
Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions

 ,
,  ,
,  ,
, 
 and
and 
Abstract
1. Introduction
The Endocannabinoid System
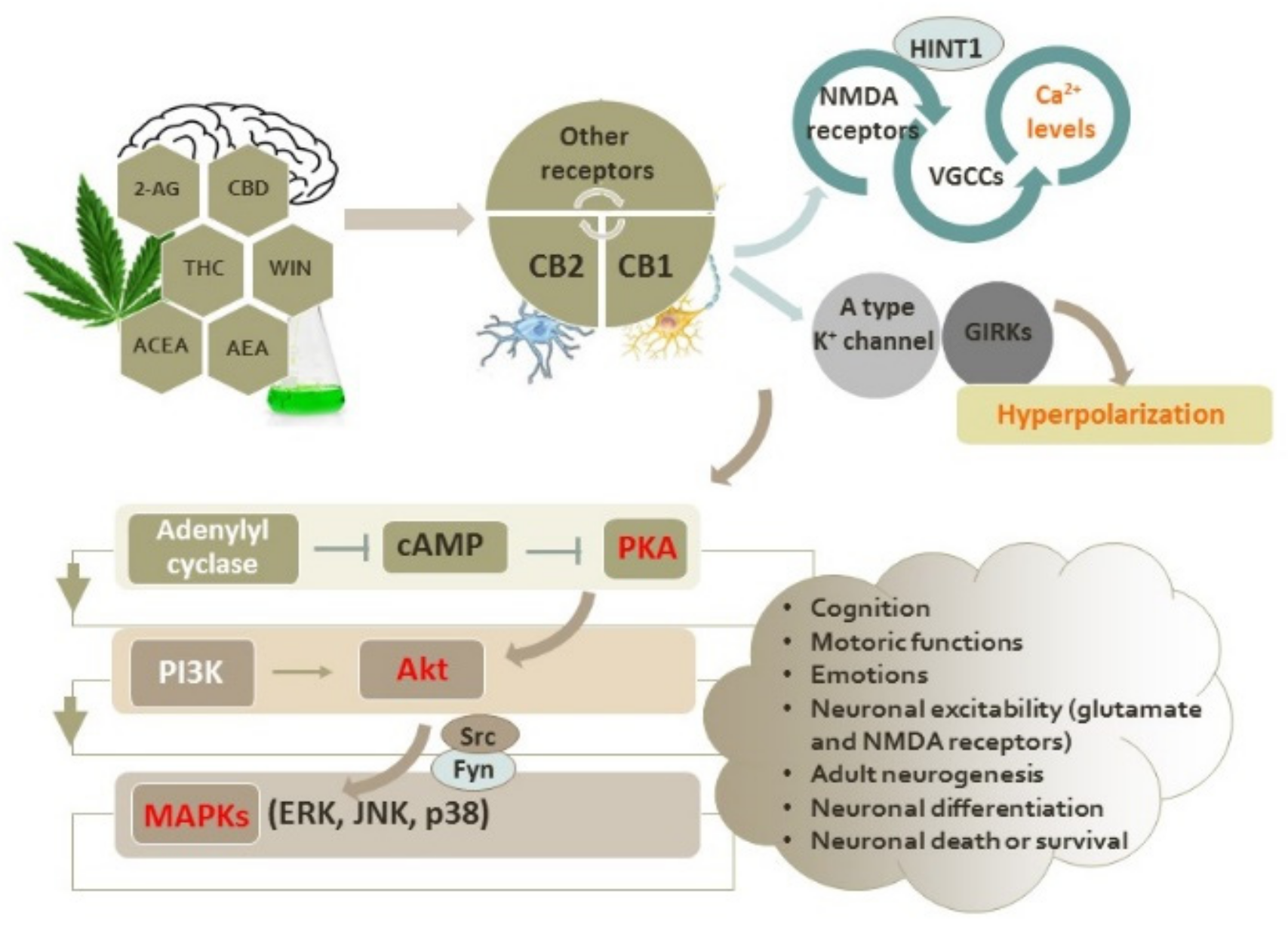
2. Intracellular Signaling Pathways Induced by Activation of CB1 and CB2 Receptors
2.1. Interactions of Cannabinoid Receptors with Other GPCRs
2.2. Functional Interplay between the Cannabinoid CB1 and CB2 Receptors and Glutamate NMDA Receptors
3. Endocannabinoid System in Neurodegenerative Diseases

{kind=link}
{kind=link}
| Disease | ECS | Observed Change | Model | Reference |
|---|---|---|---|---|
| HD | Receptors | ↓ CB1R | R6/2 mouse | [83] |
| ↓ CB1R | pre-HD mutation carriers and symptomatic HD patients | [124] | ||
| ↓ CB1R | basal ganglia of patients | [126] | ||
| ↓ CB1R | grey matter of patients | [127] | ||
| ↑ CB2R in astrocytes and reactive microglia | malonate-induced rat model | [54] | ||
| AD | EC | ↓ anandamide and its precursor | midfrontal and temporal cortices of patients | [133] |
| ↑ 2-AG | plasma of patients | [137] | ||
| ↑ 2-AG | hippocampi of rat model | [138] | ||
| Receptors | ↓ CB1R | brains of AD patients | [105] | |
| alterations of CB1R expression | mouse model of AD | [130] | ||
| unchanged levels of CB1R | hippocampi of patients | [131] | ||
| ↓ CB1R | post mortem cortical brain | [134] | ||
| CB1R deficiency | rat model of AD | [135] | ||
| EC enzymes | ↑ sn-1-DAGL α and β isoforms, no expression of ABHD6, ↓ MAGL | hippocampi of patients | [131] | |
| ↓ FAAH | frontal cortices of patients | [132] | ||
| PD | EC | ↑ anandamide | cerebrospinal fluid of patients | [143] |
| Receptors | ↑ CB1R | basal ganglia of patients | [142] | |
| ALS | EC | ↑ anandamide and 2-AG | spinal cords of SOD1 G93A mice | [146] |
| Receptors | ↑ CB2R | human spinal cord | [147] |
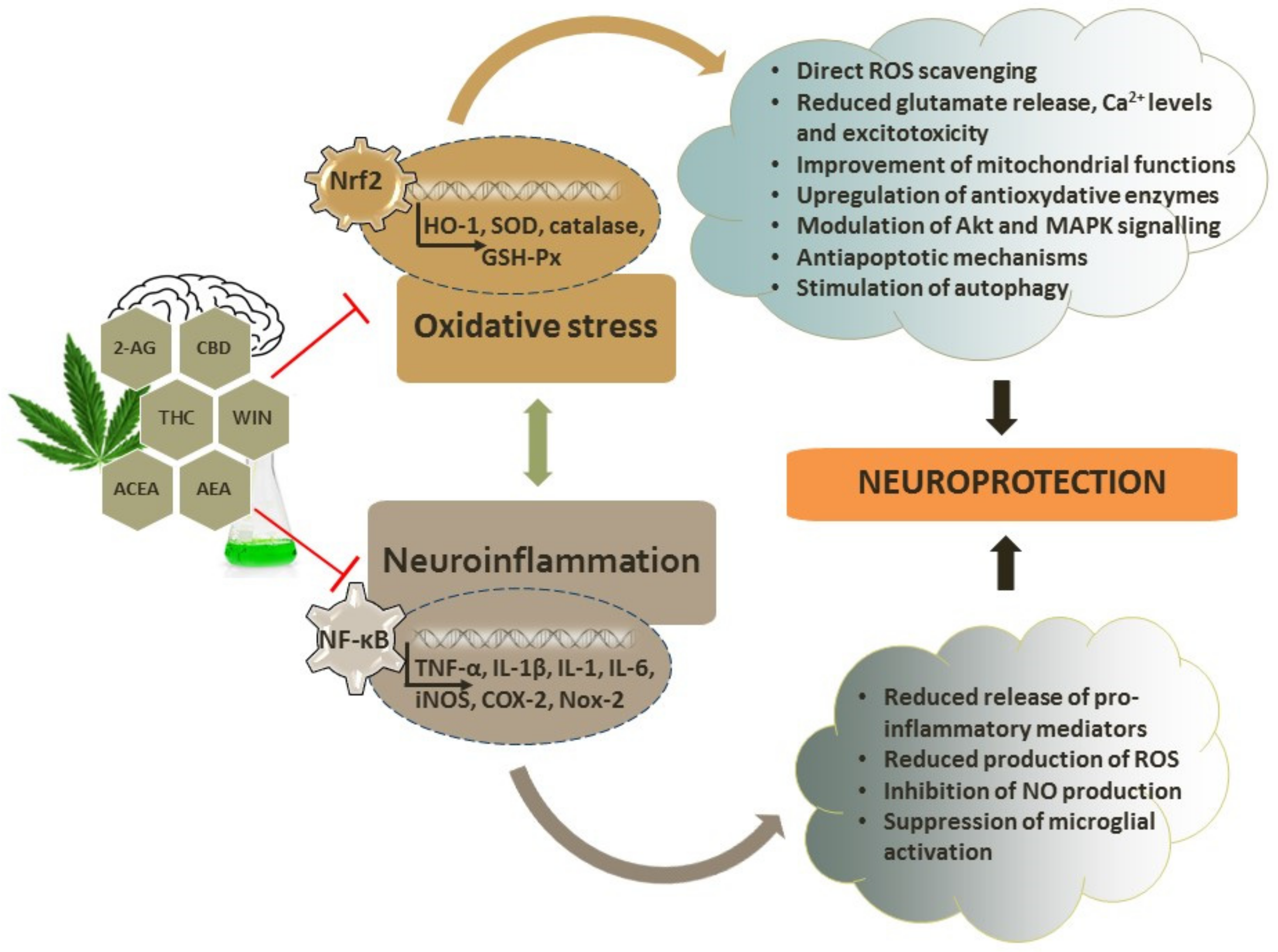
4. Antioxidant Capacity of Cannabinoids
4.1. Antioxidative and Neuroprotective Effects of Endocannabinoids
| Compound | Signaling | Effects | Model | Ref. |
|---|---|---|---|---|
| AEA | ↑ CB1R, ↓ cleaved caspase-3, ROS and Nox2, restored SOD, partially replenished GSH content and GSH/GSSG ratio | protection from H2O2-induced injury | hippocampal HT22 cells | [183] |
| AEA | Via CB1R | protection from AMPA/kainate receptor-induced lesions | perinatal rodent brain | [184] |
| AEA and 2-AG | - | failed to attenuate kainate-mediated excitotoxicity | neonatal rat spinal cord in vitro | [187] |
| OEA | PPARα agonist; ↑ IκBα, and ↓ COX-2 | neuroprotection, reduced infarct volume | cerebral ischemia mouse model | [190] |
| 2-AG | ↓ TNF-α, IL-1β and IL-6 mRNA; ↑ endogenous antioxidants | decreased BBB permeability | closed head injury mouse model | [120] |
| 2-AG | Via CB1R | reduction in brain edema, better clinical recovery, reduced infarct volume | closed head injury mouse model | [191,192] |
| MAGL inhibitor (↑ 2-AG) | ↓ inflammatory markers TNF-α, IL-1β and IL-6, ↓ Aβ formation and enzymes involved in Aβ synthesis, ↓ tau phosphorylation and aggregation of TDP-43, prevented changes in the expression of AMPA and NMDA receptor subunits | ↓ microglial and astrocytic activation, promoted recovery | repetitive mild closed head injury mouse model | [193] |
| MAGL inhibitor (↑ 2-AG) | ↓ BACE1 and accumulation of Aβ, ↓ inflammation | improved synaptic plasticity and cognitive abilities, reduced neurodegeneration | mouse model of AD | [139] |
| MAGL inhibitor (↑ 2-AG) | Via CB2R | neuroprotection from MPP+-induced toxicity | SH-SY5Y cells | [194] |
| FAAH inhibitor (↑ AEA) | ↑ Bcl-2, ↓ APP and iNOS and COX-2, via CB1R and CB2R, Akt and ERK1/2 pathways | improved motor and cognitive deficits, promoted polarization of M2 microglia, reduced neurodegeneration | traumatic brain injury mouse model | [195] |
| FAAH deletion (↑ AEA) | - | delayed signs of the disease | mouse model of ALS | [196] |
| FAAH inhibitors (↑ AEA) | Via CB1R and CB2R | prevented motor impairment, did not prevent dopamine loss, showed anti-cataleptic properties | rat model of PD | [197] |
4.2. Antioxidative and Neuroprotective Effects of Phytocannabinoids
| Compound | Signaling | Effects | Model | Ref |
|---|---|---|---|---|
| CBD | via CB1R | neurogenic effect | mouse model | [43] |
| THC | - | reduced learning without affecting adult neurogenesis | ||
| THC | greater effects in regions with higher density of CB1R | neuromodulatory effects in brain regions involved in cognitive tasks | human participants | [204] |
| CBD | ↓ LDH release | protection from tert-butyl hydroperoxide-induced toxicity | rat cortical neurons | [182] |
| THC and CBD | - | protection from glutamate toxicity via NMDA, AMPA, and kainate receptors | ||
| CBD | ↓ ROS | protection from H2O2-induced cell death | oligodendrocyte progenitor cells | |
| CBD | ↓ caspase-3 cleavage and apoptosis via mechanisms independent of CB1R, CB2R, TRPV1 or PPARγ receptors | protection from lipopolysaccharide/IFNγ-induced cytotoxicity | oligodendrocyte progenitor cells | [208] |
| ↓ phosphorylation of the eiF2α protein | protection from tunicamycin-induced ER stress | |||
| CBD | ↓ glutamate level and IL-6, ↓TNF-α, iNOS and COX-2 acting at CB2R and adenosine receptors | protection from hypoxic–ischemic damage | immature mice brain | [209] |
| THC | reduced number of AMPA receptors following CB1R activation | protection from TNF-α that promotes AMPA receptor-mediated excitotoxicity | rat hippocampal neurons | [112] |
| CBD | ↓ ROS, lipid peroxidation, intracellular calcium and induction of apoptotic cascade | neuroprotection from Aβ-mediated toxicity | PC12 neurons | [211] |
| CBD | ↓ Aβ-induced increase in pGSK-3β and tau hyperphosphorylation and reversed Aβ-induced decrease in β-catenin expression | neuroprotection from Aβ-mediated toxicity | PC12 neurons | [212] |
| CBD | ↓ nitrite production and iNOS through ↓ p38 pathway and NF-ҡB | - | Aβ-stimulated PC12 neurons | [213] |
| CBD | ↓ NO, IL-1β, TNFα, S100B, NF-ҡB pathway via PPARγ receptors | neuroprotection from Aβ and promotion of hippocampal neurogenesis | rat AD model | [214] |
| CBD | ↓ iNOS and IL-1β, NO release | prevention of microglial activation after Aβ inoculation | mouse model | [215] |
| CBD | ↓ IL-6 mRNA in cerebral cortex | improvement of cognitive deficits | Aβ-treated mice | [216] |
| THC | ↓ phosphorylated and total GSK-3β, ↓ Aβ levels, ↑ mitochondrial function | inhibition of Aβ aggregation | N2a/AβPPswe cells | [217] |
| THC | ↓ production of Aβ, ↓ formation of Aβ oligomers, ↓ GSK-3β, ↓ tau phosphorylation | improved spatial memory and cognitive decline | APP/PS1 mice | [218] |
| CBD and THC | ↑ dopamine, ↑ tyrosine hydroxylase mRNA | neuroprotection | 6-OHDA-induced mouse model of PD | [220] |
| CBD | ↑ SOD | neuroprotection when immediately administered after the lesion | 6-OHDA-induced mice model of PD | [221] |
| THCV | - | reduced loss of dopaminergic neurons, attenuated motor inhibition | 6-OHDA-induced mouse model of PD | [222] |
| CBD | ↓ 3NP-induced decrease in SOD1 and SOD2, restoration of GABA levels | neuroprotection against striatal damage | 3-NP-induced rat model of HD | [223] |
| THC | Activation of PI3K/Akt/mTORC1/BDNF pathway via CB1R | protection from NMDA-induced excitotoxicity | mouse striatal neuroblasts | [83] |
| THC-rich extracts | ↑ glucose utilization and activity of gluconeogenic enzymes; ↑ GSH, SOD and catalase activity, ↓ malondialdehyde and NO levels | improved glucose consumption | isolated rat brain | [224] |
