A.; Sarkar, F.H. Chapter 2—The Biological Significance of Zinc in Inflammation and Aging. In Inflammation, Advancing Age and Nutrition; Rahman, I., Bagchi, D., Eds.; Academic Press: San Diego, FL, USA, 2014; pp. 15–27. [Google Scholar] [CrossRef]
- Ha, K.-N.; Chen, Y.; Cai, J.; Sternberg, P., Jr. Increased Glutathione Synthesis through an ARE-Nrf2–Dependent Pathway by Zinc in the RPE: Implication for Protection against Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2709–2715. [Google Scholar] [CrossRef]
- Kaufman, Z.; Salvador, G.A.; Liu, X.; Oteiza, P.I. Zinc and the Modulation of Nrf2 in Human Neuroblastoma Cells. Free Radic. Biol. Med. 2020, 155, 1–9. [Google Scholar] [CrossRef]
- Li, B.; Cui, W.; Tan, Y.; Luo, P.; Chen, Q.; Zhang, C.; Qu, W.; Miao, L.; Cai, L. Zinc Is Essential for the Transcription Function of Nrf2 in Human Renal Tubule Cells in Vitro and Mouse Kidney in Vivo under the Diabetic Condition. J. Cell. Mol. Med. 2014, 18, 895–906. [Google Scholar] [CrossRef]
- Kocot, J.; Luchowska-Kocot, D.; Kiełczykowska, M.; Musik, I.; Kurzepa, J. Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients 2017, 9, 659. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Norbury, R.; Cutter, W.; Compton, J.; Robertson, D.; Craig, M.; Whitehead, M.; Murphy, D. The Neuroprotective Effects of Estrogen on the Aging Brain. Exp. Gerontol. 2003, 38, 109–117. [Google Scholar] [CrossRef]
- Sherwin, B.B. Estrogen Effects on Cognition in Menopausal Women. Neurology 1997, 48, S21–S26. [Google Scholar] [CrossRef]
- Sherwin, B.B. Estrogenic Effects on Memory in Women. Ann. N. Y. Acad. Sci. 1994, 743, 213–230; discussion 230–231. [Google Scholar] [CrossRef]
- Henderson, V.W.; Watt, L.; Buckwalter, J.G. Cognitive Skills Associated with Estrogen Replacement in Women with Alzheimer’s Disease. Psychoneuroendocrinology 1996, 21, 421–430. [Google Scholar] [CrossRef]
- Birge, S.J. The Role of Estrogen in the Treatment of Alzheimer’s Disease. Neurology 1997, 48, S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Fillit, H. Estrogens in the Pathogenesis and Treatment of Alzheimer’s Disease in Postmenopausal Women. Ann. N. Y. Acad. Sci. 1994, 743, 233–238; discussion 238–239. [Google Scholar] [CrossRef] [PubMed]
- Paganini-Hill, A.; Henderson, V.W. Estrogen Deficiency and Risk of Alzheimer’s Disease in Women. Am. J. Epidemiol. 1994, 140, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Fillit, H.; Weinreb, H.; Cholst, I.; Luine, V.; McEwen, B.; Amador, R.; Zabriskie, J. Observations in a Preliminary Open Trial of Estradiol Therapy for Senile Dementia-Alzheimer’s Type. Psychoneuroendocrinology 1986, 11, 337–345. [Google Scholar] [CrossRef]
- Barrett-Connor, E.; Kritz-Silverstein, D. Estrogen Replacement Therapy and Cognitive Function in Older Women. JAMA 1993, 269, 2637–2641. [Google Scholar] [CrossRef]
- Mulnard, R.A.; Cotman, C.W.; Kawas, C.; van Dyck, C.H.; Sano, M.; Doody, R.; Koss, E.; Pfeiffer, E.; Jin, S.; Gamst, A.; et al. Estrogen Replacement Therapy for Treatment of Mild to Moderate Alzheimer Disease: A Randomized Controlled Trial. Alzheimer’s Disease Cooperative Study. JAMA 2000, 283, 1007–1015. [Google Scholar] [CrossRef]
- Brenner, D.E.; Kukull, W.A.; Stergachis, A.; van Belle, G.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Larson, E.B. Postmenopausal Estrogen Replacement Therapy and the Risk of Alzheimer’s Disease: A Population-Based Case-Control Study. Am. J. Epidemiol. 1994, 140, 262–267. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Role of Oxidative Stress in the Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative Stress in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Hritcu, L.; Stoica, B.; Bild, W.; Stefanescu, C. Changes of Some Oxidative Stress Markers in the Serum of Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Neurosci. Lett. 2010, 469, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Giliberto, L.; Tamagno, E.; Tabaton, M. Oxidative Stress Mediates the Pathogenic Effect of Different Alzheimer’s Disease Risk Factors. Front. Aging Neurosci. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, A.S.; Carroll, R.T.; Bishayee, A.; Geldenhuys, W.J.; Van der Schyf, C.J. Oxidative Stress and Alzheimer’s Disease: Dietary Polyphenols as Potential Therapeutic Agents. Exp. Rev. Neurother. 2010, 10, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.M.; Moreira, W.L.; de Melo Loureiro, A.P.; de souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral Oxidative Stress Biomarkers in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.T.; Chami, B.; Youssef, P.; Witting, P.K. Oxidative Stress in Alzheimer’s Disease: Primary Villain or Physiological by-Product? Redox Rep. 2013, 18, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Chang, W.-N.; Tsai, N.-W.; Huang, C.-C.; Kung, C.-T.; Su, Y.-J.; Lin, W.-C.; Cheng, B.-C.; Su, C.-M.; Chiang, Y.-F.; et al. The Roles of Biomarkers of Oxidative Stress and Antioxidant in Alzheimer’s Disease: A Systematic Review. BioMed. Res. Int. 2014, 2014, e182303. [Google Scholar] [CrossRef]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral Rios, D.; Campos-Peña, V. Early Onset Alzheimer’s Disease and Oxidative Stress. Oxidative Med. Cell. Longev. 2014, 2014, e375968. [Google Scholar] [CrossRef]
- Bonda, D.J.; Lee, H.; Blair, J.A.; Zhu, X.; Perry, G.; Smith, M.A. Role of Metal Dyshomeostasis in Alzheimer’s Disease. Metallomics 2011, 3, 267–270. [Google Scholar] [CrossRef]
- Pohanka, M. Alzheimer´s Disease and Oxidative Stress: A Review. Curr. Med. Chem. 2014, 21, 356–364. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s Disease Hypothesis and Related Therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Luca, M.; Luca, A.; Calandra, C. The Role of Oxidative Damage in the Pathogenesis and Progression of Alzheimer’s Disease and Vascular Dementia. Oxidative Med. Cell. Longev. 2015, 2015, e504678. [Google Scholar] [CrossRef] [PubMed]
- Wojtunik-Kulesza, K.A.; Oniszczuk, A.; Oniszczuk, T.; Waksmundzka-Hajnos, M. The Influence of Common Free Radicals and Antioxidants on Development of Alzheimer’s Disease. Biomed. Pharmacother. 2016, 78, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Lefter, R.; Serban, I.L.; Stefanescu, C.; Chirita, R. The Oxidative Stress Hypothesis in Alzheimer’s Disease. Psychiatr. Danub. 2013, 25, 401–409. [Google Scholar]
- Hung, C.H.-L.; Cheng, S.S.-Y.; Cheung, Y.-T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.-S.; Lee, S.M.-Y.; Chang, R.C.-C. A Reciprocal Relationship between Reactive Oxygen Species and Mitochondrial Dynamics in Neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, Oxidative Stress and Mitochondrial Dysfunction: Cross Talk in Alzheimer’s Disease and Parkinson’s Disease. Drug. Des. Devel. 2017, 11, 797–810. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Verri, M.; Pastoris, O.; Dossena, M.; Aquilani, R.; Guerriero, F.; Cuzzoni, G.; Venturini, L.; Ricevuti, G.; Bongiorno, A.I. Mitochondrial Alterations, Oxidative Stress and Neuroinflammation in Alzheimer’s Disease. Int. J. Immunopathol. Pharm. 2012, 25, 345–353. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque Formation and the Intraneuronal Accumulation of β-Amyloid in Alzheimer’s Disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Tillement, L.; Lecanu, L.; Papadopoulos, V. Alzheimer’s Disease: Effects of β-Amyloid on Mitochondria. Mitochondrion 2011, 11, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid β Oligomers in Alzheimer’s Disease Pathogenesis, Treatment, and Diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bai, F. The Association of Tau With Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Polydoro, M.; Maury, E.A.; Nicholls, S.B.; Reddy, S.M.; Wegmann, S.; William, C.; Saqran, L.; Cagsal-Getkin, O.; Pitstick, R.; et al. Amyloid Accelerates Tau Propagation and Toxicity in a Model of Early Alzheimer’s Disease. Acta Neuropathol. Commun. 2015, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Avila, J. New Perspectives on the Role of Tau in Alzheimer’s Disease. Implications for Therapy. Biochem. Pharmacol. 2014, 88, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Malinow, R. New Developments on the Role of NMDA Receptors in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2012, 22, 559–563. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA Receptors in Alzheimer’s Disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef]
- Bordji, K.; Becerril-Ortega, J.; Buisson, A. Synapses, NMDA Receptor Activity and Neuronal Aβ Production in Alzheimer’s Disease. Gruyter 2011, 22, 285–294. [Google Scholar] [CrossRef]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional Synapse in Alzheimer’s Disease—A Focus on NMDA Receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef]
- Foster, T.C.; Kyritsopoulos, C.; Kumar, A. Central Role for NMDA Receptors in Redox Mediated Impairment of Synaptic Function during Aging and Alzheimer’s Disease. Behav. Brain Res. 2017, 322, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Beyrent, E.; Gomez, G. Oxidative Stress Differentially Induces Tau Dissociation from Neuronal Microtubules in Neurites of Neurons Cultured from Different Regions of the Embryonic Gallus Domesticus Brain. J. Neurosci. Res. 2020, 98, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative Stress in Alzheimer’s Disease: A Review on Emergent Natural Polyphenolic Therapeutics. Complement. Ther. Med. 2020, 49, 102294. [Google Scholar] [CrossRef] [PubMed]
