
Time-Course Changes in Oxidative Stress and Inflammation in the Retinas of rds Mice: A Retinitis Pigmentosa Model

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Histological and Immunofluorescence Studies
2.1.1. Hematoxylin–Eosin Assay
2.1.2. Terminal Deoxynucleotidyl Transferase (TUNEL) Assay
2.1.3. Retinal Immunohistochemistry
2.2. Western Blot Analysis
2.3. Statistical Analysis
3. Results
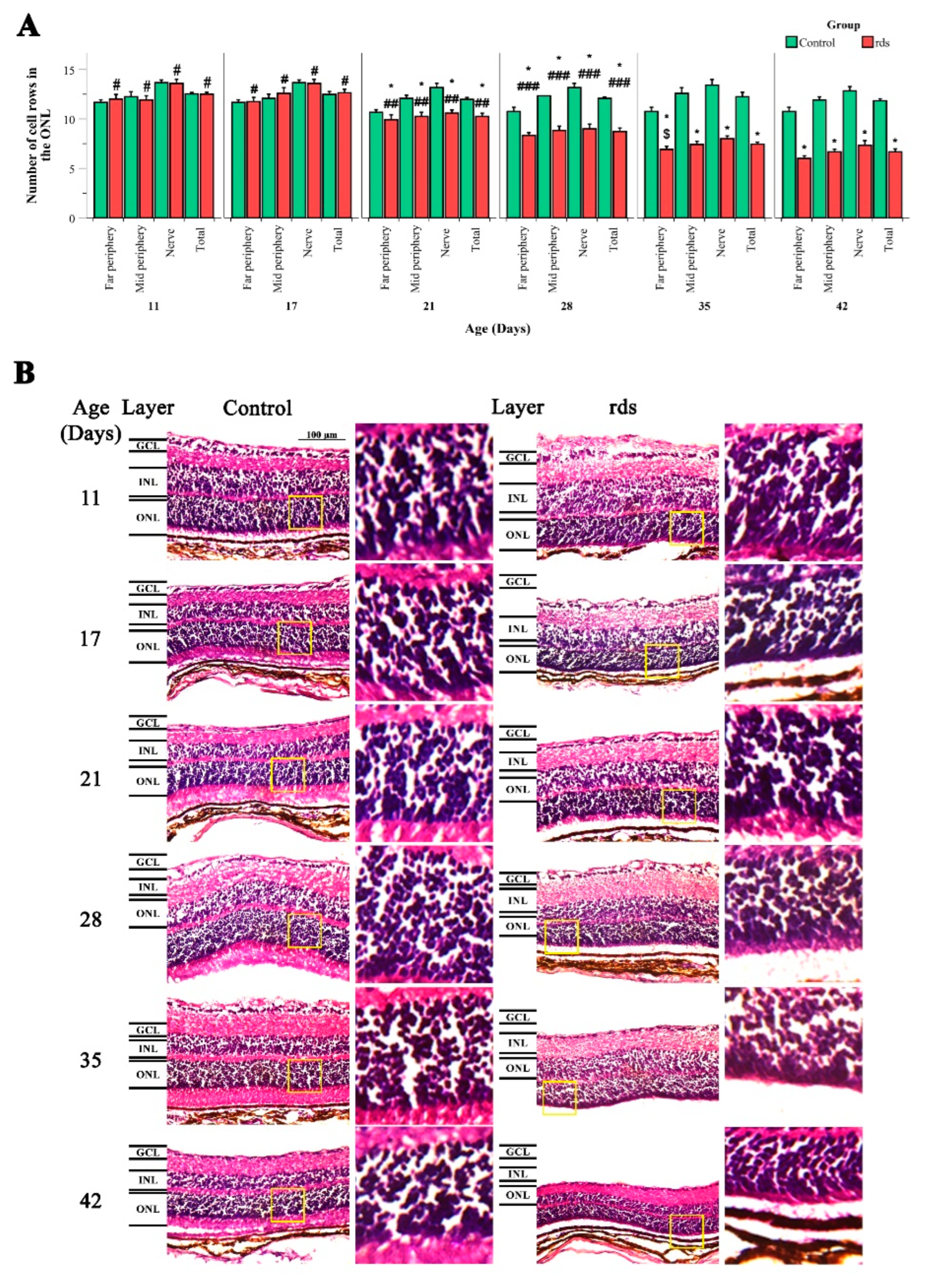
3.1. Morphological Characterization of Retinal rds Degeneration
3.2. Oxidative Stress Characterization in rds Mice
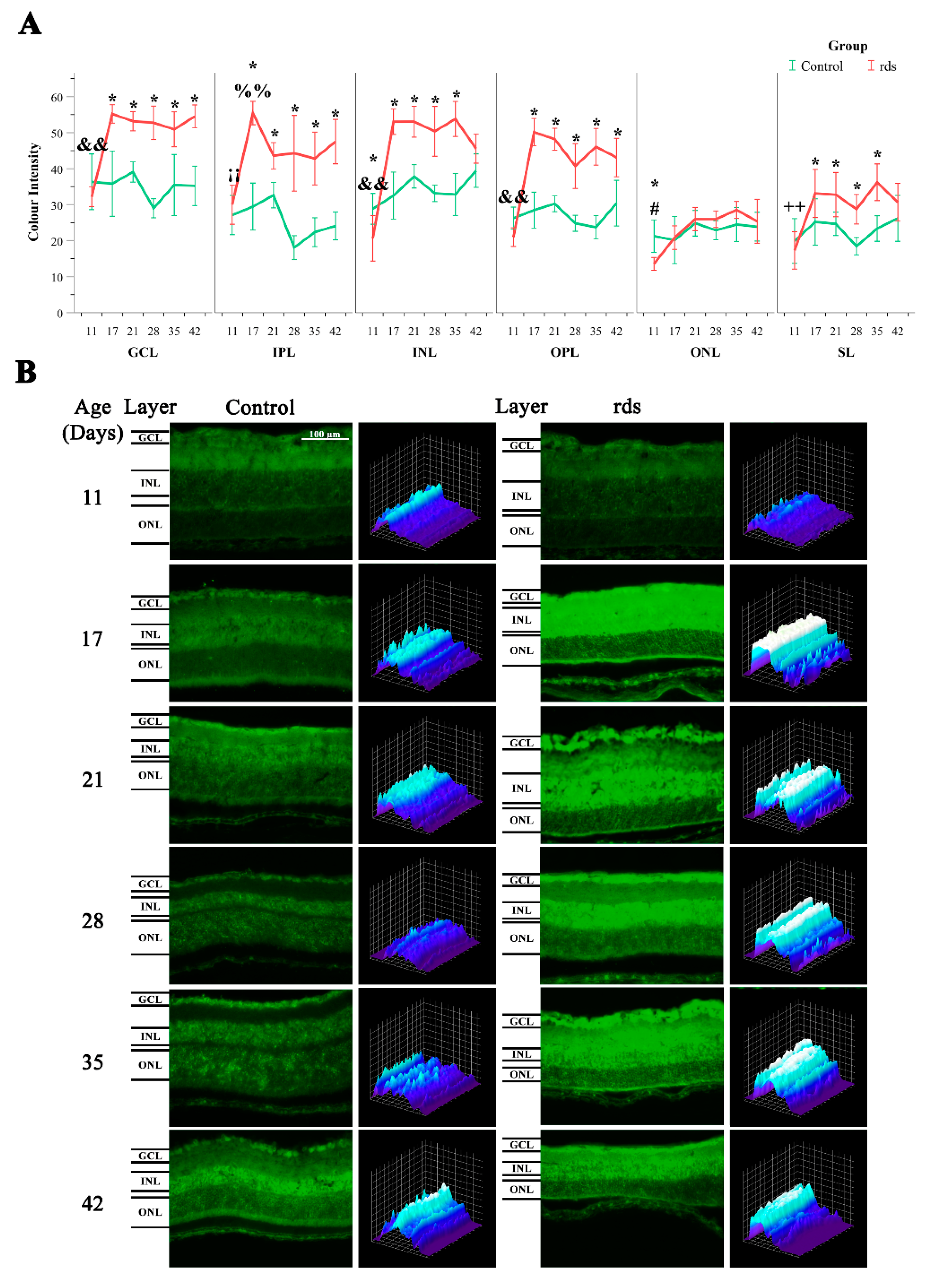
3.2.1. Higher Lipid Peroxidation in rds Retinas
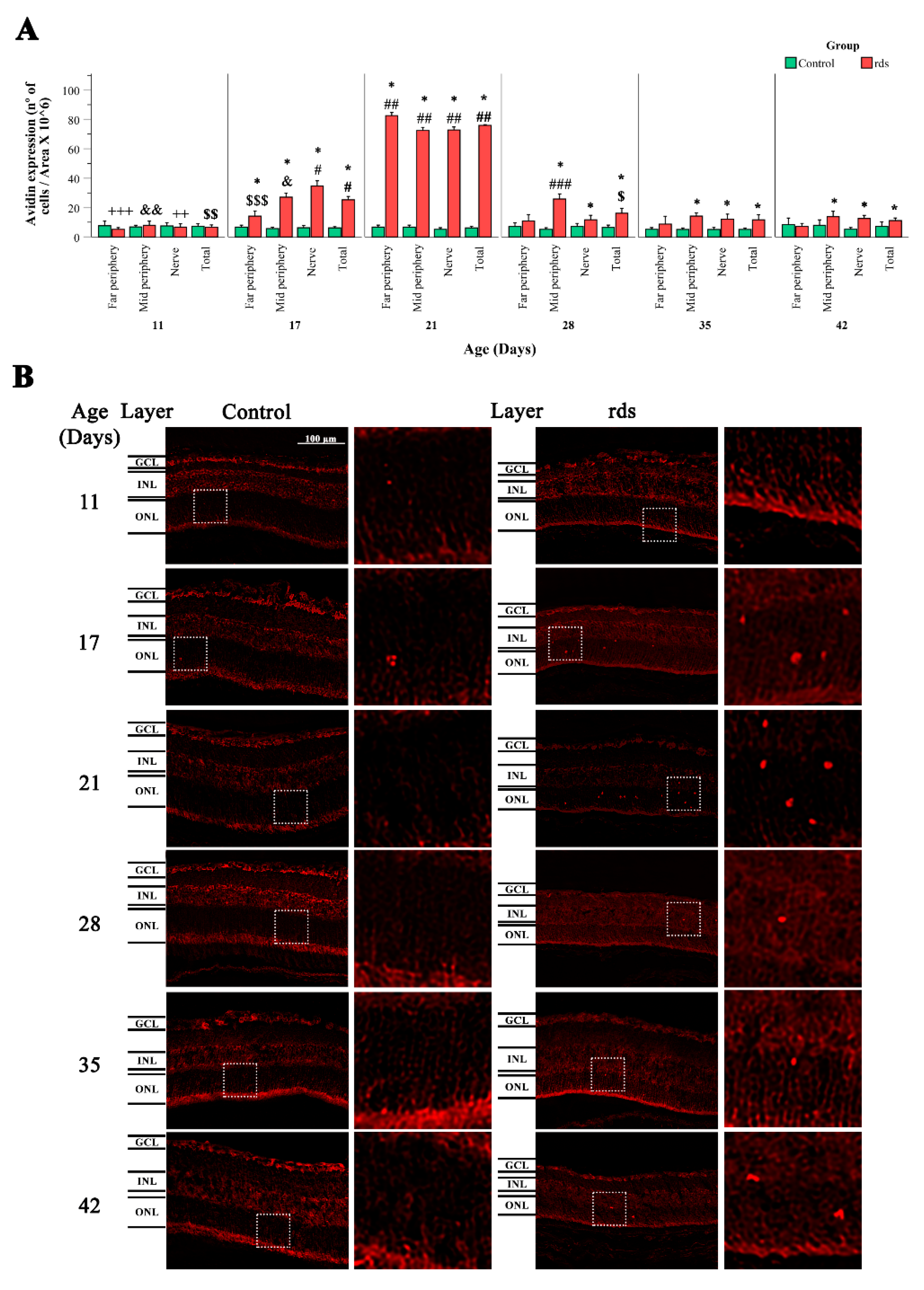
3.2.2. Increased Nuclear Oxidative Stress Damage in rds Retina
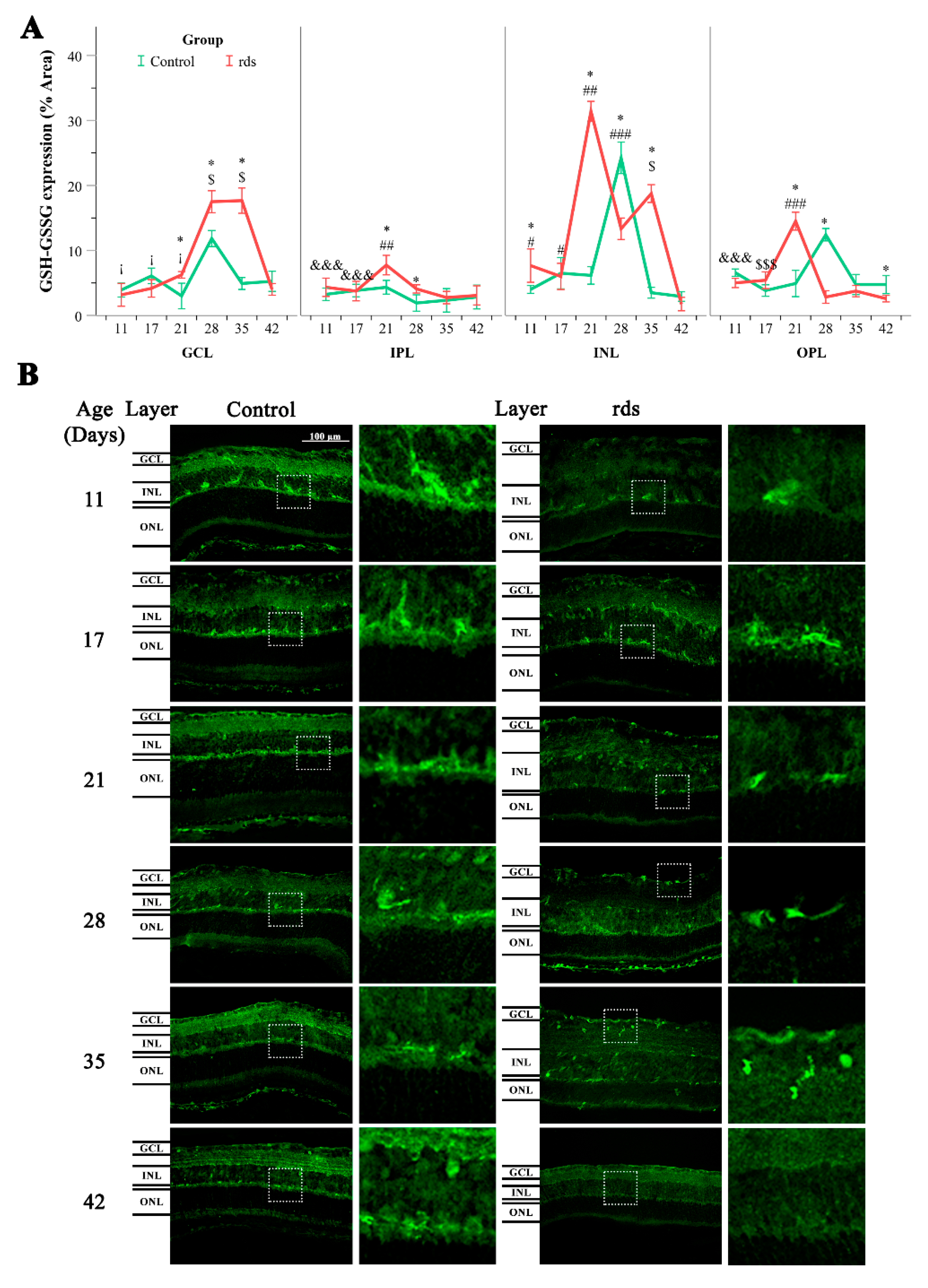
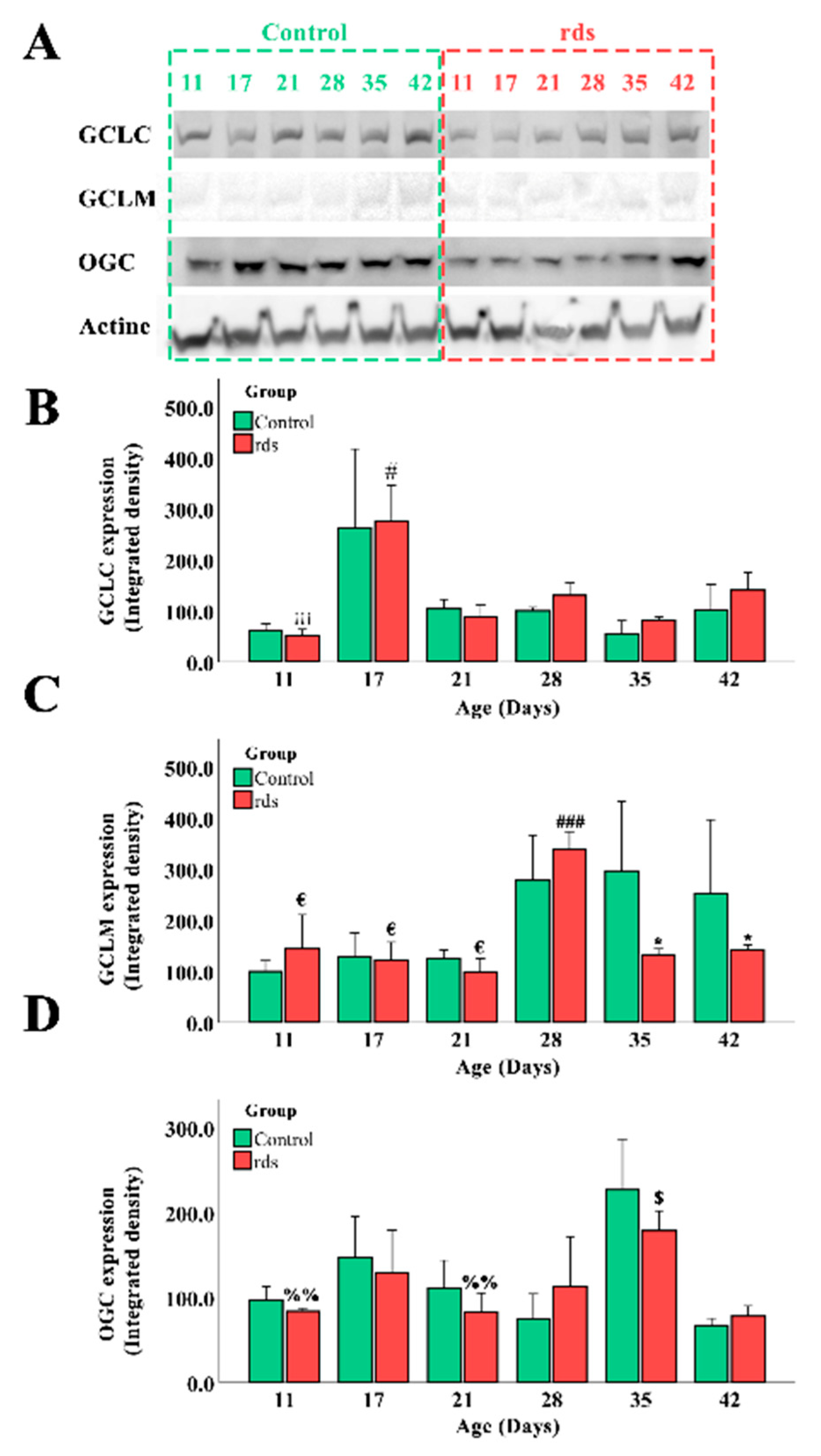
3.2.3. Glutathione Metabolism Alterations in Rds Retinas
3.2.4. Can the Alterations in Glutathione Be Explained by an Increase in the Synthesis or Transport of Glutathione?
3.3. Inflammation
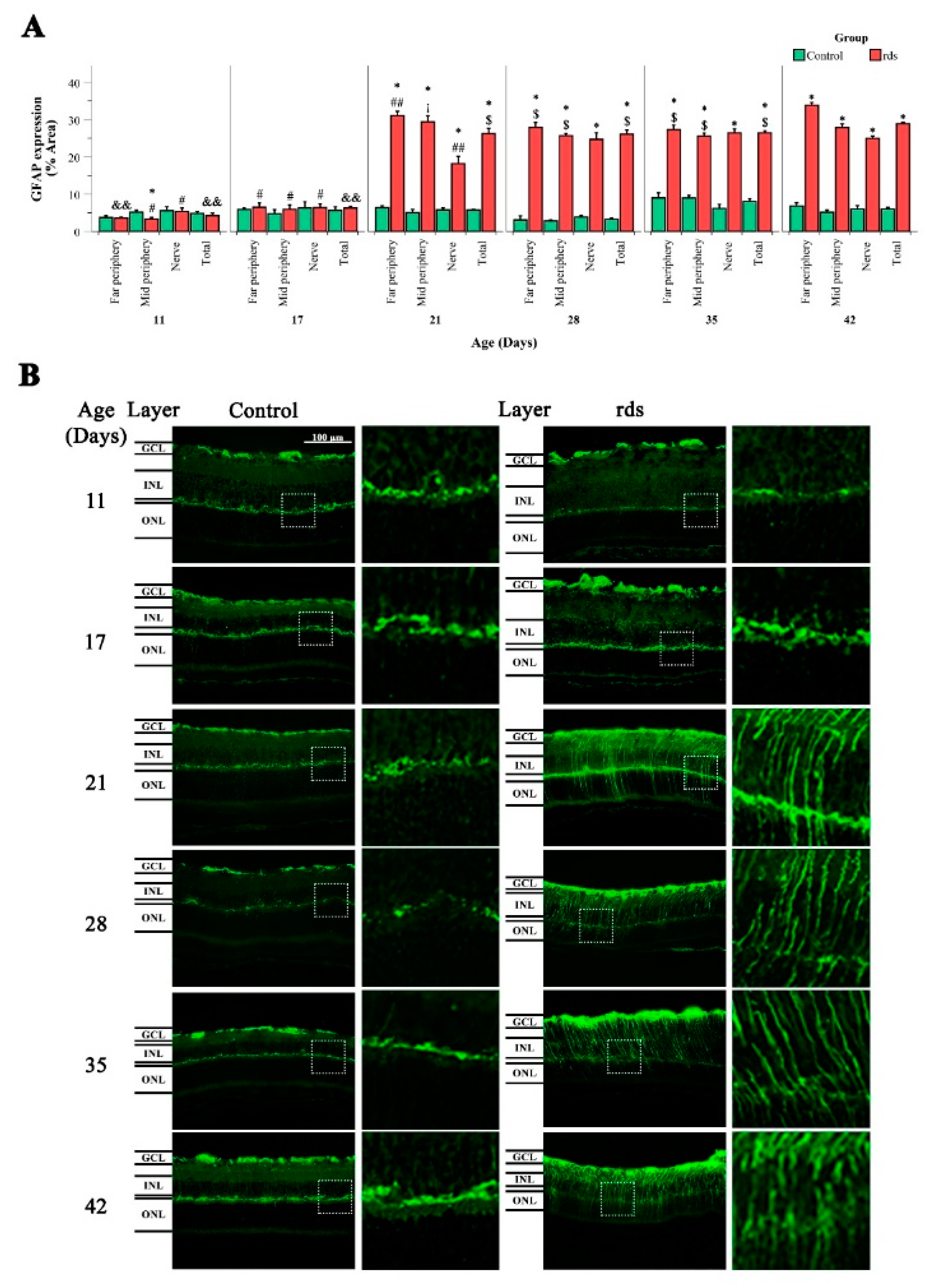
3.3.1. Macroglia Alterations with Disease Progression
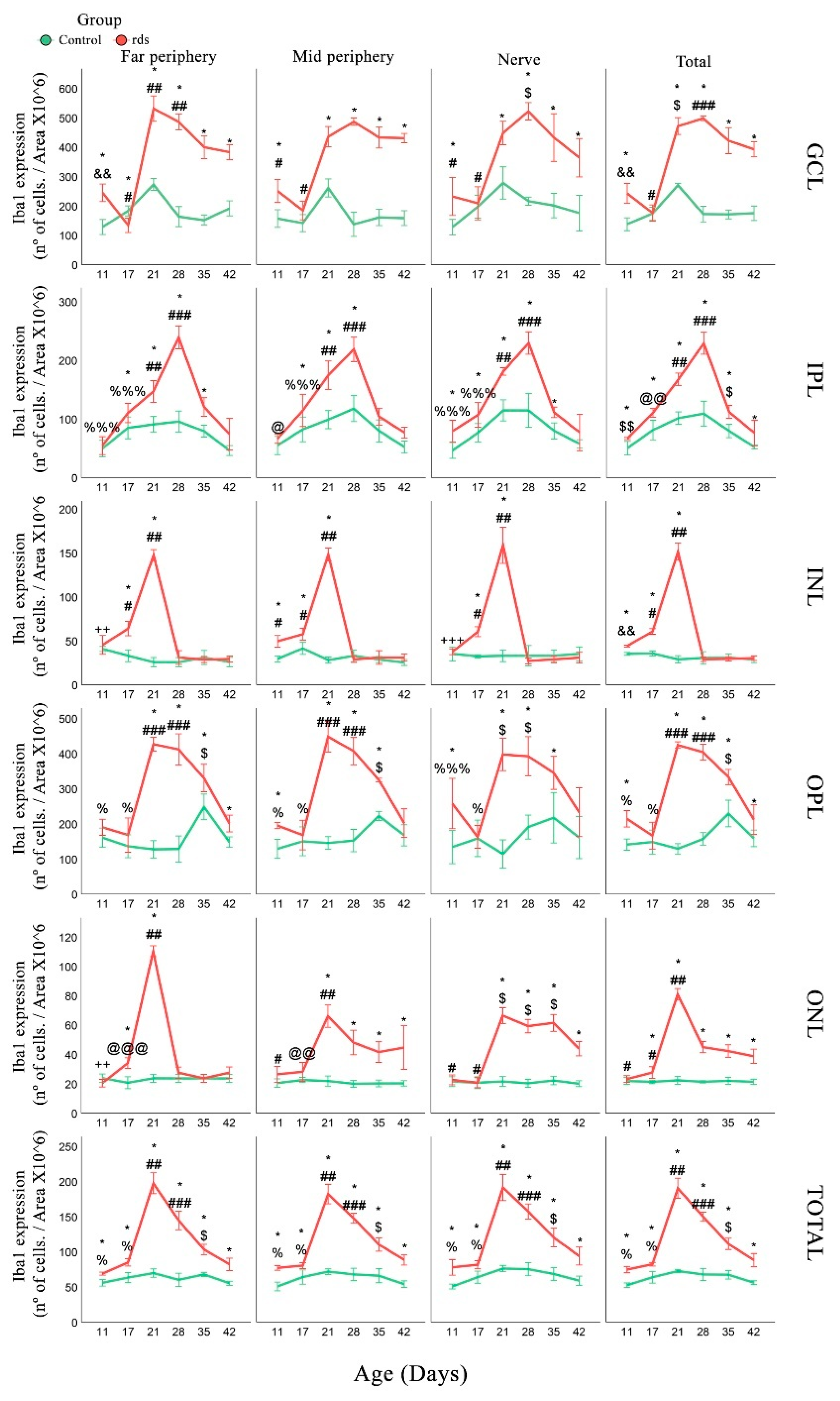
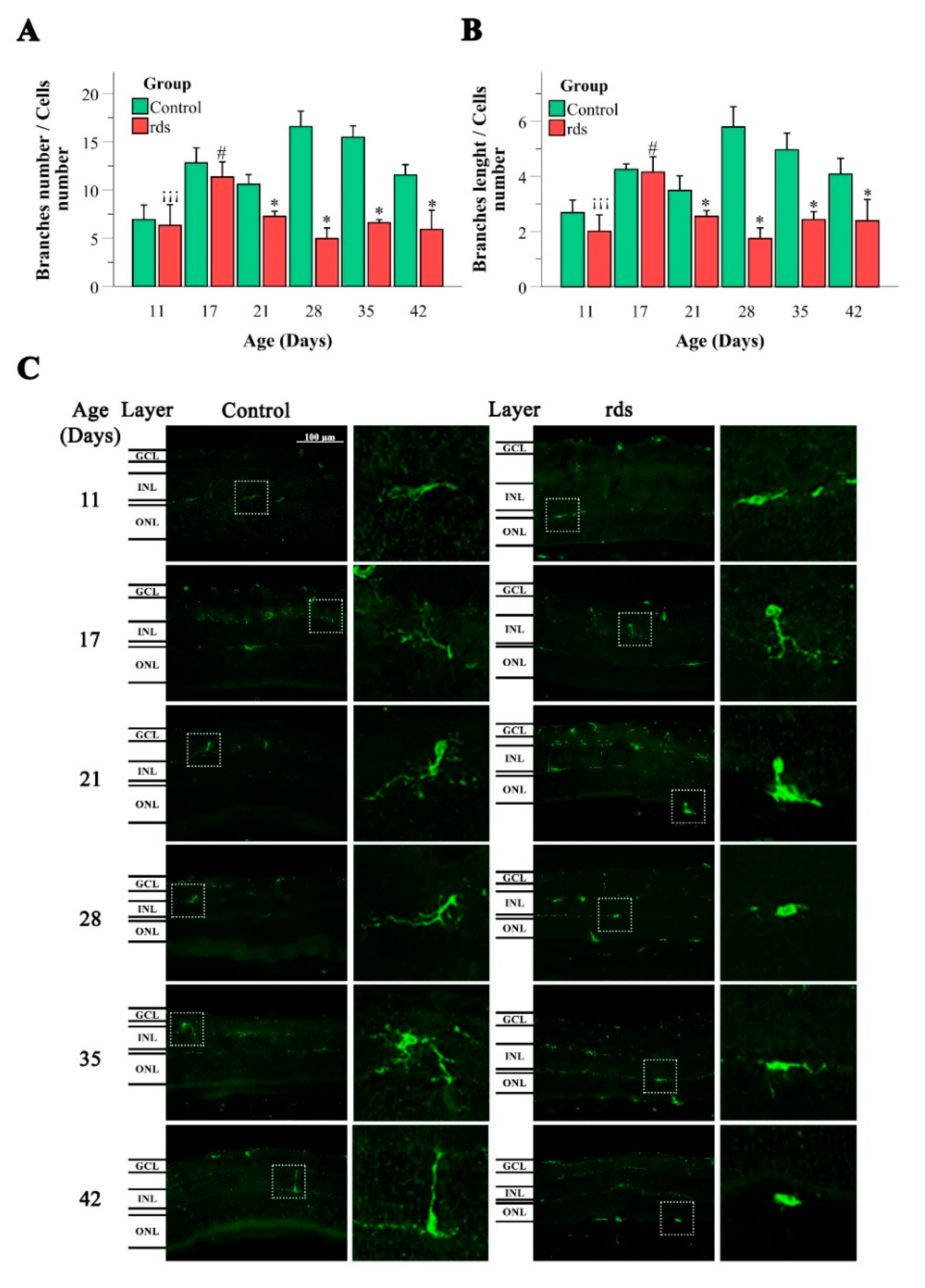
3.3.2. Changes in Microglia in rds Retinas
4. Discussion
4.1. Oxidative Damage to Macromolecules (Lipids and DNA) in the Retinas of rds Mice before the Peak of Photoreceptor Death
4.2. Microglia Activation after Oxidative Damage to Macromolecules in Retinas of Rds Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Kljavin, I.J.; Milam, A.H. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J. Neurosci. 1995, 15, 5429–5438. [Google Scholar] [CrossRef]