| THC- and CBD-enriched extract | ↓ iNOS, not mediated by CB1 and CB2R | attenuated neurodegeneration and glial activation | malonate-induced rat model of HD | [225] |
| THC- and CBD-enriched extract | ↑ CB2R | small improvements in the progression of neurological deficits | G93A-SOD1 mice model of ALS | [226] |
| Cannabis | - | moderately effective against appetite loss, depression and spasticity | patients with ALS | [227] |
4.3. Antioxidative and Neuroprotective Effects of Synthetic Cannabinoids
| Compound | Signaling | Effects | Model | Ref. |
|---|---|---|---|---|
| WIN 55,212-2 | ↓ adenylyl cyclase and ↓ intracellular calcium via CB1R and cAMP/PKA pathway | protection from NMDA-induced neuronal death | hippocampal neurons | [113] |
| WIN 55,212-2 | interfering with AMPA receptor trafficking after CB1R activation | protection from TNF-α-induced excitotoxicity | hippocampal neurons | [112] |
| WIN 55,212-2 | ↓ ROS formation through CB1R and PKA inhibition | protection from FeCl2-induced neuronal death | cortical neuron cultures | [229] |
| WIN 55,212 | activation of CB1R, ↓ PKA activity and NO production | protection from NMDA-induced cell death | cultured cortical neurons and mice brains | [230] |
| WIN 55,212-2 | ↑ mitochondrial function, ↓ ROS and lipid peroxidation | protection from organic acidemias | rat brain synaptosomes | [231] |
| WIN 55,212-2 | ↓ NMDA-induced calcium influx via IP3 signaling | protection from NMDA-induced cell death | primary dorsal root ganglia neurons and F-11 neurons | [232] |
| WIN 55,212-2 | - | prevention of cognitive impairment and microglial activation | rats administered with Aβ | [105] |
| WIN 55,212-2, HU-210, JWH-133 | ↓ TNF-α release | ↓ microglial activation | microglial cell culture | |
| WIN 55,212-2 and JWH-133 | ↓ COX-2, TNF-α mRNA, ↑ Aβ clearance | JWH-133 normalized novel object recognition and ↓ microglial activation | transgenic APP mice | [234] |
| HU-210 | - | modulation of glial function, protection from 6-OHDA-mediated toxicity | mouse cerebellar granule cells | [220] |
| ACEA | - | no neuroprotection | 6-OHDA-induced lesions in mice | [221] |
| HU-308 | - | modest neuroprotection | ||
| WIN 55,212-2 | - | no neuroprotection | ||
| WIN 55,212-2 | modulation of L-DOPA-induced ERK1/2 activation | attenuation of L-DOPA-induced motor disabilities | 6-OHDA-induced rat model of PD | [235] |
| WIN 55,212-2 and HU210 | ↓ NADPH oxidase activation, IL-1β and TNF-α release, ROS production and oxidative damage of proteins and nucleic acids | improved survival of dopaminergic neurons | MPTP-induced model of PD | [236] |
| HU210 | ↑ PI3K/Akt/mTORC1/BDNF | protection from NMDA-induced excitotoxicity | mouse striatal neurons | [83] |
| WIN 55,212-2 | ↑ dopamine levels via CB2R | ↓ microglial activation, protection against loss of dopaminergic neurons | MPTP-induced toxicity in mice | [237] |
| ACEA | via CB1R (for LPS) or via TRPV1 channel (for tunicamycin) modulation of ERK1/2 pathway | protection from neuroinflammation and endoplasmic reticulum stress | Neuro-2a cells exposed to LPS or tunicamycin | [19] |
| ACEA | ↓ ROS and apoptotic rate | improved mitochondrial function | hippocampal neurons exposed to oxygen-glucose deprivation/reoxygenation | [100] |
| WIN 55,212-2 | ↓ apoptosis via CB1R, PI3K/Akt and ERK pathways | protection from ceramide-induced apoptosis in vitro and in vivo | astrocytes | [86] |
| WIN 55,212-2 | - | no appearance of the clinical signs of the disease | G93A-SOD1 mice model of ALS | [196] |
| AM-1241 | - | ↑ survival interval after disease onset | G93A-SOD1 mice model of ALS | [238] |
| HU-308 | ↓ TNF-α and gliosis via CB2R | neuroprotection from malonate-induced toxicity | malonate-induced rat model of HD | [54] |
| JWH133 | via CB2R | neuroprotection from MPP+-induced toxicity | SH-SY5Y cells | [194] |
| JWH133 | prevented suppression of Akt signaling, reversed Bcl-2/Bax ratio and prevented caspase-3 increase | protection from Aβ-induced injury | hippocampal neurons | [239] |
| NP137 | ↓ BACE1 activity | neuroprotection from Aβ | Aβ-treated primary cortical neurons | [241] |
| - | improvement of spatial navigation | TgAPP mice | ||
| NITyr | via CB1R, ↓ ROS generation, and ↑ autophagy-related proteins and autophagy | attenuated H2O2-induced neurotoxic effects | rat pheochromocytoma PC12 cells | [185] |
| NITyr | ↑ BDNF and autophagy by CB2/AMPK/mTOR/ULK1 | attenuated Aβ-induced toxicity | primary cortical neurons | [186] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman, D.; Sirven, J.I. Historical perspective on the medical use of cannabis for epilepsy: Ancient times to the 1980s. Epilepsy Behav. 2017, 70 Pt B, 298–301. [Google Scholar] [CrossRef]
- Landucci, E.; Mazzantini, C.; Lana, D.; Davolio, P.L.; Giovannini, M.G.; Pellegrini-Giampietro, D.E. Neuroprotective Effects of Cannabidiol but Not Δ9-Tetrahydrocannabinol in Rat Hippocampal Slices Exposed to Oxygen-Glucose Deprivation: Studies with Cannabis Extracts and Selected Cannabinoids. Int. J. Mol. Sci. 2021, 22, 9773. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, C.; Francavilla, M.; Ongari, G.; Petese, A.; Ghezzi, C.; Rossini, N.; Blandini, F.; Cerri, S. Neuroprotective and Symptomatic Effects of Cannabidiol in an Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 8920. [Google Scholar] [CrossRef] [PubMed]
- Maroon, J.; Bost, J. Review of the neurological benefits of phytocannabinoids. Surg. Neurol. Int. 2018, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Brigida, A.L.; Schultz, S.; Cascone, M.; Antonucci, N.; Siniscalco, D. Endocannabinoid Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations. Int. J. Mol. Sci. 2017, 18, 1425. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Walder, K.; Kloiber, S.; Amminger, P.; Berk, M.; Bortolasci, C.C.; Maes, M.; Puri, B.K.; Carvalho, A.F. The endocannabinoidome in neuropsychiatry: Opportunities and potential risks. Pharmacol. Res. 2021, 170, 105729. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Paloczi, J.; Varga, Z.V.; Hasko, G.; Pacher, P. Neuroprotection in Oxidative Stress-Related Neurodegenerative Diseases: Role of Endocannabinoid System Modulation. Antioxid. Redox Signal. 2018, 29, 75–108. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: Implications in psychosis and schizophrenia. Front. Pharmacol. 2013, 4, 169. [Google Scholar] [CrossRef]
- Rios, C.; Gomes, I.; Devi, L.A. μ opioid and CB1 cannabinoid receptor interactions: Reciprocal inhibition of receptor signaling and neuritogenesis. Br. J. Pharmacol. 2006, 148, 387–395. [Google Scholar] [CrossRef]
- Sun, Y.; Alexander, S.P.; Garle, M.J.; Gibson, C.L.; Hewitt, K.; Murphy, S.P.; Kendall, D.A.; Bennett, A.J. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br. J. Pharmacol. 2007, 152, 734–743. [Google Scholar] [CrossRef]
- Palazzos, E.; de Novellis, V.; Marabese, I.; Rossi, F.; Maione, S. Metabotropic glutamate and cannabinoid receptor crosstalk in periaqueductal grey pain processing. Curr. Neuropharmacol. 2006, 4, 225–231. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Sylantyev, S.; Jensen, T.P.; Ross, R.A.; Rusakov, D.A. Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc. Natl. Acad. Sci. USA 2013, 110, 5193–5198. [Google Scholar] [CrossRef]