- Šerý, O.; Povová, J.; Míšek, I.; Pešák, L.; Janout, V. Molecular Mechanisms of Neuropathological Changes in Alzheimer’s Disease: A Review. Folia Neuropathol. 2013, 51, 1–9. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of Oligomers at Early Stages of Tau Aggregation in Alzheimer’s Disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef]
- Evans, D.B.; Rank, K.B.; Bhattacharya, K.; Thomsen, D.R.; Gurney, M.E.; Sharma, S.K. Tau Phosphorylation at Serine 396 and Serine 404 by Human Recombinant Tau Protein Kinase II Inhibits Tau’s Ability to Promote Microtubule Assembly. J. Biol. Chem. 2000, 275, 24977–24983. [Google Scholar] [CrossRef]
- Andorfer, C. Cell-Cycle Reentry and Cell Death in Transgenic Mice Expressing Nonmutant Human Tau Isoforms. J. Neurosci. 2005, 25, 5446–5454. [Google Scholar] [CrossRef]
- Steinhilb, M.L.; Dias-Santagata, D.; Fulga, T.A.; Felch, D.L.; Feany, M.B. Tau Phosphorylation Sites Work in Concert to Promote Neurotoxicity In Vivo. MBoC 2007, 18, 5060–5068. [Google Scholar] [CrossRef]
- Alonso, A.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s Disease Hyperphosphorylated Tau Sequesters Normal Tau into Tangles of Filaments and Disassembles Microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Chung, C.-W.; Song, Y.-H.; Kim, I.-K.; Yoon, W.-J.; Ryu, B.-R.; Jo, D.-G.; Woo, H.-N.; Kwon, Y.-K.; Kim, H.-H.; Gwag, B.-J.; et al. Proapoptotic Effects of Tau Cleavage Product Generated by Caspase-3. Neurobiol. Dis. 2001, 8, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial Oxidative Stress Causes Hyperphosphorylation of Tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, W.; Pang, W.; Xiao, Z.; Jiang, Y.; Hong, Y. Dietary Lycopene Supplementation Improves Cognitive Performances in Tau Transgenic Mice Expressing P301L Mutation via Inhibiting Oxidative Stress and Tau Hyperphosphorylation. J. Alzheimer’s Dis. 2017, 57, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Lee, H.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic Oxidative Stress Causes Increased Tau Phosphorylation in M17 Neuroblastoma Cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Egaña, J.T.; Zambrano, C.; Nuñez, M.T.; Gonzalez-Billault, C.; Maccioni, R.B. Iron-Induced Oxidative Stress Modify Tau Phosphorylation Patterns in Hippocampal Cell Cultures. Biometals 2003, 16, 215–223. [Google Scholar] [CrossRef]
- Wang, D.-L.; Ling, Z.-Q.; Cao, F.-Y.; Zhu, L.-Q.; Wang, J.-Z. Melatonin Attenuates Isoproterenol-Induced Protein Kinase A Overactivation and Tau Hyperphosphorylation in Rat Brain. J. Pineal Res. 2004, 37, 11–16. [Google Scholar] [CrossRef]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein. Oxygen 2021, 1, 96–120. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal Phosphorylated Tau Induced Cognitive Decline, Dendritic Spine Loss and Mitochondrial Abnormalities in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef]
- Horiguchi, T.; Uryu, K.; Giasson, B.I.; Ischiropoulos, H.; LightFoot, R.; Bellmann, C.; Richter-Landsberg, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Nitration of Tau Protein Is Linked to Neurodegeneration in Tauopathies. Am. J. Pathol. 2003, 163, 1021–1031. [Google Scholar] [CrossRef]
- Torres, A.K.; Jara, C.; Olesen, M.A.; Tapia-Rojas, C. Pathologically Phosphorylated Tau at S396/404 (PHF-1) Is Accumulated inside of Hippocampal Synaptic Mitochondria of Aged Wild-Type Mice. Sci. Rep. 2021, 11, 4448. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, T.C.; King, M.E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative Regulation of Fatty Acid-Induced Tau Polymerization. Biochemistry 2000, 39, 14203–14210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, S.; Xiong, J.; Li, Y.; Qu, L. Luteolin Reduces Zinc-Induced Tau Phosphorylation at Ser262/356 in an ROS-Dependent Manner in SH-SY5Y Cells. Biol. Trace Elem. Res. 2012, 149, 273–279. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.-W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) Induces Aggregation of Hyperphosphorylated τ and Its Reduction to Iron (II) Reverses the Aggregation: Implications in the Formation of Neurofibrillary Tangles of Alzheimer’s Disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Yang, L.; Ksiezak-Reding, H. Ca2+ and Mg2+ Selectively Induce Aggregates of PHF-Tau but Not Normal Human Tau. J. Neurosci. Res. 1999, 55, 36–43. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Bahmani, A.; Adem, A.; Zawia, N.H. Infantile Postnatal Exposure to Lead (Pb) Enhances Tau Expression in the Cerebral Cortex of Aged Mice: Relevance to AD. Neurotoxicology 2014, 44, 114–120. [Google Scholar] [CrossRef]
- Jiang, L.-F.; Yao, T.-M.; Zhu, Z.-L.; Wang, C.; Ji, L.-N. Impacts of Cd(II) on the Conformation and Self-Aggregation of Alzheimer’s Tau Fragment Corresponding to the Third Repeat of Microtubule-Binding Domain. Biochim. Biophys. Acta BBA Proteins Proteom. 2007, 1774, 1414–1421. [Google Scholar] [CrossRef]
- Olivieri, G.; Brack, C.; Müller-Spahn, F.; Stähelin, H.B.; Herrmann, M.; Renard, P.; Brockhaus, M.; Hock, C. Mercury Induces Cell Cytotoxicity and Oxidative Stress and Increases β-Amyloid Secretion and Tau Phosphorylation in SHSY5Y Neuroblastoma Cells. J. Neurochem. 2000, 74, 231–236. [Google Scholar] [CrossRef]
- Walton, J.R. An Aluminum-Based Rat Model for Alzheimer’s Disease Exhibits Oxidative Damage, Inhibition of PP2A Activity, Hyperphosphorylated Tau, and Granulovacuolar Degeneration. J. Inorg. Biochem. 2007, 101, 1275–1284. [Google Scholar] [CrossRef]
- Prema, A.; Justin Thenmozhi, A.; Manivasagam, T.; Mohamed Essa, M.; Guillemin, G.J. Fenugreek Seed Powder Attenuated Aluminum Chloride-Induced Tau Pathology, Oxidative Stress, and Inflammation in a Rat Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, S209–S220. [Google Scholar] [CrossRef]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef]
- Yao, K.; Zhao, Y.-F.; Zu, H.-B. Melatonin Receptor Stimulation by Agomelatine Prevents Aβ-Induced Tau Phosphorylation and Oxidative Damage in PC12 Cells. Drug Des. Dev. 2019, 13, 387–396. [Google Scholar] [CrossRef]
- Busciglio, J.; Lorenzo, A.; Yeh, J. β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995, 14, 879–888. [Google Scholar] [CrossRef]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A Possible Link between Beta Amyloid Peptide and Tau Protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau Phosphorylation: The Therapeutic Challenge for Neurodegenerative Disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of Glycogen Synthase Kinase-3 by Lithium Correlates with Reduced Tauopathy and Degeneration in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Honda, T.; Yasutake, K.; Michel, G.; Murayama, O.; Murayama, M.; Ishiguro, K.; Yamaguchi, H. Activation of Tau Protein Kinase I/Glycogen Synthase Kinase-3β by Amyloid β Peptide (25–35) Enhances Phosphorylation of Tau in Hippocampal Neurons. Neurosci. Res. 1998, 31, 317–323. [Google Scholar] [CrossRef]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid Activates GSK-3β to Aggravate Neuronal Tauopathy in Bigenic Mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef]
- Ma, J.; Ma, C.; Li, J.; Sun, Y.; Ye, F.; Liu, K.; Zhang, H. Extracellular Matrix Proteins Involved in Alzheimer’s Disease. Chem. A Eur. J. 2020, 26, 12101–12110. [Google Scholar] [CrossRef]
- Debelle, L.; Tamburro, A.M. Elastin: Molecular Description and Function. Int. J. Biochem. Cell Biol. 1999, 31, 261–272. [Google Scholar] [CrossRef]
- Sandberg, L.B.; Soskel, N.T.; Leslie, J.G. Elastin Structure, Biosynthesis, and Relation to Disease States. Available online: https://www.nejm.org/doi/pdf/10.1056/NEJM198103053041004 (accessed on 29 December 2021). [CrossRef]
- Powell, J.T.; Vine, N.; Crossman, M. On the Accumulation of D-Aspartate in Elastin and Other Proteins of the Ageing Aorta. Atherosclerosis 1992, 97, 201–208. [Google Scholar] [CrossRef]
- Robert, L.; Molinari, J.; Ravelojaona, V.; Andrès, E.; Robert, A.M. Age- and Passage-Dependent Upregulation of Fibroblast Elastase-Type Endopeptidase Activity. Role of Advanced Glycation Endproducts, Inhibition by Fucose- and Rhamnose-Rich Oligosaccharides. Arch. Gerontol. Geriatr. 2010, 50, 327–331. [Google Scholar] [CrossRef]
- Fulop, T.; Khalil, A.; Larbi, A. The Role of Elastin Peptides in Modulating the Immune Response in Aging and Age-Related Diseases. Pathol. Biol. 2012, 60, 28–33. [Google Scholar] [CrossRef]
- Edgar, S.; Hopley, B.; Genovese, L.; Sibilla, S.; Laight, D.; Shute, J. Effects of Collagen-Derived Bioactive Peptides and Natural Antioxidant Compounds on Proliferation and Matrix Protein Synthesis by Cultured Normal Human Dermal Fibroblasts. Sci. Rep. 2018, 8, 10474. [Google Scholar] [CrossRef]
- Ma, C.; Su, J.; Sun, Y.; Feng, Y.; Shen, N.; Li, B.; Liang, Y.; Yang, X.; Wu, H.; Zhang, H.; et al. Significant Upregulation of Alzheimer’s β-Amyloid Levels in a Living System Induced by Extracellular Elastin Polypeptides. Angew. Chem. Int. Ed. 2019, 58, 18703–18709. [Google Scholar] [CrossRef]
- Bochicchio, B.; Lorusso, M.; Pepe, A.; Tamburro, A.M. On Enhancers and Inhibitors of Elastin-Derived Amyloidogenesis. Available online: https://www.futuremedicine.com/doi/abs/10.2217/17435889.4.1.31 (accessed on 29 December 2021).