- Fisher, S.K.; Lewis, G.P.; Linberg, K.A.; Verardo, M.R. Cellular remodeling in mammalian retina: Results from studies of experimental retinal detachment. Prog. Retin. Eye Res. 2005, 24, 395–431. [Google Scholar] [CrossRef]
- Pinilla, I.; Cuenca, N.; Sauvé, Y.; Wang, S.; Lund, R.D. Preservation of outer retina and its synaptic connectivity following subretinal injections of human RPE cells in the Royal College of Surgeons rat. Exp. Eye Res. 2007, 85, 381–392. [Google Scholar] [CrossRef]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165. [Google Scholar] [CrossRef]
- Jones, B.W.; Marc, R.E. Retinal remodeling during retinal degeneration. Exp. Eye Res. 2005, 81, 123–137. [Google Scholar] [CrossRef]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: From basic research to clinical application. Am. J. Med. 1991, 91, 31S–38S. [Google Scholar] [CrossRef]
- Njie-Mbye, Y.F.; Kulkarni-Chitnis, M.; Opere, C.A.; Barrett, A.; Ohia, S.E. Lipid peroxidation: Pathophysiological and pharmacological implications in the eye. Front. Physiol. 2013, 4, 366. [Google Scholar] [CrossRef]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxid. Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Di Pierdomenico, J.; García-Ayuso, D.; Pinilla, I.; Cuenca, N.; Vidal-Sanz, M.; Agudo-Barriuso, M.; Villegas-Pérez, M.P. Early Events in Retinal Degeneration Caused by Rhodopsin Mutation or Pigment Epithelium Malfunction: Differences and Similarities. Front. Neuroanat. 2017, 11, 14. [Google Scholar] [CrossRef]