- Soderstrom, K.; Soliman, E.; Van Dross, R. Cannabinoids Modulate Neuronal Activity and Cancer by CB1 and CB2 Receptor-Independent Mechanisms. Front. Pharmacol. 2017, 8, 720. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid ligands targeting TRP channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef]
- Vrechi, T.A.; Crunfli, F.; Costa, A.P.; Torrão, A.S. Cannabinoid Receptor Type 1 Agonist ACEA Protects Neurons from Death and Attenuates Endoplasmic Reticulum Stress-Related Apoptotic Pathway Signaling. Neurotox. Res. 2018, 33, 846–855. [Google Scholar] [CrossRef]
- Im, D.-S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. Int. J. Mol. Sci. 2021, 22, 1034. [Google Scholar] [CrossRef]
- Felder, C.C.; Briley, E.M.; Axelrod, J.; Simpson, J.T.; Mackie, K.; Devane, W.A. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. USA 1993, 90, 7656–7660. [Google Scholar] [CrossRef] [PubMed]
- Gonsiorek, W.; Lunn, C.; Fan, X.; Narula, S.; Lundell, D.; Hipkin, R.W. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: Antagonism by anandamide. Mol. Pharmacol. 2000, 57, 1045–1050. [Google Scholar] [PubMed]
- Hampson, A.J.; Hill, W.A.; Zan-Phillips, M.; Makriyannis, A.; Leung, E.; Eglen, R.M.; Bornheim, L.M. Anandamide hydroxylation by brain lipoxygenase: Metabolite structures and potencies at the cannabinoid receptor. Biochim. Biophys. Acta 1995, 1259, 173–179. [Google Scholar] [CrossRef]
- Turcotte, C.; Chouinard, F.; Lefebvre, J.S.; Flamand, N. Regulation of inflammation by cannabinoids, the endocannabinoids 2-arachidonoyl-glycerol and arachidonoyl-ethanolamide, and their metabolites. J. Leukoc. Biol. 2015, 97, 1049–1070. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Sridar, C.; Snider, N.T.; Hollenberg, P.F. Anandamide oxidation by wild-type and polymorphically expressed CYP2B6 and CYP2D6. Drug Metab. Dispos. 2011, 39, 782–788. [Google Scholar] [CrossRef]
- Snider, N.T.; Nast, J.A.; Tesmer, L.A.; Hollenberg, P.F. A cytochrome P450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol. Pharmacol. 2009, 75, 965–972. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef]
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutiérrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef]
- Clement, A.B.; Hawkins, E.G.; Lichtman, A.H.; Cravatt, B.F. Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J. Neurosci. 2003, 23, 3916–3923. [Google Scholar] [CrossRef]
- Valdeolivas, S.; Pazos, M.R.; Bisogno, T.; Piscitelli, F.; Iannotti, F.A.; Allarà, M.; Sagredo, O.; Di Marzo, V.; Fernández-Ruiz, J. The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: A potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis. 2013, 4, e862. [Google Scholar] [CrossRef]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Lombardi, M.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on the surface of microglial microvesicles. SpringerPlus 2015, 4 (Suppl. 1), L29. [Google Scholar] [CrossRef][Green Version]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef]
- Oddi, S.; Fezza, F.; Pasquariello, N.; De Simone, C.; Rapino, C.; Dainese, E.; Finazzi-Agrò, A.; Maccarrone, M. Evidence for the intracellular accumulation of anandamide in adiposomes. Cell. Mol. Life Sci. 2008, 65, 840–850. [Google Scholar] [CrossRef]
- Maccarrone, M. Metabolism of the Endocannabinoid Anandamide: Open Questions after 25 Years. Front. Mol. Neurosci. 2017, 10, 166. [Google Scholar] [CrossRef]
- Shen, M.; Piser, T.M.; Seybold, V.S.; Thayer, S.A. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J. Neurosci. 1996, 16, 4322–4334. [Google Scholar] [CrossRef]
- Szabo, B.; Schlicker, E. Effects of cannabinoids on neurotransmission. Handb. Exp. Pharmacol. 2005, 168, 327–365. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Vicente-Sánchez, A.; Garzón, J. Cannabinoid receptors couple to NMDA receptors to reduce the production of NO and the mobilization of zinc induced by glutamate. Antioxid. Redox Signal. 2013, 19, 1766–1782. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Carriba, P.; Ortiz, O.; Patkar, K.; Justinova, Z.; Stroik, J.; Themann, A.; Müller, C.; Woods, A.S.; Hope, B.T.; Ciruela, F.; et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology 2007, 32, 2249–2259. [Google Scholar] [CrossRef]
- Carlson, G.; Wang, Y.; Alger, B.E. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat. Neurosci. 2002, 5, 723–724. [Google Scholar] [CrossRef]
- Jin, K.; Xie, L.; Kim, S.H.; Parmentier-Batteur, S.; Sun, Y.; Mao, X.O.; Childs, J.; Greenberg, D.A. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol. Pharmacol. 2004, 66, 204–208. [Google Scholar] [CrossRef]
- Wolf, S.A.; Bick-Sander, A.; Fabel, K.; Leal-Galicia, P.; Tauber, S.; Ramirez-Rodriguez, G.; Müller, A.; Melnik, A.; Waltinger, T.P.; Ullrich, O.; et al. Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun. Signal. 2010, 8, 12. [Google Scholar] [CrossRef]
- Rueda, D.; Navarro, B.; Martinez-Serrano, A.; Guzman, M.; Galve-Roperh, I. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J. Biol. Chem. 2002, 277, 46645–46650. [Google Scholar] [CrossRef]
- Twitchell, W.; Brown, S.; Mackie, K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997, 78, 43–50. [Google Scholar] [CrossRef]
- Liu, J.; Gao, B.; Mirshahi, F.; Sanyal, A.J.; Khanolkar, A.D.; Makriyannis, A.; Kunos, G. Functional CB1 cannabinoid receptors in human vascular endothelial cells. Biochem. J. 2000, 346 Pt 3, 835–840. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef]
- Freundt-Revilla, J.; Kegler, K.; Baumgärtner, W.; Tipold, A. Spatial distribution of cannabinoid receptor type 1 (CB1) in normal canine central and peripheral nervous system. PLoS ONE 2017, 12, e0181064. [Google Scholar] [CrossRef]
- Croci, T.; Manara, L.; Aureggi, G.; Guagnini, F.; Rinaldi-Carmona, M.; Maffrand, J.-P.; Fur, G.; Mukenge, S.; Ferla, G. In Vitro functional evidence of neuronal cannabinoid CB 1 receptors in human ileum. Br. J. Pharmacol. 1998, 125, 1393–1395. [Google Scholar] [CrossRef]
- Kulkarni-Narla, A.; Brown, D.R. Localization of CB1-cannabinoid receptor immunoreactivity in the porcine enteric nervous system. Cell Tissue Res. 2000, 302, 73–80. [Google Scholar] [CrossRef]
- Camilleri, M. Cannabinoids and gastrointestinal motility: Pharmacology, clinical effects, and potential therapeutics in humans. Neurogastroenterol. Motil. 2018, 30, e13370. [Google Scholar] [CrossRef] [PubMed]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Komorowska-Müller, J.A.; Schmöle, A.-C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2021, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; González, S.; Aroyo, I.; Pazos, M.R.; Benito, C.; Lastres-Becker, I.; Romero, J.P.; Tolón, R.M.; Mechoulam, R.; Brouillet, E.; et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: Relevance for Huntington’s disease. Glia 2009, 57, 1154–1167. [Google Scholar] [CrossRef]
- Rivas-Santisteban, R.; Lillo, A.; Lillo, J.; Rebassa, J.B.; Contestí, J.S.; Saura, C.A.; Franco, R.; Navarro, G. N-Methyl-D-aspartate (NMDA) and cannabinoid CB2 receptors form functional complexes in cells of the central nervous system: Insights into the therapeutic potential of neuronal and microglial NMDA receptors. Alzheimers Res. Ther. 2021, 13, 184. [Google Scholar] [CrossRef]
- Rhee, M.H.; Bayewitch, M.; Avidor-Reiss, T.; Levy, R.; Vogel, Z. Cannabinoid receptor activation differentially regulates the various adenylyl cyclase isozymes. J. Neurochem. 1998, 71, 1525–1534. [Google Scholar] [CrossRef]