- Bochicchio, B.; Pepe, A.; Delaunay, F.; Lorusso, M.; Baud, S.; Dauchez, M. Amyloidogenesis of Proteolytic Fragments of Human Elastin. RSC Adv. 2013, 3, 13273–13285. [Google Scholar] [CrossRef][Green Version]
- Szychowski, K.A.; Skóra, B. Review of the Relationship between Reactive Oxygen Species (ROS) and Elastin-Derived Peptides (EDPs). Appl. Sci. 2021, 11, 8732. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The Many Faces of PPARγ. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Gmiński, J. Impact of Elastin-Derived VGVAPG Peptide on Bidirectional Interaction between Peroxisome Proliferator-Activated Receptor Gamma (Pparγ) and Beta-Galactosidase (β-Gal) Expression in Mouse Cortical Astrocytes in Vitro. Naunyn-Schmiedeberg’s Arch Pharm. 2019, 392, 405–413. [Google Scholar] [CrossRef]
- Gmiński, J.; Wȩglarz, L.; Dróżdż, M.; Goss, M. Pharmacological Modulation of the Antioxidant Enzymes Activities and the Concentration of Peroxidation Products in Fibroblasts Stimulated with Elastin Peptides. Gen. Pharmacol. Vasc. Syst. 1991, 22, 495–497. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Skóra, B.; Wójtowicz, A.K. Elastin-Derived Peptides in the Central Nervous System: Friend or Foe. Cell Mol. Neurobiol. 2021, 1–15. [Google Scholar] [CrossRef]
- Calleri, E.; Pochetti, G.; Dossou, K.S.S.; Laghezza, A.; Montanari, R.; Capelli, D.; Prada, E.; Loiodice, F.; Massolini, G.; Bernier, M.; et al. Resveratrol and Its Metabolites Bind to PPARs. Chembiochem 2014, 15, 1154–1160. [Google Scholar] [CrossRef]
- Ma, T.; Tan, M.-S.; Yu, J.-T.; Tan, L. Resveratrol as a Therapeutic Agent for Alzheimer’s Disease. BioMed. Res. Int. 2014, 2014, e350516. [Google Scholar] [CrossRef]
- Nicoloff, G.; Tzvetanov, P.; Christova, P.; Baydanoff, S. Detection of Elastin Derived Peptides in Cerebrospinal Fluid of Patients with First Ever Ischaemic Stroke. Neuropeptides 2008, 42, 277–282. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Gmiński, J. The VGVAPG Peptide Regulates the Production of Nitric Oxide Synthases and Reactive Oxygen Species in Mouse Astrocyte Cells In Vitro. Neurochem. Res. 2019, 44, 1127–1137. [Google Scholar] [CrossRef]
- van Horssen, J.; Wesseling, P.; van den Heuvel, L.P.; de Waal, R.M.; Verbeek, M.M. Heparan Sulphate Proteoglycans in Alzheimer’s Disease and Amyloid-related Disorders. Lancet Neurol. 2003, 2, 482–492. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Piperigkou, Z.; Theocharis, A.D.; Watanabe, H.; Franchi, M.; Baud, S.; Brézillon, S.; Götte, M.; Passi, A.; Vigetti, D.; et al. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem. Rev. 2018, 118, 9152–9232. [Google Scholar] [CrossRef]
- Kawecki, C.; Hézard, N.; Bocquet, O.; Poitevin, G.; Rabenoelina, F.; Kauskot, A.; Duca, L.; Blaise, S.; Romier, B.; Martiny, L.; et al. Elastin-Derived Peptides Are New Regulators of Thrombosis. Arter. Thromb. Vasc. Biol. 2014, 34, 2570–2578. [Google Scholar] [CrossRef]
- Tzvetanov, P.; Nicoloff, G.; Rousseff, R.; Christova, P. Increased Levels of Elastin-Derived Peptides in Cerebrospinal Fluid of Patients with Lacunar Stroke. Clin. Neurol. Neurosurg. 2008, 110, 239–244. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Xia, Z.; Dudek, H.; Miranti, C.K.; Greenberg, M.E. Calcium Influx via the NMDA Receptor Induces Immediate Early Gene Transcription by a MAP Kinase/ERK-Dependent Mechanism. J. Neurosci. 1996, 16, 5425–5436. [Google Scholar] [CrossRef]
- Evans, R.C.; Morera-Herreras, T.; Cui, Y.; Du, K.; Sheehan, T.; Kotaleski, J.H.; Venance, L.; Blackwell, K.T. The Effects of NMDA Subunit Composition on Calcium Influx and Spike Timing-Dependent Plasticity in Striatal Medium Spiny Neurons. PLoS Comput. Biol. 2012, 8, e1002493. [Google Scholar] [CrossRef]
- Paoletti, P.; Neyton, J. NMDA Receptor Subunits: Function and Pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Ali, S.; Najmi, A.K.; Nath, C. Okadaic Acid-Induced Tau Phosphorylation in Rat Brain: Role of NMDA Receptor. Neuroscience 2013, 238, 97–113. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal Calcium Mishandling and the Pathogenesis of Alzheimer’s Disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef]
- Ahmed, H.A.; Ismael, S.; Mirzahosseini, G.; Ishrat, T. Verapamil Prevents Development of Cognitive Impairment in an Aged Mouse Model of Sporadic Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3374–3387. [Google Scholar] [CrossRef]
- Jomsky, M.; Kerna, N. Nifedipine: Can This Calcium Channel Blocker Be Used “Off Label” to Inhibit the Development and Symptoms of Alzheimer’s Disease and Related Beta-Amyloid-Producing Syndromes? EC Pharmacol. Toxicol. 2019. [Google Scholar] [CrossRef]
- Pithadia, A.S.; Lim, M.H. Metal-Associated Amyloid-β Species in Alzheimer’s Disease. Curr. Opin. Chem. Biol. 2012, 16, 67–73. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal Dyshomeostasis and Oxidative Stress in Alzheimer’s Disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Raymick, J.; Sarkar, S. Role of Metals in Alzheimer’s Disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef]
- Tiiman, A.; Palumaa, P.; Tõugu, V. The Missing Link in the Amyloid Cascade of Alzheimer’s Disease—Metal Ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current Understanding of Metal Ions in the Pathogenesis of Alzheimer’s Disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Strodel, B.; Coskuner-Weber, O. Transition Metal Ion Interactions with Disordered Amyloid-β Peptides in the Pathogenesis of Alzheimer’s Disease: Insights from Computational Chemistry Studies. J. Chem. Inf. Model. 2019, 59, 1782–1805. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446. [Google Scholar] [CrossRef]
- Kepp, K.P. Alzheimer’s Disease: How Metal Ions Define β-Amyloid Function. Coord. Chem. Rev. 2017, 351, 127–159. [Google Scholar] [CrossRef]
- Watt, A.D.; Villemagne, V.L.; Barnham, K.J. Metals, Membranes, and Amyloid-β Oligomers: Key Pieces in the Alzheimer’s Disease Puzzle? J. Alzheimer’s Dis. 2013, 33, S283–S293. [Google Scholar] [CrossRef]
- Roberts, B.R.; Ryan, T.M.; Bush, A.I.; Masters, C.L.; Duce, J.A. The Role of Metallobiology and Amyloid-β Peptides in Alzheimer’s Disease. J. Neurochem. 2012, 120, 149–166. [Google Scholar] [CrossRef]
- Tõugu, V.; Tiiman, A.; Palumaa, P. Interactions of Zn(Ii) and Cu(Ii) Ions with Alzheimer’s Amyloid-Beta Peptide. Metal Ion Binding, Contribution to Fibrillization and Toxicity. Metallomics 2011, 3, 250–261. [Google Scholar] [CrossRef]
- Dahms, S.O.; Könnig, I.; Roeser, D.; Gührs, K.-H.; Mayer, M.C.; Kaden, D.; Multhaup, G.; Than, M.E. Metal Binding Dictates Conformation and Function of the Amyloid Precursor Protein (APP) E2 Domain. J. Mol. Biol. 2012, 416, 438–452. [Google Scholar] [CrossRef]
- Kozin, S.A.; Mezentsev, Y.V.; Kulikova, A.A.; Indeykina, M.I.; Golovin, A.V.; Ivanov, A.S.; Tsvetkov, P.O.; Makarov, A.A. Zinc-Induced Dimerization of the Amyloid-β Metal-Binding Domain 1–16 Is Mediated by Residues 11–14. Mol. Biosyst. 2011, 7, 1053–1055. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Girvan, P.; Teng, X.; Brooks, N.J.; Baldwin, G.S.; Ying, L. Redox Kinetics of the Amyloid-β-Cu Complex and Its Biological Implications. Biochemistry 2018, 57, 6228–6233. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-Wide Analysis Reveals Mechanisms Modulating Autophagy in Normal Brain Aging and in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Kaur, U.; Banerjee, P.; Bir, A.; Sinha, M.; Biswas, A.; Chakrabarti, S. Reactive Oxygen Species, Redox Signaling and Neuroinflammation in Alzheimer’s Disease: The NF-ΚB Connection. Curr. Top. Med. Chem. 2015, 15, 446–457. [Google Scholar] [CrossRef]
- Mota, S.I.; Costa, R.O.; Ferreira, I.L.; Santana, I.; Caldeira, G.L.; Padovano, C.; Fonseca, A.C.; Baldeiras, I.; Cunha, C.; Letra, L.; et al. Oxidative Stress Involving Changes in Nrf2 and ER Stress in Early Stages of Alzheimer’s Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1428–1441. [Google Scholar] [CrossRef]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive Oxygen Species: Stuck in the Middle of Neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S357–S367. [Google Scholar] [CrossRef]
- Caldeira, G.L.; Ferreira, I.L.; Rego, A.C. Impaired Transcription in Alzheimer’s Disease: Key Role in Mitochondrial Dysfunction and Oxidative Stress. J. Alzheimer’s Dis. 2013, 34, 115–131. [Google Scholar] [CrossRef]
- Nesi, G.; Sestito, S.; Digiacomo, M.; Rapposelli, S. Oxidative Stress, Mitochondrial Abnormalities and Proteins Deposition: Multitarget Approaches in Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 3062–3079. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer’s Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Hemmelgarn, B.T.; Chuang, C.-C.; Best, T.M. The Role of Oxidative Stress-Induced Epigenetic Alterations in Amyloid-β Production in Alzheimer’s Disease. Oxid. Med. Cell Longev. 2015, 2015, 604658. [Google Scholar] [CrossRef]