- Cuenca, N.; Fernández-Sánchez, L.; Campello, L.; Maneu, V.; De la Villa, P.; Lax, P.; Pinilla, I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog. Retin. Eye Res. 2014, 43, 17–75. [Google Scholar] [CrossRef]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef]
- Roche, S.L.; Ruiz-Lopez, A.M.; Moloney, J.N.; Byrne, A.M.; Cotter, T.G. Microglial-induced Müller cell gliosis is attenuated by progesterone in a mouse model of retinitis pigmentosa. Glia 2018, 66, 295–310. [Google Scholar] [CrossRef]
- Luna, G.; Lewis, G.P.; Banna, C.D.; Skalli, O.; Fisher, S.K. Expression profiles of nestin and synemin in reactive astrocytes and Müller cells following retinal injury: A comparison with glial fibrillar acidic protein and vimentin. Mol. Vis. 2010, 16, 2511–2523. [Google Scholar]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.B.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef] [PubMed]
- Trachsel-Moncho, L.; Benlloch-Navarro, S.; Fernández-Carbonell, Á.; Ramírez-Lamelas, D.T.; Olivar, T.; Silvestre, D.; Poch, E.; Miranda, M. Oxidative stress and autophagy-related changes during retinal degeneration and development. Cell Death Dis. 2018, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Gimeno-Hernández, R.; Cantó, A.; Fernández-Carbonell, A.; Olivar, T.; Hernández-Rabaza, V.; Almansa, I.; Miranda, M. Thioredoxin Delays Photoreceptor Degeneration, Oxidative and Inflammation Alterations in Retinitis Pigmentosa. Front. Pharmacol. 2020, 11, 590572. [Google Scholar] [CrossRef] [PubMed]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Salom, D.; González-García, E.; Soriano Del Castillo, J.M.; Rodrigo, R. Nutraceutical Supplementation Ameliorates Visual Function, Retinal Degeneration, and Redox Status in rd10 Mice. Antioxidants 2021, 10, 1033. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, e5–e12. [Google Scholar] [CrossRef]