- Zhou, J.; Noori, H.; Burkovskiy, I.; Lafreniere, J.D.; Kelly, M.E.M.; Lehmann, C. Modulation of the Endocannabinoid System Following Central Nervous System Injury. Int. J. Mol. Sci. 2019, 20, 388. [Google Scholar] [CrossRef]
- Kibret, B.G.; Ishiguro, H.; Horiuchi, Y.; Onaivi, E.S. New Insights and Potential Therapeutic Targeting of CB2 Cannabinoid Receptors in CNS Disorders. Int. J. Mol. Sci. 2022, 23, 975. [Google Scholar] [CrossRef]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target. Front. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Zhang, J.; Hoffert, C.; Vu, H.K.; Groblewski, T.; Ahmad, S.; O’Donnell, D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur. J. Neurosci. 2003, 17, 2750–2754. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2005, 2, 29. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef]
- Nogueras-Ortiz, C.; Yudowski, G.A. The multiple waves of cannabinoid 1 receptor signaling. Mol. Pharmacol. 2016, 90, 620–626. [Google Scholar] [CrossRef]
- Howlett, A.; Qualy, J.M.; Khachatrian, L.L. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol. Pharmacol. 1986, 29, 307–313. [Google Scholar]
- Childers, S.R.; Deadwyler, S.A. Role of cyclic AMP in the actions of cannabinoid receptors. Biochem. Pharmacol. 1996, 52, 819–827. [Google Scholar] [CrossRef]
- Hampson, A.J.; Grimaldi, M. Cannabinoid receptor activation and elevated cyclic AMP reduce glutamate neurotoxicity. Eur. J. Neurosci. 2001, 13, 1529–1536. [Google Scholar] [CrossRef]
- Huang, C.C.; Chen, Y.L.; Lo, S.W.; Hsu, K.S. Activation of cAMP-dependent protein kinase suppresses the presynaptic cannabinoid inhibition of glutamatergic transmission at corticostriatal synapses. Mol. Pharmacol. 2002, 61, 578–585. [Google Scholar] [CrossRef]
- Gyombolai, P.; Boros, E.; Hunyady, L.; Turu, G. Differential β-arrestin 2 requirements for constitutive and agonist-induced internalization of the CB1 cannabinoid receptor. Mol. Cell. Endocrinol. 2013, 372, 116–127. [Google Scholar] [CrossRef]
- Ahn, K.H.; Mahmoud, M.M.; Shim, J.-Y.; Kendall, D.A. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J. Biol. Chem. 2013, 288, 9790–9800. [Google Scholar] [CrossRef]
- Kang, K.A.; Wang, Z.H.; Zhang, R.; Piao, M.J.; Kim, K.C.; Kang, S.S.; Kim, Y.W.; Lee, J.; Park, D.; Hyun, J.W. Myricetin protects cells against oxidative stress-induced apoptosis via regulation of PI3K/Akt and MAPK signaling pathways. Int. J. Mol. Sci. 2010, 11, 4348–4360. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Radovanović, V.; Vlainić, J.; Vuković, L.; Hanžić, N. Neuroprotective effect of zolpidem against glutamate-induced toxicity is mediated via the PI3K/Akt pathway and inhibited by PK11195. Toxicology 2018, 406, 58–69. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Vlainić, J.; Čadež, V.; Šegota, S. Atomic force microscopy reveals new biophysical markers for monitoring subcellular changes in oxidative injury: Neuroprotective effects of quercetin at the nanoscale. PLoS ONE 2018, 13, e0200119. [Google Scholar] [CrossRef]
- Zubčić, K.; Radovanović, V.; Vlainić, J.; Hof, P.R.; Oršolić, N.; Šimić, G.; Jazvinšćak Jembrek, M. PI3K/Akt and ERK1/2 Signalling Are Involved in Quercetin-Mediated Neuroprotection against Copper-Induced Injury. Oxid. Med. Cell. Longev. 2020, 2020, 9834742. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, H.; Wu, J.; Yin, L.; Yan, L.J.; Zhang, C. Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/p38 MAPK Pathway. Int. J. Mol. Sci. 2018, 19, 2982. [Google Scholar] [CrossRef]
- Zakharova, I.O.; Sokolova, T.V.; Bayunova, L.V.; Zorina, I.I.; Rychkova, M.P.; Shpakov, A.O.; Avrova, N.F. The Protective Effect of Insulin on Rat Cortical Neurons in Oxidative Stress and Its Dependence on the Modulation of Akt, GSK-3beta, ERK1/2, and AMPK Activities. Int. J. Mol. Sci. 2019, 20, 3702. [Google Scholar] [CrossRef]
- Rueda, D.; Galve-Roperh, I.; Haro, A.; Guzmán, M. The CB(1) cannabinoid receptor is coupled to the activation of c-Jun N-terminal kinase. Mol. Pharmacol. 2000, 58, 814–820. [Google Scholar] [CrossRef]
- Davis, M.I.; Ronesi, J.; Lovinger, D.M. A predominant role for inhibition of the adenylate cyclase/protein kinase A pathway in ERK activation by cannabinoid receptor 1 in N1E-115 neuroblastoma cells. J. Biol. Chem. 2003, 278, 48973–48980. [Google Scholar] [CrossRef]
- Derkinderen, P.; Valjent, E.; Toutant, M.; Corvol, J.C.; Enslen, H.; Ledent, C.; Trzaskos, J.; Caboche, J.; Girault, J.A. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J. Neuroci. 2003, 23, 2371–2382. [Google Scholar] [CrossRef]
- Bouaboula, M.; Poinot-Chazel, C.; Bourrié, B.; Canat, X.; Calandra, B.; Rinaldi-Carmona, M.; Le Fur, G.; Casellas, P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem. J. 1995, 312 Pt 2, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, O.; Mangoura, D. Cannabinoid receptor 1 induces a biphasic ERK activation via multiprotein signaling complex formation of proximal kinases PKCε, Src, and Fyn in primary neurons. Neurochem. Int. 2011, 58, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I.; et al. The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lv, X.A.; Dai, Q.; Ge, Y.Q.; Xu, J. Acute upregulation of neuronal mitochondrial type-1 cannabinoid receptor and it’s role in metabolic defects and neuronal apoptosis after TBI. Mol. Brain 2016, 9, 75. [Google Scholar] [CrossRef]
- Viscomi, M.T.; Oddi, S.; Latini, L.; Pasquariello, N.; Florenzano, F.; Bernardi, G.; Molinari, M.; Maccarrone, M. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J. Neurosci. 2009, 29, 4564–4570. [Google Scholar] [CrossRef]
- Gómez Del Pulgar, T.; De Ceballos, M.L.; Guzmán, M.; Velasco, G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef]
- Velasco, G.; Galve-Roperh, I.; Sánchez, C.; Blázquez, C.; Haro, A.; Guzmán, M. Cannabinoids and ceramide: Two lipids acting hand-by-hand. Life Sci. 2005, 77, 1723–1731. [Google Scholar] [CrossRef]
- Carracedo, A.; Geelen, M.J.H.; Diez, M.; Hanada, K.; Guzmán, M.; Velasco, G. Ceramide sensitizes astrocytes to oxidative stress: Protective role of cannabinoids. Biochem. J. 2004, 380, 435–440. [Google Scholar] [CrossRef]
- Guo, J.; Ikeda, S.R. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol. Pharmacol. 2004, 65, 665–674. [Google Scholar] [CrossRef]
- Kwan Cheung, K.A.; Peiris, H.; Wallace, G.; Holland, O.J.; Mitchell, M.D. The Interplay between the Endocannabinoid System, Epilepsy and Cannabinoids. Int. J. Mol. Sci. 2019, 20, 6079. [Google Scholar] [CrossRef]
- Kearn, C.S.; Blake-Palmer, K.; Daniel, E.; Mackie, K.; Glass, M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: A mechanism for receptor cross-talk? Mol. Pharmacol. 2005, 67, 1697–1704. [Google Scholar] [CrossRef]
- Chiang, Y.-C.; Lo, Y.-N.; Chen, J.-C. Crosstalk between dopamine D2 receptors and cannabinoid CB1 receptors regulates CNR1 promoter activity via ERK1/2 signaling. J. Neurochem. 2013, 127, 163–176. [Google Scholar] [CrossRef]
- Cinar, R.; Freund, T.F.; Katona, I.; Mackie, K.; Szucs, M. Reciprocal inhibition of G-protein signaling is induced by CB1 cannabinoid and GABAB receptor interactions in rat hippocampal membranes. Neurochem. Int. 2008, 52, 1402–1409. [Google Scholar] [CrossRef]