- Lin, H.-C.; Hsieh, H.-M.; Chen, Y.-H.; Hu, M.-L. S-Adenosylhomocysteine Increases Beta-Amyloid Formation in BV-2 Microglial Cells by Increased Expressions of Beta-Amyloid Precursor Protein and Presenilin 1 and by Hypomethylation of These Gene Promoters. Neurotoxicology 2009, 30, 622–627. [Google Scholar] [CrossRef]
- Sung, H.Y.; Choi, E.N.; Ahn Jo, S.; Oh, S.; Ahn, J.-H. Amyloid Protein-Mediated Differential DNA Methylation Status Regulates Gene Expression in Alzheimer’s Disease Model Cell Line. Biochem. Biophys. Res. Commun. 2011, 414, 700–705. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative Stress Increases BACE1 Protein Levels through Activation of the PKR-EIF2α Pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of EIF2α Kinases Alleviates AD-Related Synaptic Plasticity and Spatial Memory Deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Klann, E. EIF2-Dependent Translation Initiation: Memory Consolidation and Disruption in Alzheimer’s Disease. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-Specific Epigenetic Drift in Late-Onset Alzheimer’s Disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, S.; Cavallaro, R.A.; D’Anselmi, F.; Fuso, A. Gene Silencing through Methylation: An Epigenetic Intervention on Alzheimer Disease. J. Alzheimers Dis. 2006, 9, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Lithner, C.U.; Hernandez, C.; Sweatt, J.D.; Nordberg, A. O3-05-05: Epigenetic Effects of Aβ and the Implication on the Pathophysiology in Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, S508. [Google Scholar] [CrossRef]
- Marques, S.C.F.; Lemos, R.; Ferreiro, E.; Martins, M.; de Mendonça, A.; Santana, I.; Outeiro, T.F.; Pereira, C.M.F. Epigenetic Regulation of BACE1 in Alzheimer’s Disease Patients and in Transgenic Mice. Neuroscience 2012, 220, 256–266. [Google Scholar] [CrossRef]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.M.; Coleman, P.D.; Rutten, B.P.F.; van den Hove, D.L.A. Consistent Decrease in Global DNA Methylation and Hydroxymethylation in the Hippocampus of Alzheimer’s Disease Patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef]
- Gu, X.; Sun, J.; Li, S.; Wu, X.; Li, L. Oxidative Stress Induces DNA Demethylation and Histone Acetylation in SH-SY5Y Cells: Potential Epigenetic Mechanisms in Gene Transcription in Aβ Production. Neurobiol. Aging 2013, 34, 1069–1079. [Google Scholar] [CrossRef]
- Luque-Contreras, D.; Carvajal, K.; Toral-Rios, D.; Franco-Bocanegra, D.; Campos-Peña, V. Oxidative Stress and Metabolic Syndrome: Cause or Consequence of Alzheimer’s Disease? Oxidative Med. Cell. Longev. 2014, 2014, e497802. [Google Scholar] [CrossRef]
- Massaad, C.A. Neuronal and Vascular Oxidative Stress in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef]
- Hamilton, A.; Holscher, C. The Effect of Ageing on Neurogenesis and Oxidative Stress in the APPswe/PS1deltaE9 Mouse Model of Alzheimer’s Disease. Brain Res. 2012, 1449, 83–93. [Google Scholar] [CrossRef]
- Sultana, R.; Mecocci, P.; Mangialasche, F.; Cecchetti, R.; Baglioni, M.; Butterfield, D.A. Increased Protein and Lipid Oxidative Damage in Mitochondria Isolated from Lymphocytes from Patients with Alzheimer’s Disease: Insights into the Role of Oxidative Stress in Alzheimer’s Disease and Initial Investigations into a Potential Biomarker for This Dementing Disorder. J. Alzheimer’s Dis. 2011, 24, 77–84. [Google Scholar] [CrossRef]
- Readnower, R.D.; Sauerbeck, A.D.; Sullivan, P.G. Mitochondria, Amyloid β, and Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011, e104545. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The Key Peptide in the Pathogenesis of Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal Mitochondrial Dynamics and Synaptic Degeneration as Early Events in Alzheimer’s Disease: Implications to Mitochondria-Targeted Antioxidant Therapeutics. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic Exposure to Sub-Lethal Beta-Amyloid (Abeta) Inhibits the Import of Nuclear-Encoded Proteins to Mitochondria in Differentiated PC12 Cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef]
- Crouch, P.J.; Blake, R.; Duce, J.A.; Ciccotosto, G.D.; Li, Q.X.; Barnham, K.J.; Curtain, C.C.; Cherny, R.A.; Cappai, R.; Dyrks, T.; et al. Copper-Dependent Inhibition of Human Cytochrome c Oxidase by a Dimeric Conformer of Amyloid-Beta(1-42). J. Neurosci. 2005, 25, 672–679. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-β-Induced Mitochondrial Dysfunction. J. Alzheimers Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Bajouco, L.M.; Mota, S.I.; Auberson, Y.P.; Oliveira, C.R.; Rego, A.C. Amyloid Beta Peptide 1-42 Disturbs Intracellular Calcium Homeostasis through Activation of GluN2B-Containing N-Methyl-d-Aspartate Receptors in Cortical Cultures. Cell Calcium. 2012, 51, 95–106. [Google Scholar] [CrossRef]
- Shirwany, N.A.; Payette, D.; Xie, J.; Guo, Q. The Amyloid Beta Ion Channel Hypothesis of Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 597–612. [Google Scholar]
- Magi, S.; Castaldo, P.; Macrì, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular Calcium Dysregulation: Implications for Alzheimer’s Disease. Biomed. Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Brewer, G.J. Amyloid Beta as a Modulator of Synaptic Plasticity. J. Alzheimers Dis. 2010, 22, 741–763. [Google Scholar] [CrossRef] [PubMed]
- Granold, M.; Moosmann, B.; Staib-Lasarzik, I.; Arendt, T.; Del Rey, A.; Engelhard, K.; Behl, C.; Hajieva, P. High Membrane Protein Oxidation in the Human Cerebral Cortex. Redox Biol. 2015, 4, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of Amyloid β-Peptide-Associated Oxidative Stress and Brain Protein Modifications in the Pathogenesis of Alzheimer’s Disease and Mild Cognitive Impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Mattson, M.P. Adverse Stress, Hippocampal Networks, and Alzheimer’s Disease. Neuromol. Med. 2010, 12, 56–70. [Google Scholar] [CrossRef]
- Venkateshappa, C.; Harish, G.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Elevated Oxidative Stress and Decreased Antioxidant Function in the Human Hippocampus and Frontal Cortex with Increasing Age: Implications for Neurodegeneration in Alzheimer’s Disease. Neurochem. Res. 2012, 37, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.; Eckert, A.; Kurz, C.; Eckert, G.P.; Leuner, K. Mitochondrial Dysfunction: Common Final Pathway in Brain Aging and Alzheimer’s Disease—Therapeutic Aspects. Mol. Neurobiol. 2010, 41, 159–171. [Google Scholar] [CrossRef]
- Sivanandam, T.M.; Thakur, M.K. Traumatic Brain Injury: A Risk Factor for Alzheimer’s Disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic Brain Injury and Amyloid-β Pathology: A Link to Alzheimer’s Disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef]
- Government of Canada, H.C. Drug Product Database Online Query. Available online: https://health-products.canada.ca/dpd-bdpp/info.do?lang=en&code=93258 (accessed on 28 December 2021).
- Government of Canada, H.C. Drug Product Database Online Query. Available online: https://health-products.canada.ca/dpd-bdpp/info.do?lang=en&code=80954 (accessed on 28 December 2021).
- Hasselmo, M.E. The Role of Acetylcholine in Learning and Memory. Curr. Opin. Neurobiol. 2006, 16, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gupta, V.; Sharma, S. Donepezil. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Müller, T. Rivastigmine in the Treatment of Patients with Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Razay, G.; Wilcock, G.K. Galantamine in Alzheimer’s Disease. Exp. Rev. Neurother. 2008, 8, 9–17. [Google Scholar] [CrossRef]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of Action of Memantine. Curr. Opin. Pharm. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J.; Memantine Study Group. Memantine in Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef]
- Deardorff, W.J.; Grossberg, G.T. A Fixed-Dose Combination of Memantine Extended-Release and Donepezil in the Treatment of Moderate-to-Severe Alzheimer’s Disease. Drug Des. Dev. 2016, 10, 3267–3279. [Google Scholar] [CrossRef]
- InformedHealth.org. Non-Drug Interventions for Alzheimer’s Disease; Institute for Quality and Efficiency in Health Care (IQWiG): Cologne, Germany, 2017. [Google Scholar]
- Non-pharmacological Treatment of Alzheimer’s|IntechOpen. Available online: https://www.intechopen.com/chapters/65852 (accessed on 28 December 2021).