- Zabel, M.K.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.N.; Wong, W.T. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef]
- Sanyal, S.; De Ruiter, A.; Hawkins, R.K. Development and degeneration of retina in rds mutant mice: Light microscopy. J. Comp. Neurol. 1980, 194, 193–207. [Google Scholar] [CrossRef]
- Nir, I.; Kedzierski, W.; Chen, J.; Travis, G.H. Expression of Bcl-2 protects against photoreceptor degeneration in retinal degeneration slow (rds) mice. J. Neurosci. 2000, 20, 2150–2154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bradford, M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Jansen, H.G.; Sanyal, S. Development and degeneration of retina in rds mutant mice: Electron microscopy. J. Comp. Neurol. 1984, 224, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández de la Cámara, C.; Hernández-Pinto, A.M.; Olivares-González, L.; Cuevas-Martín, C.; Sánchez-Aragó, M.; Hervás, D.; Salom, D.; Cuezva, J.M.; de la Rosa, E.J.; Millán, J.M.; et al. Adalimumab Reduces Photoreceptor Cell Death in A Mouse Model of Retinal Degeneration. Sci. Rep. 2015, 5, 11764. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.M.; Johnson, L.E.; Ahuja, S.; Ekström, P.A.; Romero, J.; van Veen, T. Significant photoreceptor rescue by treatment with a combination of antioxidants in an animal model for retinal degeneration. Neuroscience 2007, 145, 1120–1129. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Hippert, C.; Graca, A.B.; Barber, A.C.; West, E.L.; Smith, A.J.; Ali, R.R.; Pearson, R.A. Müller glia activation in response to inherited retinal degeneration is highly varied and disease-specific. PLoS ONE 2015, 10, e0120415. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Ferrington, D.A.; Kannan, R. Glutathione Metabolism and the Novel Role of Mitochondrial GSH in Retinal Degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef]
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, 136, 57648. [Google Scholar] [CrossRef]
- Noailles, A.; Fernández-Sánchez, L.; Lax, P.; Cuenca, N. Microglia activation in a model of retinal degeneration and TUDCA neuroprotective effects. J. Neuroinflammation 2014, 11, 186. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Cohen, G.; Shamni, O.; Sasson, S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E879–E886. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.R.; Evans, M.D.; Hu, C.W.; Ji, Y.; Møller, P.; Rossner, P.; Cooke, M.S. Biomarkers of nucleic acid oxidation—A summary state-of-the-art. Redox Biol. 2021, 42, 101872. [Google Scholar] [CrossRef]
- Struthers, L.; Patel, R.; Clark, J.; Thomas, S. Direct detection of 8-oxodeoxyguanosine and 8-oxoguanine by avidin and its analogues. Anal. Biochem. 1998, 255, 20–31. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Zhou, J.; Cai, B. DNA is a target of the photodynamic effects elicited in A2E-laden RPE by blue-light illumination. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2245–2251. [Google Scholar] [CrossRef] [PubMed]
- Conners, R.; Hooley, E.; Clarke, A.R.; Thomas, S.; Brady, R.L. Recognition of oxidatively modified bases within the biotin-binding site of avidin. J. Mol. Biol. 2006, 357, 263–274. [Google Scholar] [CrossRef]