- Callén, L.; Moreno, E.; Barroso-Chinea, P.; Moreno-Delgado, D.; Cortés, A.; Mallol, J.; Casadó, V.; Lanciego, J.L.; Franco, R.; Lluis, C.; et al. Cannabinoid receptors CB 1 and CB 2 form functional heteromers in brain. J. Biol. Chem. 2012, 287, 20851–20865. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB₁ receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 5, 558–564. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Reguero, L.; Puente, N.; Lutz, B.; Chaouloff, F.; Rossignol, R.; Piazza, P.V.; Benard, G.; Grandes, P.; Marsicano, G. Cannabinoid control of brain bioenergetics: Exploring the subcellular localization of the CB1 receptor. Mol. Metab. 2014, 3, 495–504. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Naoi, M.; Wu, Y.; Shamoto-Nagai, M.; Maruyama, W. Mitochondria in Neuroprotection by Phytochemicals: Bioactive Polyphenols Modulate Mitochondrial Apoptosis System, Function and Structure. Int. J. Mol. Sci. 2019, 20, 2451. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Niu, W.; Jiang, T.; Zhai, Q.; Yang, L.; Bai, F.; Wang, Q.; Xiong, L. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 2015, 5, 12440. [Google Scholar] [CrossRef]
- Khajehali, E.; Malone, D.T.; Glass, M.; Sexton, P.M.; Christopoulos, A.; Leach, K. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol. Pharmacol. 2015, 88, 368–379. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol. Pharmacol. 2016, 89, 364–375. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Ramírez, B.G.; Blázquez, C.; Gómez del Pulgar, T.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Jhaveri, A.; Maron, B.A.; Wands, J.R. Nitric oxide synthase 3-mediated neurodegeneration after intracerebral gene delivery. J. Neuropathol. Exp. Neurol. 2007, 66, 272–283. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef]
- Ebadi, M.; Sharma, S.K.; Ghafourifar, P.; Brown-Borg, H.; El Refaey, H. Peroxynitrite in the pathogenesis of Parkinson’s disease and the neuroprotective role of metallothioneins. Methods Enzymol. 2005, 396, 276–298. [Google Scholar] [CrossRef]
- Leathem, A.; Ortiz-Cerda, T.; Dennis, J.M.; Witting, P.K. Evidence for Oxidative Pathways in the Pathogenesis of PD: Are Antioxidants Candidate Drugs to Ameliorate Disease Progression? Int. J. Mol. Sci. 2022, 23, 6923. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef]
- Bono-Yagüe, J.; Gómez-Escribano, A.P.; Millán, J.M.; Vázquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Leonoudakis, D.; Abood, M.E.; Beattie, E.C. Cannabinoid receptor activation reduces TNFalpha-induced surface localization of AMPAR-type glutamate receptors and excitotoxicity. Neuropharmacology 2010, 58, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.Y.; Bridges, D.; Grigorenko, E.; McCloud, S.; Boon, A.; Hampson, R.E.; Deadwyler, S.A. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology 2005, 48, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Hampson, A.J.; Bornheim, L.M.; Scanziani, M.; Yost, C.S.; Gray, A.T.; Hansen, B.M.; Leonoudakis, D.J.; Bickler, P.E. Dual effects of anandamide on NMDA receptor-mediated responses and neurotransmission. J. Neurochem. 1998, 70, 671–676. [Google Scholar] [CrossRef]
- Khaspekov, L.G.; Brenz Verca, M.S.; Frumkina, L.E.; Hermann, H.; Marsicano, G.; Lutz, B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 2004, 19, 1691–1698. [Google Scholar] [CrossRef]
- Vicente-Sánchez, A.; Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Mol. Brain 2013, 6, 42. [Google Scholar] [CrossRef]
- Kreutz, S.; Koch, M.; Ghadban, C.; Korf, H.W.; Dehghani, F. Cannabinoids and neuronal damage: Differential effects of THC, AEA and 2-AG on activated microglial cells and degenerating neurons in excitotoxically lesioned rat organotypic hippocampal slice cultures. Exp. Neurol. 2007, 203, 246–257. [Google Scholar] [CrossRef]
- Kreutz, S.; Koch, M.; Böttger, C.; Ghadban, C.; Korf, H.W.; Dehghani, F. 2-Arachidonoylglycerol elicits neuroprotective effects on excitotoxically lesioned dentate gyrus granule cells via abnormal-cannabidiol-sensitive receptors on microglial cells. Glia 2009, 57, 286–294. [Google Scholar] [CrossRef]
- Netzeband, J.G.; Conroy, S.M.; Parsons, K.L.; Gruol, D.L. Cannabinoids enhance NMDA-elicited Ca2+ signals in cerebellar granule neurons in culture. J. Neuroscienci. 1999, 19, 8765–8777. [Google Scholar] [CrossRef]
- Panikashvili, D.; Shein, N.A.; Mechoulam, R.; Trembovler, V.; Kohen, R.; Alexandrovich, A.; Shohami, E. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis. 2006, 22, 257–264. [Google Scholar] [CrossRef]
- Yu, S.J.; Reiner, D.; Shen, H.; Wu, K.J.; Liu, Q.R.; Wang, Y. Time-dependent protection of CB2 receptor agonist in stroke. PLoS ONE 2015, 10, e0132487. [Google Scholar] [CrossRef]
- Vasincu, A.; Rusu, R.-N.; Ababei, D.-C.; Larion, M.; Bild, W.; Stanciu, G.D.; Solcan, C.; Bild, V. Endocannabinoid Modulation in Neurodegenerative Diseases: In Pursuit of Certainty. Biology 2022, 11, 440. [Google Scholar] [CrossRef]
- Armeli, F.; Bonucci, A.; Maggi, E.; Pinto, A.; Businaro, R. Mediterranean Diet and Neurodegenerative Diseases: The Neglected Role of Nutrition in the Modulation of the Endocannabinoid System. Biomolecules 2021, 11, 790. [Google Scholar] [CrossRef]
- Ceccarini, J.; Ahmad, R.; Van De Vliet, L.; Casteels, C.; Vandenbulcke, M.; Vandenberghe, W.; Van Laere, K. Behavioral symptoms in premanifest Huntington disease correlate with reduced frontal CB 1 R levels. J. Nucl. Med. 2019, 60, 115–121. [Google Scholar] [CrossRef]
- Glass, M.; Dragunow, M.; Faull, R.L. The pattern of neurodegeneration in Huntington’s disease: A comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 2000, 97, 505–519. [Google Scholar] [CrossRef]
- Van Laere, K.; Casteels, C.; Dhollander, I.; Goffin, K.; Grachev, I.; Bormans, G.; Vandenberghe, W. Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. J. Nucl. Med. 2010, 51, 1413–1417. [Google Scholar] [CrossRef]
- López-Sendón Moreno, J.L.; García Caldentey, J.; Trigo Cubillo, P.; Ruiz Romero, C.; García Ribas, G.; Alonso Arias, M.A.A.; García de Yébenes, M.J.; Tolón, R.M.; Galve-Roperh, I.; Sagredo, O.; et al. A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J. Neurol. 2016, 263, 1390–1400. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Bizat, N.; Boyer, F.; Hantraye, P.; Brouillet, E.; Fernández-Ruiz, J. Effects of cannabinoids in the rat model of Huntington’s disease generated by an intrastriatal injection of malonate. Neuroreport 2003, 14, 813–816. [Google Scholar] [CrossRef]
- Dhopeshwarkar, A.; Mackie, K. Functional Selectivity of CB2 Cannabinoid Receptor Ligands at a Canonical and Noncanonical Pathway. J. Pharm. Exp. Ther. 2016, 358, 342–351. [Google Scholar] [CrossRef]
- Bedse, G.; Romano, A.; Cianci, S.; Lavecchia, A.M.; Lorenzo, P.; Elphick, M.R.; Laferla, F.M.; Vendemiale, G.; Grillo, C.; Altieri, F.; et al. Altered expression of the CB1 cannabinoid receptor in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 701–712. [Google Scholar] [CrossRef]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134 Pt 4, 1041–1060. [Google Scholar] [CrossRef]
- Pascual, A.C.; Martín-Moreno, A.M.; Giusto, N.M.; de Ceballos, M.L.; Pasquaré, S.J. Normal aging in rats and pathological aging in human Alzheimer’s disease decrease FAAH activity: Modulation by cannabinoid agonists. Exp. Gerontol. 2014, 60, 92–99. [Google Scholar] [CrossRef]