- Bastrup, J.; Hansen, K.H.; Poulsen, T.B.G.; Kastaniegaard, K.; Asuni, A.A.; Christensen, S.; Belling, D.; Helboe, L.; Stensballe, A.; Volbracht, C. Anti-Aβ Antibody Aducanumab Regulates the Proteome of Senile Plaques and Closely Surrounding Tissue in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 79, 249–265. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Leinenga, G.; Koh, W.K.; Götz, J. A Comparative Study of the Effects of Aducanumab and Scanning Ultrasound on Amyloid Plaques and Behavior in the APP23 Mouse Model of Alzheimer Disease. Alzheimers Res. 2021, 13, 76. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to Demonstrate Efficacy of Aducanumab: An Analysis of the EMERGE and ENGAGE Trials as Reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Boonruamkaew, P.; Chonpathompikunlert, P.; Vong, L.B.; Sakaue, S.; Tomidokoro, Y.; Ishii, K.; Tamaoka, A.; Nagasaki, Y. Chronic Treatment with a Smart Antioxidative Nanoparticle for Inhibition of Amyloid Plaque Propagation in Tg2576 Mouse Model of Alzheimer’s Disease. Sci. Rep. 2017, 7, 3785. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, N.S.; Ghoneum, M.H. Antia, a Natural Antioxidant Product, Attenuates Cognitive Dysfunction in Streptozotocin-Induced Mouse Model of Sporadic Alzheimer’s Disease by Targeting the Amyloidogenic, Inflammatory, Autophagy, and Oxidative Stress Pathways. Oxid. Med. Cell Longev. 2020, 2020, 4386562. [Google Scholar] [CrossRef] [PubMed]
- Nicolakakis, N.; Aboulkassim, T.; Ongali, B.; Lecrux, C.; Fernandes, P.; Rosa-Neto, P.; Tong, X.-K.; Hamel, E. Complete Rescue of Cerebrovascular Function in Aged Alzheimer’s Disease Transgenic Mice by Antioxidants and Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma Agonist. J. Neurosci. 2008, 28, 9287–9296. [Google Scholar] [CrossRef] [PubMed]
- Veurink, G.; Perry, G.; Singh, S.K. Role of Antioxidants and a Nutrient Rich Diet in Alzheimer’s Disease. Open Biol. 2020, 10, 200084. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.; Puścion-Jakubik, A.; Markiewicz-Żukowska, R.; Socha, K. Vitamins in Alzheimer’s Disease—Review of the Latest Reports. Nutrients 2020, 12, 3458. [Google Scholar] [CrossRef]
- Fenech, M. Vitamins Associated with Brain Aging, Mild Cognitive Impairment, and Alzheimer Disease: Biomarkers, Epidemiological and Experimental Evidence, Plausible Mechanisms, and Knowledge Gaps. Adv. Nutr. 2017, 8, 958–970. [Google Scholar] [CrossRef]
- Antibody–Drug Conjugates: A Comprehensive Review|Molecular Cancer Research. Available online: https://mcr.aacrjournals.org/content/18/1/3 (accessed on 28 December 2021).
- Barroso-Sousa, R.; Tolaney, S.M. Clinical Development of New Antibody–Drug Conjugates in Breast Cancer: To Infinity and Beyond. BioDrugs 2021, 35, 159–174. [Google Scholar] [CrossRef]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. DESTINY-Lung01 Trial Investigators. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2021, 386, 241–251. [Google Scholar] [CrossRef]
- Calo, C.A.; O’Malley, D.M. Antibody-Drug Conjugates for the Treatment of Ovarian Cancer. Exp. Opin. Biol. 2021, 21, 875–887. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Pandey, M.K.; Tyagi, A.K.; Deb, L. Oxidative Stress and Cancer: Advances and Challenges. Oxidative Med. Cell. Longev. 2016, 2016, e5010423. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative Stress and Cancer: An Overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Andrisic, L.; Dudzik, D.; Barbas, C.; Milkovic, L.; Grune, T.; Zarkovic, N. Short Overview on Metabolomics Approach to Study Pathophysiology of Oxidative Stress in Cancer. Redox Biol. 2018, 14, 47–58. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative Stress and Lipid Peroxidation Products in Cancer Progression and Therapy. ISRN Oncol. 2012, 2012, 1–21. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. Int. J. Mol. Sci. 2015, 16, 193–217. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of Reactive Oxygen Species: An Emerging Approach for Cancer Therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Chen, P.; Luo, X.; Nie, P.; Wu, B.; Xu, W.; Shi, X.; Chang, H.; Li, B.; Yu, X.; Zou, Z. CQ Synergistically Sensitizes Human Colorectal Cancer Cells to SN-38/CPT-11 through Lysosomal and Mitochondrial Apoptotic Pathway via P53-ROS Cross-Talk. Free Radic. Biol. Med. 2017, 104, 280–297. [Google Scholar] [CrossRef]
- Dewangan, J.; Tandon, D.; Srivastava, S.; Verma, A.K.; Yapuri, A.; Rath, S.K. Novel Combination of Salinomycin and Resveratrol Synergistically Enhances the Anti-Proliferative and pro-Apoptotic Effects on Human Breast Cancer Cells. Apoptosis 2017, 22, 1246–1259. [Google Scholar] [CrossRef]
- Zhao, Y.; Qu, T.; Wang, P.; Li, X.; Qiang, J.; Xia, Z.; Duan, H.; Huang, J.; Zhu, L. Unravelling the Relationship between Macroautophagy and Mitochondrial ROS in Cancer Therapy. Apoptosis 2016, 21, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 Autophosphorylation Is Promoted by Mitochondrial ROS and Is Essential for RIP3 Recruitment into Necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, R.; Tian, X.; Zhang, Z.; Zhang, P.; Li, C.; Feng, Z. Synergistic Anti-Tumor Activity of Nimotuzumab in Combination with Trastuzumab in HER2-Positive Breast Cancer. Biochem. Biophys. Res. Commun. 2017, 489, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Santoro, V.; Jia, R.; Thompson, H.; Nijhuis, A.; Jeffery, R.; Kiakos, K.; Silver, A.R.; Hartley, J.A.; Hochhauser, D. Role of Reactive Oxygen Species in the Abrogation of Oxaliplatin Activity by Cetuximab in Colorectal Cancer. JNCI J. Natl. Cancer Inst. 2016, 108, djv394. [Google Scholar] [CrossRef]
- Combined Oridonin with Cetuximab Treatment Shows Synergistic Anticancer Effects on Laryngeal Squamous Cell Carcinoma: Involvement of Inhibition of EGFR and Activation of Reactive Oxygen Species-Mediated JNK Pathway. Available online: https://www.spandidos-publications.com/10.3892/ijo.2016.3696 (accessed on 29 December 2021).
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab Treatment Induces Metabolic Adaptation toward Anaerobic Metabolism in Glioblastomas. Acta Neuropathol. 2015, 129, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, D.; Sun, K.; Wang, J.; Liu, Y.; Song, J.; Zhao, Q.; Zhang, S.; Deng, W.; Zhao, X.; et al. Inhibition of Autophagy Enhances Anticancer Effects of Bevacizumab in Hepatocarcinoma. J. Mol. Med. 2013, 91, 473–483. [Google Scholar] [CrossRef]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat Synergizes with EGFR Inhibitors in NSCLC Cells by Increasing ROS via Up-Regulation of the Major Mitochondrial Porin VDAC1 and Modulation of the c-Myc-NRF2-KEAP1 Pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef]
- Abounader, R.; Schiff, D. The Blood-Brain Barrier Limits the Therapeutic Efficacy of Antibody-Drug Conjugates in Glioblastoma. Neuro. Oncol. 2021, 23, 1993–1994. [Google Scholar] [CrossRef]
- Frontiers|Antibody Drug Conjugates in Glioblastoma—Is There a Future for Them?|Oncology. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2021.718590/full (accessed on 28 December 2021).
- Banks, W.A. Characteristics of Compounds That Cross the Blood-Brain Barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef]
- Cavaco, M.; Frutos, S.; Oliete, P.; Valle, J.; Andreu, D.; Castanho, M.A.R.B.; Vila-Perelló, M.; Neves, V. Conjugation of a Blood Brain Barrier Peptide Shuttle to an Fc Domain for Brain Delivery of Therapeutic Biomolecules. ACS Med. Chem. Lett. 2021, 12, 1663–1668. [Google Scholar] [CrossRef]
- Oldendorf, W.H. Brain Uptake of Radiolabeled Amino Acids, Amines, and Hexoses after Arterial Injection. Am. J. Physiol. 1971, 221, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.M. The Impact of Efflux Transporters in the Brain on the Development of Drugs for CNS Disorders. Clin. Pharm. 2002, 41, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Zlokovic, B. The Role of the Cell Surface LRP and Soluble LRP in Blood-Brain Barrier Abeta Clearance in Alzheimers Disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef]
- Frontiers|Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease|Cellular Neuroscience. Available online: https://www.frontiersin.org/articles/10.3389/fncel.2021.695479/full (accessed on 28 December 2021).
- Sarma, B.K.; Mugesh, G. Glutathione Peroxidase (GPx)-like Antioxidant Activity of the Organoselenium Drug Ebselen: Unexpected Complications with Thiol Exchange Reactions. J. Am. Chem. Soc. 2005, 127, 11477–11485. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.K.; Boyd, R.J. Effect of Substituents on the GPx-like Activity of Ebselen: Steric versus Electronic. J. Phys. Chem. A 2008, 112, 1013–1017. [Google Scholar] [CrossRef]
- Asbaghi, O.; Zakeri, N.; Rezaei Kelishadi, M.; Naeini, F.; Mirzadeh, E. Selenium Supplementation and Oxidative Stress: A Review. PharmaNutrition 2021, 17, 100263. [Google Scholar] [CrossRef]
- Xie, Y.; Tan, Y.; Zheng, Y.; Du, X.; Liu, Q. Ebselen Ameliorates β-Amyloid Pathology, Tau Pathology, and Cognitive Impairment in Triple-Transgenic Alzheimer’s Disease Mice. J. Biol. Inorg. Chem. 2017, 22, 851–865. [Google Scholar] [CrossRef]
- Luo, Z.; Sheng, J.; Sun, Y.; Lu, C.; Yan, J.; Liu, A.; Luo, H.-B.; Huang, L.; Li, X. Synthesis and Evaluation of Multi-Target-Directed Ligands against Alzheimer’s Disease Based on the Fusion of Donepezil and Ebselen. J. Med. Chem. 2013, 56, 9089–9099. [Google Scholar] [CrossRef]
- Terra, B.; da Silva, P.; Tramarin, A.; Franco, L.; da Cunha, E.; Macedo Junior, F.; Ramalho, T.; Bartolini, M.; Bolognesi, M.; de Fátima, Â. Two Novel Donepezil-Lipoic Acid Hybrids: Synthesis, Anticholinesterase and Antioxidant Activities and Theoretical Studies. J. Braz. Chem. Soc. 2017, 29, 738–747. [Google Scholar] [CrossRef]
- Pi, R.; Mao, X.; Chao, X.; Cheng, Z.; Liu, M.; Duan, X.; Ye, M.; Chen, X.; Mei, Z.; Liu, P.; et al. Tacrine-6-Ferulic Acid, a Novel Multifunctional Dimer, Inhibits Amyloid-β-Mediated Alzheimer’s Disease-Associated Pathogenesis In Vitro and In Vivo. PLoS ONE 2012, 7, e31921. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cruz, K.; Moncada-Basualto, M.; Morales-Valenzuela, J.; Barriga-González, G.; Navarrete-Encina, P.; Núñez-Vergara, L.; Squella, J.A.; Olea-Azar, C. Synthesis and Antioxidant Study of New Polyphenolic Hybrid-Coumarins. Arab. J. Chem. 2018, 11, 525–537. [Google Scholar] [CrossRef]
- Trang, N.V.; Thuy, P.T.; Thanh, D.T.M.; Son, N.T. Benzofuran–Stilbene Hybrid Compounds: An Antioxidant Assessment—A DFT Study. RSC Adv. 2021, 11, 12971–12980. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2013, 2013, e316523. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A Controlled Trial of Selegiline, Alpha-Tocopherol, or Both as Treatment for Alzheimer’s Disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Alzheimer’s Disease Cooperative Study Group. Vitamin E and Donepezil for the Treatment of Mild Cognitive Impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of Vitamin E and Memantine on Functional Decline in Alzheimer Disease: The TEAM-AD VA Cooperative Randomized Trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Alzheimer’s Disease Cooperative Study. Antioxidants for Alzheimer Disease: A Randomized Clinical Trial with Cerebrospinal Fluid Biomarker Measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. Alzheimer Disease Cooperative Study. High-Dose B Vitamin Supplementation and Cognitive Decline in Alzheimer Disease: A Randomized Controlled Trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef]
- Annweiler, C.; Fantino, B.; Parot-Schinkel, E.; Thiery, S.; Gautier, J.; Beauchet, O. Alzheimer’s Disease--Input of Vitamin D with MEmantine Assay (AD-IDEA Trial): Study Protocol for a Randomized Controlled Trial. Trials 2011, 12, 230. [Google Scholar] [CrossRef]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A Phase II Randomized Clinical Trial of a Nutritional Formulation for Cognition and Mood in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.W.; Grossman, H.; Neugroschl, J.; Parker, S.; Burden, A.; Luo, X.; Sano, M. A Randomized, Double-Blind, Placebo-Controlled Trial of Resveratrol with Glucose and Malate (RGM) to Slow the Progression of Alzheimer’s Disease: A Pilot Study. Alzheimers Dement. 2018, 4, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol Regulates Neuro-Inflammation and Induces Adaptive Immunity in Alzheimer’s Disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral Curcumin for Alzheimer’s Disease: Tolerability and Efficacy in a 24-Week Randomized, Double Blind, Placebo-Controlled Study. Alzheimers Res. 2012, 4, 43. [Google Scholar] [CrossRef]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients with Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef]
- Pilot Study to Investigate the Safety and Feasibility of Senolytic Therapy to Modulate Progression of Alzheimer’s Disease (SToMP-AD). ClinicalTrials.gov Identifier: NCT04063124. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04063124 (accessed on 18 December 2021).