- Lohr, H.R.; Kuntchithapautham, K.; Sharma, A.K.; Rohrer, B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp. Eye Res. 2006, 83, 380–389. [Google Scholar] [CrossRef]
- Pow, D.V.; Crook, D.K. Immunocytochemical evidence for the presence of high levels of reduced glutathione in radial glial cells and horizontal cells in the rabbit retina. Neurosci. Lett. 1995, 193, 25–28. [Google Scholar] [CrossRef]
- Badiei, A.; Sudharsan, R.; Santana, E.; Dunaief, J.L.; Aguirre, G.D. Comparative localization of cystathionine beta synthases and cystathionine gamma lyase in canine, non-human primate and human retina. Exp. Eye Res. 2019, 181, 72–84. [Google Scholar] [CrossRef]
- Vlachantoni, D.; Bramall, A.N.; Murphy, M.P.; Taylor, R.W.; Shu, X.; Tulloch, B.; Van Veen, T.; Turnbull, D.M.; McInnes, R.R.; Wright, A.F. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Hum. Mol. Genet. 2011, 20, 322–335. [Google Scholar] [CrossRef]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Roesch, K.; Stadler, M.B.; Cepko, C.L. Gene expression changes within Müller glial cells in retinitis pigmentosa. Mol. Vis. 2012, 18, 1197–1214. [Google Scholar] [PubMed]
- Canto, A.; Martínez-González, J.; Miranda, M.; Olivar, T.; Almansa, I.; Hernández-Rabaza, V. Sulforaphane Modulates the Inflammation and Delays Neurodegeneration on a Retinitis Pigmentosa Mice Model. Front. Pharmacol. 2022, 13, 811257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Liu, S.L.; Hu, D.P.; Xing, Y.Q.; Shen, Y. N-methyl-N-nitrosourea-induced retinal degeneration in mice. Exp. Eye Res. 2014, 121, 102–113. [Google Scholar] [CrossRef]