- Jung, K.M.; Astarita, G.; Yasar, S.; Vasilevko, V.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. An amyloid β42-dependent deficit in anandamide mobilization is associated with cognitive dysfunction in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1522–1532. [Google Scholar] [CrossRef]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 791–804. [Google Scholar] [CrossRef]
- Stumm, C.; Hiebel, C.; Hanstein, R.; Purrio, M.; Nagel, H.; Conrad, A.; Lutz, B.; Behl, C.; Clement, A.B. Cannabinoid receptor 1 deficiency in a mouse model of Alzheimer’s disease leads to enhanced cognitive impairment despite of a reduction in amyloid deposition. Neurobiol. Aging 2013, 34, 2574–2584. [Google Scholar] [CrossRef]
- Altamura, C.; Ventriglia, M.; Martini, M.G.; Montesano, D.; Errante, Y.; Piscitelli, F.; Scrascia, F.; Quattrocchi, C.; Palazzo, P.; Seccia, S.; et al. Elevation of Plasma 2-Arachidonoylglycerol Levels in Alzheimer’s Disease Patients as a Potential Protective Mechanism against Neurodegenerative Decline. J. Alzheimers Dis. 2015, 46, 497–506. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; De Filippis, D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. 2006, 63, 1410–1424. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflammation 2015, 12, 81. [Google Scholar] [CrossRef]
- Elmazoglu, Z.; Rangel-López, E.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Túnez, I.; Aschner, M.; Santamaría, A.; Karasu, Ç. Cannabinoid-profiled agents improve cell survival via reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Aβ1-42 peptide in rat hippocampal neurons. Neurochem. Int. 2020, 140, 104817. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Cebeira, M.; De Ceballos, M.L.; Zeng, B.Y.; Jenner, P.; Ramos, J.A.; Fernández-Ruiz, J.J. Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson’s syndrome and of MPTP-treated marmosets. Eur. J. Neurosci. 2001, 14, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Pisani, V.; Moschella, V.; Bari, M.; Fezza, F.; Galati, S.; Bernardi, G.; Stanzione, P.; Pisani, A.; Maccarrone, M. Dynamic changes of anandamide in the cerebrospinal fluid of Parkinson’s disease patients. Mov. Disord. 2010, 25, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Lotan, I.; Treves, T.A.; Roditi, Y.; Djaldetti, R. Cannabis (medical marijuana) treatment for motor and non-motor symptoms of Parkinson disease: An open-label observational study. Clin. Neuropharmacol. 2014, 37, 41–44. [Google Scholar] [CrossRef]
- Chung, Y.C.; Shin, W.H.; Baek, J.Y.; Cho, E.J.; Baik, H.H.; Kim, S.R.; Won, S.Y.; Jin, B.K. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson’s disease. Exp. Mol. Med. 2016, 48, e205. [Google Scholar] [CrossRef]
- Witting, A.; Weydt, P.; Hong, S.; Kliot, M.; Moller, T.; Stella, N. Endocannabinoids accumulate in spinal cord of SOD1 G93A transgenic mice. J. Neurochem. 2004, 89, 1555–1557. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef]
- Romigi, A.; Bari, M.; Placidi, F.; Marciani, M.G.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, C.; Zannino, S.; Corte, F.; et al. Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef]
- Rocha, L.; Cinar, R.; Guevara-Guzmán, R.; Alonso-Vanegas, M.; San-Juan, D.; Martínez-Juárez, I.; Castañeda-Cabral, J.L.; Carmona-Cruz, F. Endocannabinoid System and Cannabinoid 1 Receptors in Patients With Pharmacoresistant Temporal Lobe Epilepsy and Comorbid Mood Disorders. Front. Behav. Neurosci. 2020, 14, 52. [Google Scholar] [CrossRef]
- Ludányi, A.; Eross, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef]
- Perucca, E. Cannabinoids in the Treatment of Epilepsy: Hard Evidence at Last? J. Epilepsy Res. 2017, 7, 61–76. [Google Scholar] [CrossRef]
- Funada, M.; Takebayashi-Ohsawa, M. Synthetic cannabinoid AM2201 induces seizures: Involvement of cannabinoid CB1 receptors and glutamatergic transmission. Toxicol. Appl. Pharmacol. 2018, 338, 1–8. [Google Scholar] [CrossRef]
- Knowles, M.D.; de la Tremblaye, P.B.; Azogu, I.; Plamondon, H. Endocannabinoid CB1 receptor activation upon global ischemia adversely impact recovery of reward and stress signaling molecules, neuronal survival and behavioral impulsivity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 66, 8–21. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, A.B.; Siopi, E.; Finn, D.P.; Marchand-Leroux, C.; Garcia-Segura, L.M.; Jafarian-Tehrani, M.; Viveros, M.P. CB1 and CB2 cannabinoid receptor antagonists prevent minocycline-induced neuroprotection following traumatic brain injury in mice. Cereb. Cortex. 2015, 25, 35–45. [Google Scholar] [CrossRef]
- Behl, T.; Makkar, R.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Bungau, S.; Andronie-Cioara, F.L.; Munteanu, M.A.; Brisc, M.C.; et al. Current trends in neurodegeneration: Cross talks between oxidative stress, cell death, and inflammation. Int. J. Mol. Sci. 2021, 22, 7432. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Long. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Oršolić, N.; Mandić, L.; Sadžak, A.; Šegota, S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration. Antioxidants 2021, 10, 1628. [Google Scholar] [CrossRef]
- Domanskyi, A.; Parlato, R. Oxidative Stress in Neurodegenerative Diseases. Antioxidants 2022, 11, 504. [Google Scholar] [CrossRef]
- Piccirillo, S.; Magi, S.; Preziuso, A.; Serfilippi, T.; Cerqueni, G.; Orciani, M.; Amoroso, S.; Lariccia, V. The Hidden Notes of Redox Balance in Neurodegenerative Diseases. Antioxidants 2022, 11, 1456. [Google Scholar] [CrossRef]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Zubčić, K.; Hof, P.R.; Šimić, G.; Jazvinšćak Jembrek, M. The Role of Copper in Tau-Related Pathology in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 572308. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Petrovic, S.; Arsic, A.; Ristic-Medic, D.; Cvetkovic, Z.; Vucic, V. Lipid Peroxidation and Antioxidant Supplementation in Neurodegenerative Diseases: A Review of Human Studies. Antioxidants 2020, 9, 1128. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- De Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef]
- Kosyreva, A.M.; Sentyabreva, A.V.; Tsvetkov, I.S.; Makarova, O.V. Alzheimer’s Disease and Inflammaging. Brain Sci. 2022, 12, 1237. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.; Yousefian-Jazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Musetti, B.; González-Ramos, H.; González, M.; Bahnson, E.M.; Varela, J.; Thomson, L. Cannabis sativa extracts protect LDL from Cu2+-mediated oxidation. J. Cannabis Res. 2020, 2, 37. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.S.; Batista, J., Jr.; Viana, R.B.; Baetas, A.C.; Orestes, E.; Andrade, M.A.; Honório, K.M.; Da Silva, A.B.F. Understanding the Molecular Aspects of Tetrahydrocannabinol and Cannabidiol as Antioxidants. Molecules 2013, 18, 12663–12674. [Google Scholar] [CrossRef]
- Duncan, R.S.; Riordan, S.M.; Hall, C.W.; Payne, A.J.; Chapman, K.D.; Koulen, P. N-acylethanolamide metabolizing enzymes are upregulated in human neural progenitor-derived neurons exposed to sub-lethal oxidative stress. Front. Cell Neurosci. 2022, 16, 902278. [Google Scholar] [CrossRef]
- Parga, J.A.; Rodriguez-Perez, A.I.; Garcia-Garrote, M.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. NRF2 Activation and Downstream Effects: Focus on Parkinson’s Disease and Brain Angiotensin. Antioxidants 2021, 10, 1649. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-ҡB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef]