- Phase II Clinical Trial to Evaluate the Safety and Feasibility of Senolytic Therapy in Alzheimer’s Disease. ClinicalTrials.gov Identifier: NCT04685590. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04685590 (accessed on 18 December 2021).
- Sunphenon EGCg (Epigallocatechin-Gallate) in the Early Stage of Alzheimer´s Disease. ClinicalTrials.gov Identifier: NCT00951834. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00951834 (accessed on 18 December 2021).
- Prevention of Cognitive Decline in ApoE4 Carriers with Subjective Cognitive Decline After EGCG and a Multimodal Intervention. ClinicalTrials.gov Identifier: NCT03978052. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03978052 (accessed on 18 December 2021).
- Vina, J. Effect of Activation of the Receptor PPARg/RXR as a Possible Treatment for Alzheimer’s Disease. Role of Genistein. Clinical Trial Registration NCT01982578; clinicaltrials.gov, 2021. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01982578 (accessed on 18 December 2021).
- Gleason, C.E.; Fischer, B.L.; Dowling, N.M.; Setchell, K.D.R.; Atwood, C.S.; Carlsson, C.M.; Asthana, S. Cognitive Effects of Soy Isoflavones in Patients with Alzheimer’s Disease. J. Alzheimers. Dis. 2015, 47, 1009–1019. [Google Scholar] [CrossRef]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A Randomized Placebo-Controlled Pilot Trial of Omega-3 Fatty Acids and Alpha Lipoic Acid in Alzheimer’s Disease. J. Alzheimers Dis. 2014, 38, 111–120. [Google Scholar] [CrossRef]
- Treatment with Copper in Patients with Mild Alzheimer´s Dementia—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00608946 (accessed on 28 December 2021).
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Wilson, R.S.; Scherr, P.A. Dietary Intake of Antioxidant Nutrients and the Risk of Incident Alzheimer Disease in a Biracial Community Study. JAMA 2002, 287, 3230–3237. [Google Scholar] [CrossRef]
- Grundman, M.; Grundman, M.; Delaney, P. Antioxidant Strategies for Alzheimer’s Disease. Proc. Nutr. Soc. 2002, 61, 191–202. [Google Scholar] [CrossRef]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.S.; Cache County Study Group. Reduced Risk of Alzheimer Disease in Users of Antioxidant Vitamin Supplements: The Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Praticò, D. Early Vitamin E Supplementation in Young but Not Aged Mice Reduces Abeta Levels and Amyloid Deposition in a Transgenic Model of Alzheimer’s Disease. FASEB J. 2004, 18, 323–325. [Google Scholar] [CrossRef]
- Nakashima, H.; Ishihara, T.; Yokota, O.; Terada, S.; Trojanowski, J.Q.; Lee, V.M.-Y.; Kuroda, S. Effects of Alpha-Tocopherol on an Animal Model of Tauopathies. Free Radic. Biol. Med. 2004, 37, 176–186. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and Tau Toxicities in Alzheimer’s Are Linked via Oxidative Stress-Induced P38 Activation: Protective Role of Vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef]
- Veinbergs, I.; Mallory, M.; Sagara, Y.; Masliah, E. Vitamin E Supplementation Prevents Spatial Learning Deficits and Dendritic Alterations in Aged Apolipoprotein E-Deficient Mice. Eur. J. Neurosci. 2000, 12, 4541–4546. [Google Scholar]
- Ishihara, Y.; Itoh, K.; Mitsuda, Y.; Shimada, T.; Kubota, T.; Kato, C.; Song, S.Y.; Kobayashi, Y.; Mori-Yasumoto, K.; Sekita, S.; et al. Involvement of Brain Oxidation in the Cognitive Impairment in a Triple Transgenic Mouse Model of Alzheimer’s Disease: Noninvasive Measurement of the Brain Redox State by Magnetic Resonance Imaging. Free Radic. Res. 2013, 47, 731–739. [Google Scholar] [CrossRef]
- Moore, J.J.; Saadabadi, A. Selegiline. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Filip, V.; Kolibás, E. Selegiline in the Treatment of Alzheimer’s Disease: A Long-Term Randomized Placebo-Controlled Trial. Czech and Slovak Senile Dementia of Alzheimer Type Study Group. J. Psychiatry Neurosci. 1999, 24, 234–243. [Google Scholar]
- Birks, J.; Flicker, L. Selegiline for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2003, CD000442. [Google Scholar] [CrossRef]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the Nervous System: Current Knowledge of the Biochemical Modes of Action and Synergies of Thiamine, Pyridoxine, and Cobalamin. CNS Neurosci. 2019, 26, 5–13. [Google Scholar] [CrossRef]
- Kim, H.; Kim, G.; Jang, W.; Kim, S.Y.; Chang, N. Association between Intake of B Vitamins and Cognitive Function in Elderly Koreans with Cognitive Impairment. Nutr. J. 2014, 13, 118. [Google Scholar] [CrossRef]
- Morris, M.S. Homocysteine and Alzheimer’s Disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef]
- Yazdi, D.S.; Bar-Yosef, D.L.; Adsi, H.; Kreiser, T.; Sigal, S.; Bera, S.; Zaguri, D.; Shaham-Niv, S.; Oluwatoba, D.S.; Levy, D.; et al. Homocysteine Fibrillar Assemblies Display Cross-Talk with Alzheimer’s Disease β-Amyloid Polypeptide. Proc. Natl. Acad. Sci. USA 2021, 118, e2017575118C. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and Dementia: An International Consensus Statement1. J. Alzheimers Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.F.; Wolf, P.A. Plasma Homocysteine as a Risk Factor for Dementia and Alzheimer’s Disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef]
- Strain, J.J.; Dowey, L.; Ward, M.; Pentieva, K.; McNulty, H. B-Vitamins, Homocysteine Metabolism and CVD. Proc. Nutr. Soc. 2004, 63, 597–603. [Google Scholar] [CrossRef]