- Zhao, T.; Li, Y.; Weng, C.; Yin, Z. The changes of potassium currents in RCS rat Müller cell during retinal degeneration. Brain Res. 2012, 1427, 78–87. [Google Scholar] [CrossRef]
- Alambiaga-Caravaca, A.M.; Cantó, A.; Rodilla, V.; Miranda, M.; López-Castellano, A. Topical Ocular Administration of Progesterone Decreases Photoreceptor Cell Death in Retinal Degeneration Slow (rds) Mice. Pharmaceuticals 2022, 15, 328. [Google Scholar] [CrossRef]
- Iandiev, I.; Biedermann, B.; Bringmann, A.; Reichel, M.B.; Reichenbach, A.; Pannicke, T. Atypical gliosis in Müller cells of the slowly degenerating rds mutant mouse retina. Exp. Eye Res. 2006, 82, 449–457. [Google Scholar] [CrossRef]
- Hughes, E.H.; Schlichtenbrede, F.C.; Murphy, C.C.; Sarra, G.M.; Luthert, P.J.; Ali, R.R.; Dick, A.D. Generation of activated sialoadhesin-positive microglia during retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2229–2234. [Google Scholar] [CrossRef]
- Peng, B.; Xiao, J.; Wang, K.; So, K.F.; Tipoe, G.L.; Lin, B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef]
- Dammak, A.; Huete-Toral, F.; Carpena-Torres, C.; Martin-Gil, A.; Pastrana, C.; Carracedo, G. From Oxidative Stress to Inflammation in the Posterior Ocular Diseases: Diagnosis and Treatment. Pharmaceutics 2021, 13, 1376. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.M.; Naash, M.I. Gene therapy for PRPH2-associated ocular disease: Challenges and prospects. Cold Spring Harb. Perspect. Med. 2014, 4, a017376. [Google Scholar] [CrossRef] [PubMed]