- Galán-Ganga, M.; Del Río, R.; Jiménez-Moreno, N.; Díaz-Guerra, M.; Lastres-Becker, I. Cannabinoid CB2 Receptor Modulation by the Transcription Factor NRF2 is Specific in Microglial Cells. Cell. Mol. Neurobiol. 2020, 40, 167–177. [Google Scholar] [CrossRef]
- Chianese, G.; Sirignano, C.; Benetti, E.; Marzaroli, V.; Collado, J.A.; de la Vega, L.; Appendino, G.; Muñoz, E.; Taglialatela-Scafati, O. A Nrf-2 Stimulatory Hydroxylated Cannabidiol Derivative from Hemp (Cannabis sativa). J. Nat. Prod. 2022, 85, 1089–1097. [Google Scholar] [CrossRef]
- Hampson, A.J.; Grimaldi, M.; Axelrod, J.; Wink, D. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. USA 1998, 95, 8268–8273. [Google Scholar] [CrossRef]
- Jia, J.; Ma, L.; Wu, M.; Zhang, L.; Zhang, X.; Zhai, Q.; Jiang, T.; Wang, Q.; Xiong, L. Anandamide protects HT22 cells exposed to hydrogen peroxide by inhibiting CB1 receptor-mediated type 2 NADPH oxidase. Oxid. Med. Cell. Longev. 2014, 2014, 893516. [Google Scholar] [CrossRef]
- Shouman, B.; Fontaine, R.H.; Baud, O.; Schwendimann, L.; Keller, M.; Spedding, M.; Lelièvre, V.; Gressens, P. Endocannabinoids potently protect the newborn brain against AMPA-kainate receptor-mediated excitotoxic damage. Br. J. Pharmacol. 2006, 148, 442–451. [Google Scholar] [CrossRef]
- Liu, X.; Wu, Y.; Zhou, D.; Xie, Y.; Zhou, Y.; Lu, Y.; Yang, R.; Liu, S. N-linoleyltyrosine protects PC12 cells against oxidative damage via autophagy: Possible involvement of CB1 receptor regulation. Int. J. Mol. Med. 2020, 46, 1827–1837. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Z.X.; Liu, Y.T.; Xu, Z.C.; Hu, Y.; Lv, W.; Yang, Z.Y.; Sheng, Y.M.; Liu, S. N-linoleyltyrosine protects neurons against Aβ1-40-induced cell toxicity via autophagy involving the CB2/AMPK/mTOR/ULK1 pathway. Brain Res. Bull. 2022, 188, 203–213. [Google Scholar] [CrossRef]
- Veeraraghavan, P.; Dekanic, A.; Nistri, A. A study of cannabinoid-1 receptors during the early phase of excitotoxic damage to rat spinal locomotor networks in vitro. Neuroscience 2016, 333, 214–228. [Google Scholar] [CrossRef]
- Hassan, F.-u.; Nadeem, A.; Li, Z.; Javed, M.; Liu, Q.; Azhar, J.; Rehman, M.S.-u.; Cui, K.; Rehman, S.u. Role of Peroxisome Proliferator-Activated Receptors (PPARs) in Energy Homeostasis of Dairy Animals: Exploiting Their Modulation through Nutrigenomic Interventions. Int. J. Mol. Sci. 2021, 22, 12463. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Płonka, P.M.; Pierzchalski, P. The Role of PPAR Alpha in the Modulation of Innate Immunity. Int. J. Mol. Sci. 2021, 22, 10545. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Ma, A.; Zhang, X.; Li, W.; Yang, W.; Chen, C.; Jin, X. Orally administered oleoylethanolamide protects mice from focal cerebral ischemic injury by activating peroxisome proliferator-activated receptor α. Neuropharmacology 2012, 63, 242–249. [Google Scholar] [CrossRef]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef]
- Panikashvili, D.; Mechoulam, R.; Beni, S.M.; Alexandrovich, A.; Shohami, E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J. Cereb. Blood Flow Metab. 2005, 25, 477–484. [Google Scholar] [CrossRef]
- Zhang, J.; Teng, Z.; Song, Y.; Hu, M.; Chen, C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J. Cereb. Blood Flow Metab. 2015, 35, 443–453. [Google Scholar] [CrossRef]
- Aymerich, M.S.; Rojo-Bustamante, E.; Molina, C.; Celorrio, M.; Sánchez-Arias, J.A.; Franco, R. Neuroprotective Effect of JZL184 in MPP(+)-Treated SH-SY5Y Cells Through CB2 Receptors. Mol. Neurobiol. 2016, 53, 2312–2319. [Google Scholar] [CrossRef]
- Tchantchou, F.; Tucker, L.B.; Fu, A.H.; Bluett, R.J.; McCabe, J.T.; Patel, S.; Zhang, Y. The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 2014, 85, 427–439. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Dick, J.R.; Pryce, G.; Petrosino, S.; Di Marzo, V.; Baker, D.; Greensmith, L. Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. FASEB J. 2006, 20, 1003–1005. [Google Scholar] [CrossRef]
- Celorrio, M.; Fernández-Suárez, D.; Rojo-Bustamante, E.; Echeverry-Alzate, V.; Ramírez, M.J.; Hillard, C.J.; López-Moreno, J.A.; Maldonado, R.; Oyarzábal, J.; Franco, R.; et al. Fatty acid amide hydrolase inhibition for the symptomatic relief of Parkinson’s disease. Brain Behav. Immun. 2016, 57, 94–105. [Google Scholar] [CrossRef]
- Bergamaschi, M.M.; Queiroz, R.H.; Zuardi, A.W.; Crippa, J.A. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf. 2011, 6, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Iffland, K.; Grotenhermen, F. An Update on Safety and Side Effects of Cannabidiol: A Review of Clinical Data and Relevant Animal Studies. Cannabis Cannabinoid Res. 2016, 2, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Papagianni, E.P.; Stevenson, C.W. Cannabinoid Regulation of Fear and Anxiety: An Update. Curr. Psychiatry Rep. 2019, 21, 38. [Google Scholar] [CrossRef]
- García-Gutiérrez, M.S.; Navarrete, F.; Gasparyan, A.; Austrich-Olivares, A.; Sala, F.; Manzanares, J. Cannabidiol: A Potential New Alternative for the Treatment of Anxiety, Depression, and Psychotic Disorders. Biomolecules 2020, 10, 1575. [Google Scholar] [CrossRef]
- Campos, A.C.; Fogaça, M.V.; Sonego, A.B.; Guimarães, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef]
- Gunasekera, B.; Davies, C.; Blest-Hopley, G.; Veronese, M.; Ramsey, N.F.; Bossong, M.G.; Radua, J.; Bhattacharyya, S.; CBE Consortium. Task-independent acute effects of delta-9-tetrahydrocannabinol on human brain function and its relationship with cannabinoid receptor gene expression: A neuroimaging meta-regression analysis. Neurosci. Biobehav. Rev. 2022, 140, 104801. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Masteikova, R.; Lazauskas, R.; Bernatoniene, J. Cannabis sativa L. Bioactive Compounds and Their Protective Role in Oxidative Stress and Inflammation. Antioxidants 2022, 11, 660. [Google Scholar] [CrossRef]
- Nadal, X.; Del Río, C.; Casano, S.; Palomares, B.; Ferreiro-Vera, C.; Navarrete, C.; Sánchez-Carnerero, C.; Cantarero, I.; Bellido, M.L.; Meyer, S.; et al. Tetrahydrocannabinolic acid is a potent PPARγ agonist with neuroprotective activity. Br. J. Pharmacol. 2017, 174, 4263–4276. [Google Scholar] [CrossRef]
- Verhoeckx, K.C.; Korthout, H.A.; van Meeteren-Kreikamp, A.P.; Ehlert, K.A.; Wang, M.; van der Greef, J.; Rodenburg, R.J.; Witkamp, R.F. Unheated Cannabis sativa extracts and its major compound THC-acid have potential immuno-modulating properties not mediated by CB1 and CB2 receptor coupled pathways. Int. Immunopharmacol. 2006, 6, 656–665. [Google Scholar] [CrossRef]
- Mecha, M.; Torrao, A.S.; Mestre, L.; Carrillo-Salinas, F.J.; Mechoulam, R.; Guaza, C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012, 3, e331. [Google Scholar] [CrossRef]
- Castillo, A.; Tolón, M.R.; Fernández-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef]
- Vrechi, T.; Leão, A.; Morais, I.; Abílio, V.C.; Zuardi, A.W.; Hallak, J.; Crippa, J.A.; Bincoletto, C.; Ureshino, R.P.; Smaili, S.S.; et al. Cannabidiol induces autophagy via ERK1/2 activation in neural cells. Sci. Rep. 2021, 11, 5434. [Google Scholar] [CrossRef]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; Di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L., Jr.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef]