- Remington, R.; Chan, A.; Paskavitz, J.; Shea, T.B. Efficacy of a Vitamin/Nutriceutical Formulation for Moderate-Stage to Later-Stage Alzheimer’s Disease: A Placebo-Controlled Pilot Study. Am. J. Alzheimers Dis. Other Demen. 2009, 24, 27–33. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms, and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharm. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Prasad, K.N.; Hovland, A.R.; Cole, W.C.; Prasad, K.C.; Nahreini, P.; Edwards-Prasad, J.; Andreatta, C.P. Multiple Antioxidants in the Prevention and Treatment of Alzheimer Disease: Analysis of Biologic Rationale. Clin. Neuropharmacol. 2000, 23, 2–13. [Google Scholar] [CrossRef]
- Salehi, B.; Martorell, M.; Arbiser, J.L.; Sureda, A.; Martins, N.; Maurya, P.K.; Sharifi-Rad, M.; Kumar, P.; Sharifi-Rad, J. Antioxidants: Positive or Negative Actors? Biomolecules 2018, 8, 124. [Google Scholar] [CrossRef]
- Figueira, I.; Garcia, G.; Pimpão, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols Journey through Blood-Brain Barrier towards Neuronal Protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Andrus, P.K.; Smith, S.L.; Fleck, T.J.; Scherch, H.M.; Lutzke, B.S.; Sawada, G.A.; Althaus, J.S.; Vonvoigtlander, P.F.; Padbury, G.E.; et al. Pyrrolopyrimidines: Novel Brain-Penetrating Antioxidants with Neuroprotective Activity in Brain Injury and Ischemia Models. J. Pharm. Exp. 1997, 281, 895–904. [Google Scholar]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C Crosses the Blood-Brain Barrier in the Oxidized Form through the Glucose Transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Johanson, C.E. Vitamin Transport and Homeostasis in Mammalian Brain: Focus on Vitamins B and E. J. Neurochem. 2007, 103, 425–438. [Google Scholar] [CrossRef]
- Figueira, I.; Tavares, L.; Jardim, C.; Costa, I.; Terrasso, A.P.; Almeida, A.F.; Govers, C.; Mes, J.J.; Gardner, R.; Becker, J.D.; et al. Blood–Brain Barrier Transport and Neuroprotective Potential of Blackberry-Digested Polyphenols: An in Vitro Study. Eur. J. Nutr. 2019, 58, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Milbury, P.E.; Kalt, W. Xenobiotic Metabolism and Berry Flavonoid Transport across the Blood-Brain Barrier. J. Agric. Food Chem. 2010, 58, 3950–3956. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Negut, I.; Grumezescu, V.; Grumezescu, A.M.; Teleanu, R.I. Nanomaterials for Drug Delivery to the Central Nervous System. Nanomaterials 2019, 9, 371. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, N.L.; Manickam, D.S.; Brynskikh, A.M.; Uglanova, S.V.; Li, S.; Higginbotham, S.M.; Bronich, T.K.; Batrakova, E.V.; Kabanov, A.V. Cross-Linked Antioxidant Nanozymes for Improved Delivery to CNS. Nanomedicine 2012, 8, 119–129. [Google Scholar] [CrossRef]
- Khalil, I.; Yehye, W.A.; Etxeberria, A.E.; Alhadi, A.A.; Dezfooli, S.M.; Julkapli, N.B.M.; Basirun, W.J.; Seyfoddin, A. Nanoantioxidants: Recent Trends in Antioxidant Delivery Applications. Antioxidants 2019, 9, 24. [Google Scholar] [CrossRef]




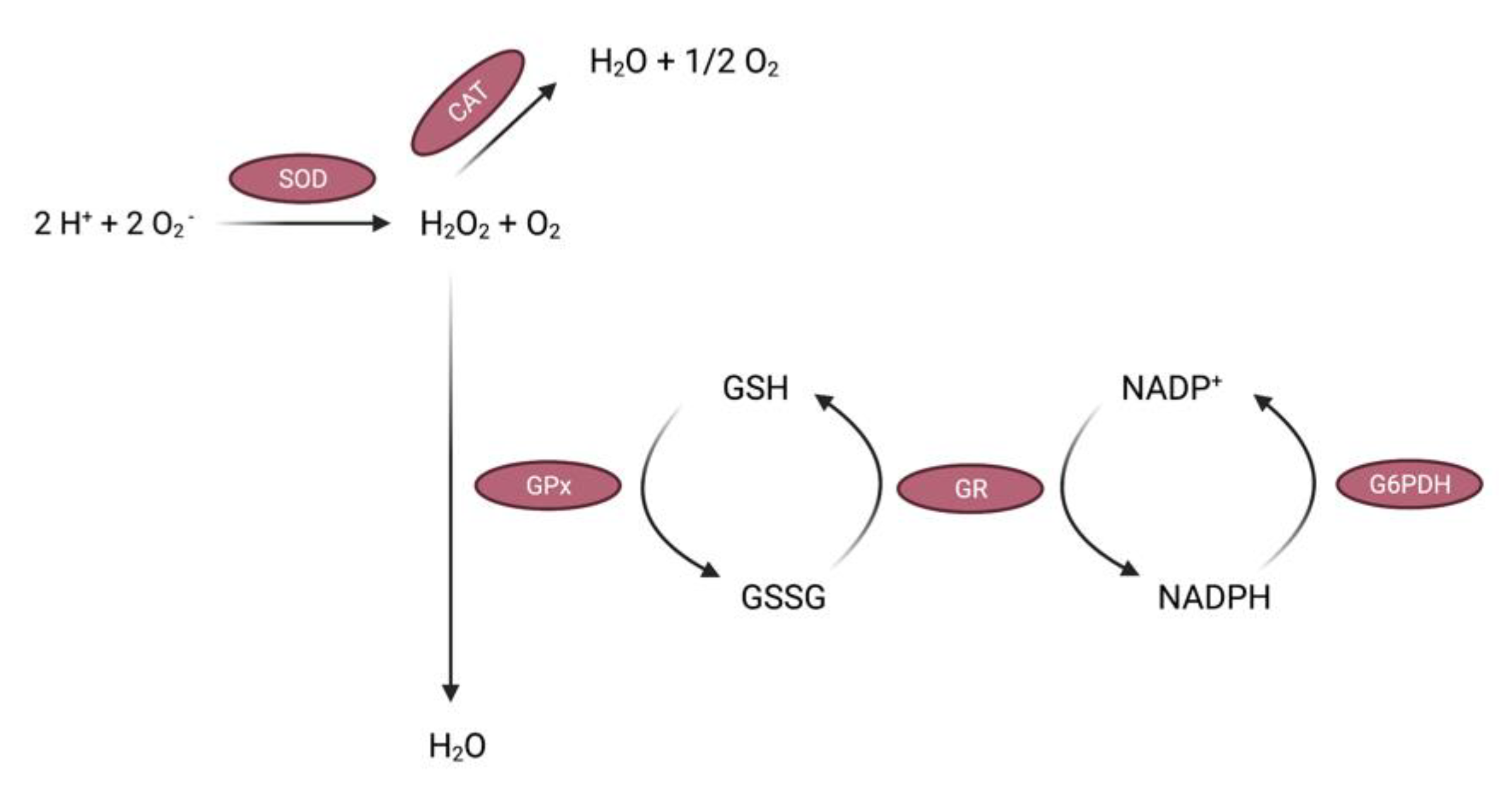{kind=link}
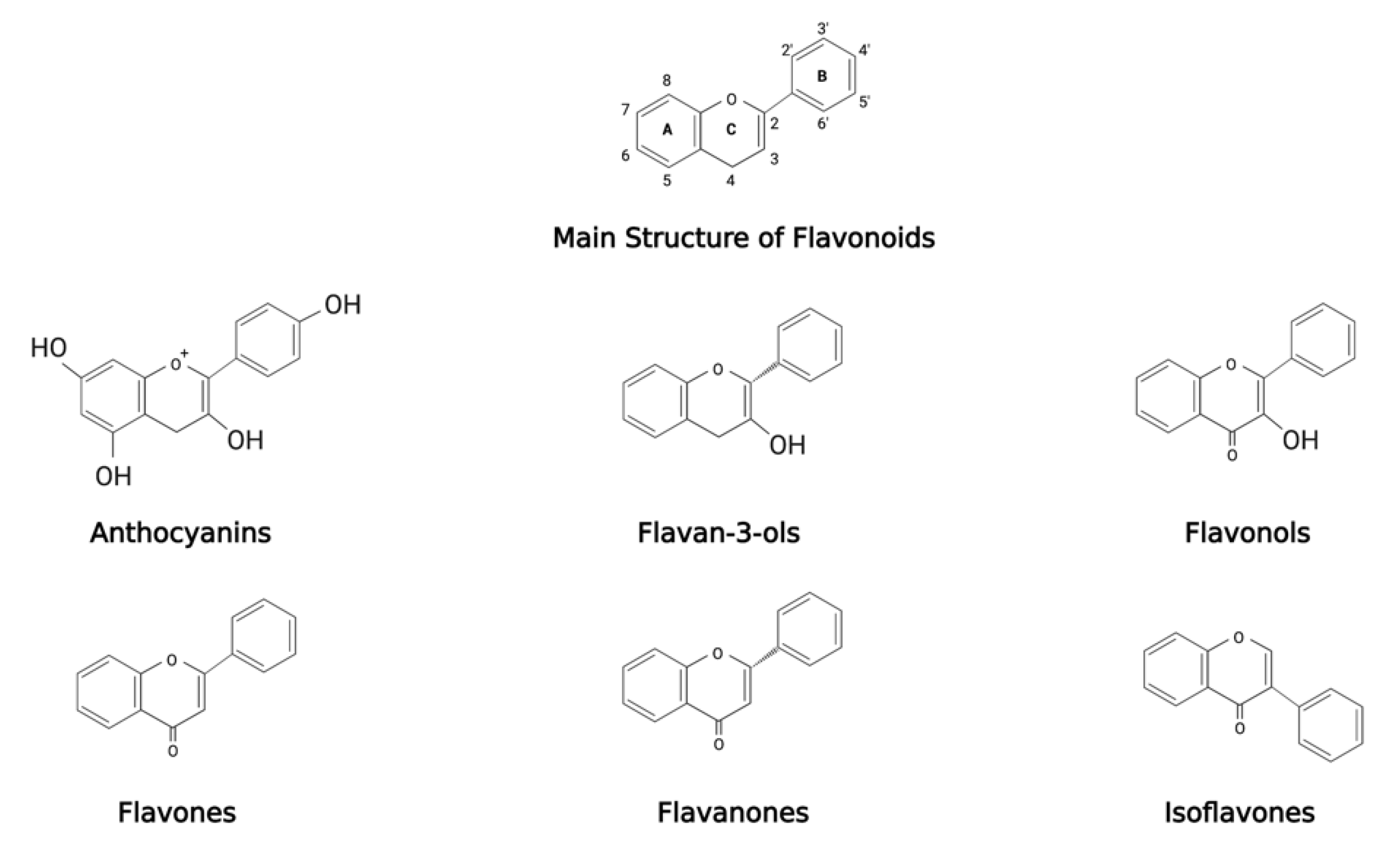{kind=link}
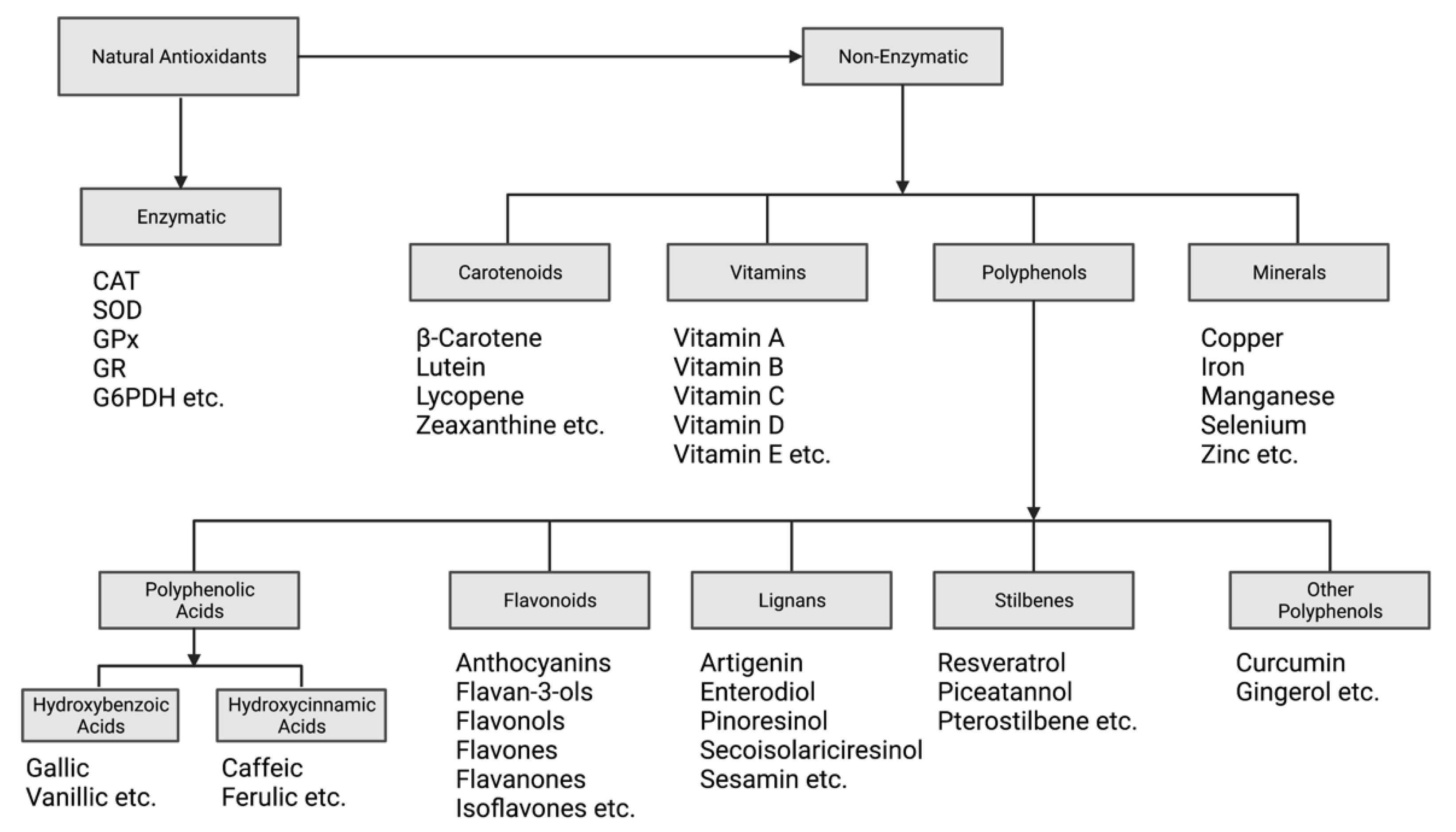{kind=link}
| Disease or Condition |
|---|
| Alzheimer’s disease [4,5] |
| Amyotrophic lateral sclerosis [6,7] |
| Corticobasal degeneration [8] |
| Creutzfeldt-Jakob disease (Prion disease) [9] |
| Down syndrome [10] |
| Diabetic neuropathy [11,12] |
| Friedreich’s ataxia [13] |
| Huntington’s disease [14,15,16] |
| Lewy body disease [17] |
| Multiple sclerosis [18,19] |
| Neiman-Pick C disease [20,21] |
| Neuromyelitis optica [22] |
| Parkinson’s disease [23,24,25] |
| Progressive supranuclear palsy [26] |
| Spinocerebellar ataxia [27,28,29] |
| Stroke [30,31,32] |
| Traumatic brain injury [33,34,35] |
| Classification | Compound(s) | Participants | Intervention | Primary Outcome Measures | Main Results | In-Text Reference |
|---|---|---|---|---|---|---|
| Vitamins | Vitamin E + Selegiline | 341 patients with moderate AD | 2000 IU vitamin E, 10 mg selegiline, both or placebo daily for 2 years | Time until occurrence of death, institutionalization, loss of ability to perform activities of daily living, or severe dementia | Treatment with vitamin E or selegiline slowed the progression of disease in patients with moderately severe impairment from AD | [415] |
| Vitamin E + Donepezil | 790 patients with mild cognitive impairment (MCI) | 2000 IU vitamin E, 10 mg donepezil or placebo, daily for 3 years | Clinically possible or probable AD | Vitamin E had no benefit. Donepezil was associated with a lower rate of progression in first 12 months | [416] | |
| Vitamin E + Memantine | 613 patients with mild to moderate AD | 2000 IU vita-min E, 20 mg memantine, both or placebo daily for 5 years | ADCS-ADL | 2000 IU/day of vitamin E compared to placebo slowed functional decline. No difference between groups receiving memantine alone or memantine + vitamin E | [417] | |
| Vitamin E + Vitamin C + Alpha-Lipoic Acid | 75 patients with mild to moderate AD | 800 IU vitamin E + 500 mg vitamin C + 900 mg alpha-lipoic acid, 400 mg coenzyme Q10 3 times/day or placebo daily for 16 weeks | Changes in cerebral spinal fluid (CSF) biomarkers related to AD and oxidative stress, cognition and function | Antioxidants did not influence CSF biomarkers related to amyloid or tau pathology | [418] | |
| B Vitamins | 340 patients with mild to moderate AD | 5 mg folate + 25 mg vitamin B6 + 1 mg vitamin B12 or placebo daily for 18 months | Changes in the cognitive subscale of the ADAS-Cog | Regimen of high-dose B vitamin supplements does not slow cognitive decline in individuals with mild to moderate AD | [419] | |
| Vitamin D + Memantine | 90 patients with moderate AD | 100,000 IU vitamin D3 (every 4 weeks) + 20 mg memantine or placebo daily for 24 weeks | Change of cognitive performance | Ongoing | [420] | |
| Multivitamin | 135 patients with AD or MCI | Nutraceutical formulation (NF) of: 400 ug folic acid, 6 ug vitamin B12, 30 IU vitamin E, 400 mg S-adenosylmethionine, 600 mg N-acetyl cysteine, 500 mg acetyl-l-carnitine daily for 1 year | Cognitive improvement or maintenance of cognitive performance | NF maintained or improved cognitive performance and mood/behaviour | [421] | |
| Polyphenols | Resveratrol | 39 patients with mild to moderate AD | 5 mg resveratrol + 5 mg dextrose + 5 mg malate or placebo twice daily for 1 year | Evaluate the safety, tolerability and efficacy of resveratrol, glucose and malate in slowing the progression of AD | Low-dose resveratrol is safe and well-tolerated | [422] |
| Resveratrol | 119 patients with mild to moderate AD | Up to 1 mg resveratrol twice daily or placebo for 52 weeks | Safety and tolerability of treatment with resveratrol and change in ADL | Resveratrol decreases CSF biomarkers, modulates neuro-inflammation and induces adaptive immunity | [423] | |
| Curcumin | 36 patients with mild to moderate AD | 2 g or 4 g curcumin or placebo daily for 24 weeks | Examine safety and tolerability of curcumin and determine its side effects on patients | Curcumin well-tolerated. Unable to demonstrate clinical or biochemical evidence of efficacy of curcumin C3 complex. Data suggest limited bioavailability | [424] | |
| Curcumin | 36 patients with dementia, presumed AD | 1 g curcumin + 120 mg ginkgo leaf extract, 4 g curcumin + ginkgo leaf extract or placebo daily for 6 months | Change in isoprostane levels in plasma and change in beta-amyloid levels in serum | Serum beta-amyloid rose on curcumin. Fewer adverse events reported | [425] | |
| Quercetin | 48 patients with MCI or early AD | 1000 mg quercetin or 100 mg dasatinib or placebo daily for 2 days | Serious adverse events and adverse events, and change in cellular senescence blood markers | Ongoing | [426] | |
| Quercetin | Recruiting patients with early AD | Quercetin + dasatinib for 2 days on, 14 days off for 12 weeks (6 cycles) | Brain penetrance of dasatinib and quercetin | Ongoing | [427] | |
| EGCG | 21 patients with early AD | 200 mg, 400 mg, 600 mg and 800 mg EGCG tri-monthly or placebo for 18 months | ADAS-Cog | Ongoing | [428] | |
| EGCG | 200 patients with AD carrying ApoE4 allele | 260–520 mg EGCG + personalized intervention or placebo + non personalized intervention or 260–520 mg EGCG + non personalized intervention or placebo to personalized intervention, daily for 15 months | Evaluate the efficacy of multimodal intervention (dietary, physical and cognition) combined with EGCG in slowing down cognitive decline | Ongoing | [429] | |
| Genistein | 27 patients with mild AD | 60 mg genistein or placebo daily for 360 days | Changes in amyloid beta concentration of CSF | Ongoing | [430] | |
| Genistein + Daidzein | 72 patients with AD | 100 mg of soy isoflavones or placebo daily for 6 months | Cognitive outcomes: language execution function, verbal memory and recall, attention, visual memory and planning | Six months of 100 mg/day isoflavones did not benefit cognition in older men and women with AD | [431] | |
| Alpha-Lipoic Acid + Omega-3 Fatty Acids | 39 patients with mild AD | 600 mg alpha-lipoic acid + 3 g fish oil, 3 g fish oil alone or placebo daily for 12 months | Peripheral F2-isoprostane levels (oxidative stress measure) | Combination of alpha lipoic acid with omega-3 fatty acids slowed cognitive and functional decline | [432] | |
| Minerals | Copper | 68 patients with mild to moderate AD | 8 mg copper or placebo daily for 1 year | Change in cognitive function, measured by ADAS-Cog | Results not yet published | [433] |
| Selenium + Vitamin E | 7540 participants with dementia | 200 μg Selenium + 400 IU Vitamin E, 200 μg selenium + placebo or 400 IU vitamin E + placebo or placebo + placebo daily for 7–12 years | Incidence of dementia (including AD) | Neither supplement prevented dementia | [434] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, A.E.; Saleh, T.M.; Kalisch, B.E. Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease. Antioxidants 2022, 11, 213. https://doi.org/10.3390/antiox11020213
Collins AE, Saleh TM, Kalisch BE. Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease. Antioxidants. 2022; 11(2):213. https://doi.org/10.3390/antiox11020213
Chicago/Turabian StyleCollins, Andrila E., Tarek M. Saleh, and Bettina E. Kalisch. 2022. "Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease" Antioxidants 11, no. 2: 213. https://doi.org/10.3390/antiox11020213
APA StyleCollins, A. E., Saleh, T. M., & Kalisch, B. E. (2022). Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease. Antioxidants, 11(2), 213. https://doi.org/10.3390/antiox11020213