- Cao, C.; Li, Y.; Liu, H.; Bai, G.; Mayl, J.; Lin, X.; Sutherland, K.; Nabar, N.; Cai, J. The potential therapeutic effects of THC on Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 973–984. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, Y.; Yan, J.; Brown, B.; Lin, X.; Zhang, X.; Shen, N.; Li, M.; Cai, J.; Gordon, M.; et al. Low-Dose Delta-9-Tetrahydrocannabinol as Beneficial Treatment for Aged APP/PS1 Mice. Int. J. Mol. Sci. 2022, 23, 2757. [Google Scholar] [CrossRef]
- Fihurka, O.; Hong, Y.; Yan, J.; Brown, B.; Lin, X.; Shen, N.; Wang, Y.; Zhao, H.; Gordon, M.N.; Morgan, D.; et al. The Memory Benefit to Aged APP/PS1 Mice from Long-Term Intranasal Treatment of Low-Dose THC. Int. J. Mol. Sci. 2022, 23, 4253. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Molina-Holgado, F.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson’s disease. Neurobiol. Dis. 2005, 19, 96–107. [Google Scholar] [CrossRef]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef] [PubMed]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.; Pertwee, R.; Fernández-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ⁹-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol. 2011, 163, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; Ramos, J.A.; Decio, A.; Mechoulam, R.; Fernández-Ruiz, J. Cannabidiol reduced the striatal atrophy caused 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur. J. Neurosci. 2007, 26, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Erukainure, O.L.; Matsabisa, M.G.; Salau, V.F.; Islam, M.S. Tetrahydrocannabinol-Rich Extracts from Cannabis sativa L. Improve Glucose Consumption and Modulate Metabolic Complications Linked to Neurodegenerative Diseases in Isolated Rat Brains. Front. Pharmacol. 2020, 11, 592981. [Google Scholar] [CrossRef]
- Valdeolivas, S.; Satta, V.; Pertwee, R.G.; Fernández-Ruiz, J.; Sagredo, O. Sativex-like combination of phytocannabinoids is neuroprotective in malonate-lesioned rats, an inflammatory model of Huntington’s disease: Role of CB1 and CB2 receptors. ACS Chem. Neurosci. 2012, 3, 400–406. [Google Scholar] [CrossRef]
- Moreno-Martet, M.; Espejo-Porras, F.; Fernández-Ruiz, J.; de Lago, E. Changes in endocannabinoid receptors and enzymes in the spinal cord of SOD1(G93A) transgenic mice and evaluation of a Sativex(®) -like combination of phytocannabinoids: Interest for future therapies in amyotrophic lateral sclerosis. CNS Neurosci. Ther. 2014, 20, 809–815. [Google Scholar] [CrossRef]
- Amtmann, D.; Weydt, P.; Johnson, K.L.; Jensen, M.P.; Carter, G.T. Survey of cannabis use in patients with amyotrophic lateral sclerosis. Am. J. Hosp. Paliat. Care 2004, 21, 95–104. [Google Scholar] [CrossRef]
- Paes-Colli, Y.; Aguiar, A.; Isaac, A.R.; Ferreira, B.K.; Campos, R.; Trindade, P.; de Melo Reis, R.A.; Sampaio, L.S. Phytocannabinoids and Cannabis-Based Products as Alternative Pharmacotherapy in Neurodegenerative Diseases: From Hypothesis to Clinical Practice. Front. Cell. Neurosci. 2022, 16, 917164. [Google Scholar] [CrossRef]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Involvement of protein kinase A in cannabinoid receptor-mediated protection from oxidative neuronal injury. J. Pharmacol. Exp. Ther. 2005, 313, 88–94. [Google Scholar] [CrossRef]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol. Pharmacol. 2006, 69, 691–696. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Paz-Loyola, A.L.; Serratos, I.N.; Seminotti, B.; Ribeiro, C.A.; Leipnitz, G.; Souza, D.O.; Wajner, M.; Santamaría, A. The effect of WIN 55,212-2 suggests a cannabinoid-sensitive component in the early toxicity induced by organic acids accumulating in glutaric acidemia type I and in related disorders of propionate metabolism in rat brain synaptosomes. Neuroscience 2015, 310, 578–588. [Google Scholar] [CrossRef]
- Liu, Q.; Bhat, M.; Bowen, W.D.; Cheng, J. Signaling pathways from cannabinoid receptor-1 activation to inhibition of N-methyl-D-aspartic acid mediated calcium influx and neurotoxicity in dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 2009, 331, 1062–1070. [Google Scholar] [CrossRef]
- Rycroft, B.K.; Gibb, A.J. Inhibitory interactions of calcineurin (phosphatase 2B) and calmodulin on rat hippocampal NMDA receptors. Neuropharmacology 2004, 47, 505–514. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; García-García, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; de Ceballos, M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation 2012, 9, 8. [Google Scholar] [CrossRef]
- Song, L.; Yang, X.; Ma, Y.; Wu, N.; Liu, Z. The CB1 cannabinoid receptor agonist reduces L-DOPA-induced motor fluctuation and ERK1/2 phosphorylation in 6-OHDA-lesioned rats. Drug Des. Devel. Ther. 2014, 8, 2173–2179. [Google Scholar] [CrossRef][Green Version]
- Chung, Y.C.; Bok, E.; Huh, S.H.; Park, J.Y.; Yoon, S.H.; Kim, S.R.; Kim, Y.S.; Maeng, S.; Park, S.H.; Jin, B.K. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 2011, 187, 6508–6517. [Google Scholar] [CrossRef]
- Price, D.A.; Martinez, A.A.; Seillier, A.; Koek, W.; Acosta, Y.; Fernandez, E.; Strong, R.; Lutz, B.; Marsicano, G.; Roberts, J.L.; et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 2177–2186. [Google Scholar] [CrossRef]
- Shoemaker, J.L.; Seely, K.A.; Reed, R.L.; Crow, J.P.; Prather, P.L. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J. Neurochem. 2007, 101, 87–98. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; Liu, W.; Ma, Z.; Wu, J. Activation of cannabinoid receptor 2 protects rat hippocampal neurons against Aβ-induced neuronal toxicity. Neurosci. Lett. 2020, 735, 135207. [Google Scholar] [CrossRef]
- Eshhar, N.; Striem, S.; Kohen, R.; Tirosh, O.; Biegon, A. Neuroprotective and antioxidant activities of HU-211, a novel NMDA receptor antagonist. Eur. J. Pharmacol. 1995, 283, 19–29. [Google Scholar] [CrossRef]
- Nuñez-Borque, E.; González-Naranjo, P.; Bartolomé, F.; Alquézar, C.; Reinares-Sebastián, A.; Pérez, C.; Ceballos, M.L.; Páez, J.A.; Campillo, N.E.; Martín-Requero, Á. Targeting Cannabinoid Receptor Activation and BACE-1 Activity Counteracts TgAPP Mice Memory Impairment and Alzheimer’s Disease Lymphoblast Alterations. Mol. Neurobiol. 2020, 57, 1938–1951. [Google Scholar] [CrossRef]
- Li, L.; Xu, Y.; Zhao, M.; Gao, Z. Neuro-protective roles of long non-coding RNA MALAT1 in Alzheimer’s disease with the involvement of the microRNA-30b/CNR1 network and the following PI3K/AKT activation. Exp. Mol. Pathol. 2020, 117, 104545. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tadijan, A.; Vlašić, I.; Vlainić, J.; Đikić, D.; Oršolić, N.; Jazvinšćak Jembrek, M. Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions. Antioxidants 2022, 11, 2049. https://doi.org/10.3390/antiox11102049
Tadijan A, Vlašić I, Vlainić J, Đikić D, Oršolić N, Jazvinšćak Jembrek M. Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions. Antioxidants. 2022; 11(10):2049. https://doi.org/10.3390/antiox11102049
Chicago/Turabian StyleTadijan, Ana, Ignacija Vlašić, Josipa Vlainić, Domagoj Đikić, Nada Oršolić, and Maja Jazvinšćak Jembrek. 2022. "Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions" Antioxidants 11, no. 10: 2049. https://doi.org/10.3390/antiox11102049
APA StyleTadijan, A., Vlašić, I., Vlainić, J., Đikić, D., Oršolić, N., & Jazvinšćak Jembrek, M. (2022). Intracellular Molecular Targets and Signaling Pathways Involved in Antioxidative and Neuroprotective Effects of Cannabinoids in Neurodegenerative Conditions. Antioxidants, 11(10), 2049. https://doi.org/10.3390/antiox11102049