- Tebbe, L.; Kakakhel, M.; Makia, M.S.; Al-Ubaidi, M.R.; Naash, M.I. The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells 2020, 9, 784. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Abbas, S.N. Diabetes-induced mitochondrial dysfunction in the retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef]
- Mao, X.W.; Crapo, J.D.; Gridley, D.S. Mitochondrial oxidative stress-induced apoptosis and radioprotection in proton-irradiated rat retina. Radiat. Res. 2012, 178, 118–125. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Franklin, T.; Ghaley, N.; Yang, J.; Brands, M.W.; Caldwell, R.B.; Behzadian, M.A. Diabetes-induced superoxide anion and breakdown of the blood-retinal barrier: Role of the VEGF/uPAR pathway. PLoS ONE 2013, 8, e71868. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
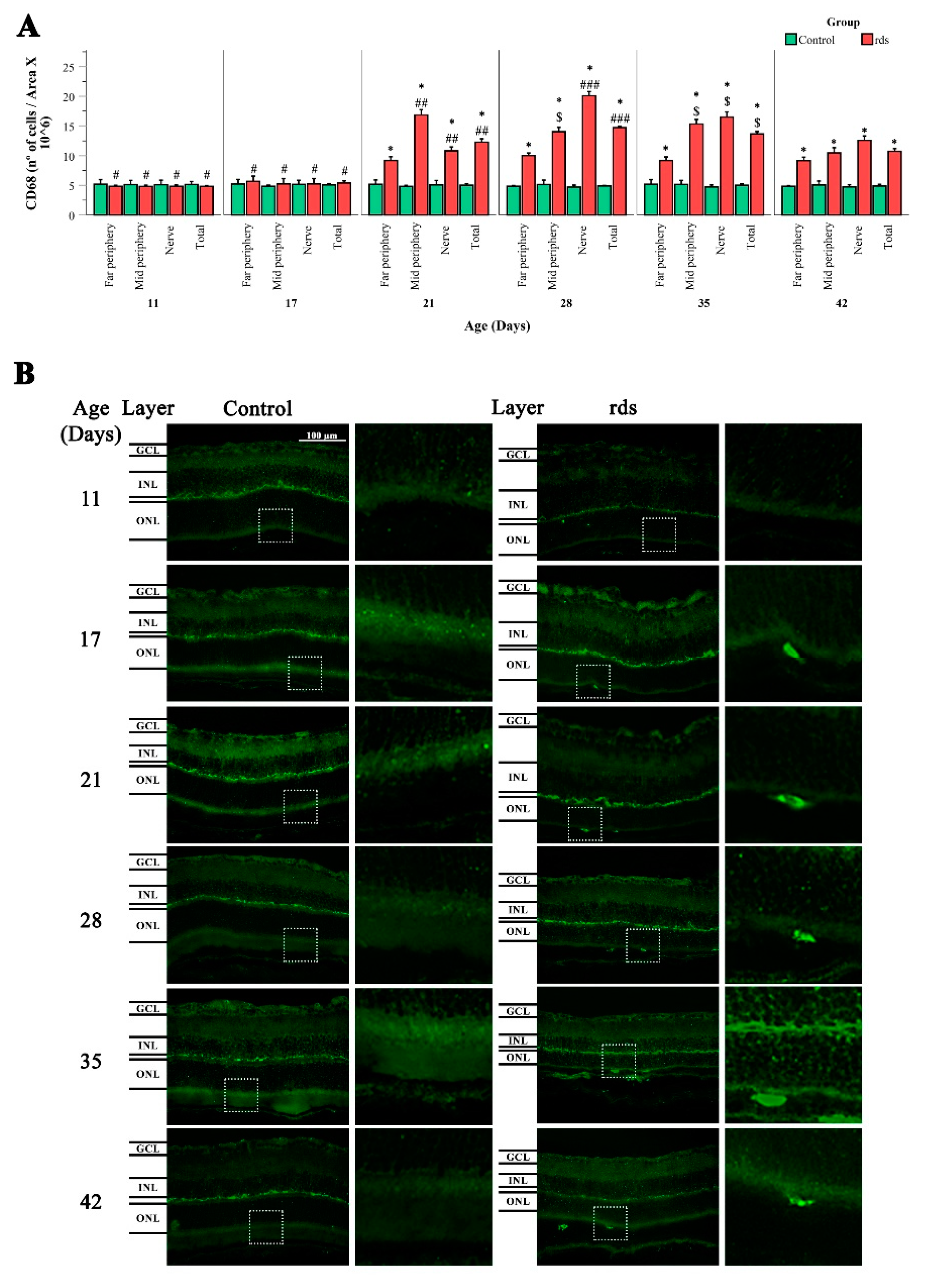{kind=link}
{kind=link}
| Antibody | Reference Number | Working Concentration |
|---|---|---|
| Alexa Fluor© (594) Conjugated Anti-Rhodopsin 4D1 | sc-57432 (Santa Cruz, CA, USA) | 1:100 |
| Anti-Cone Arrestin | AB15282 (Merck Millipore, Burlington, MA, USA) | 1:500 |
| Rabbit Anti-HNE Antiserum | HNE11-S (Alpha Diagnostic, San Antonio, TX, USA) | 1:100 |
| Texas Red Conjugated Avidin | A820 (Molecular Probes Inc., Eugene, OR, USA) | 1:200 |
| Rb X Glutathione | AB5010 (Merck Millipore) | 1:100 |
| Polyclonal Rabbit Anti-Glial Fibrillar Acidic Protein | Z0334 (Dako, Santa Clara, CA, USA) | 1:500 |
| Anti-Iba-1 Rabbit | 019-19741 (Wako, Wako, Japan) | 1:2000 |
| Rabbit Anti-CD68 | ab 125212 (Abcam, Cambridge, UK) | 1:500 |
| Alexa Fluor© 488 goat Anti-rabbit IgG | A11008 (Invitrogen, Waltham, MA, USA) | 1:200 |
| Antibody | Reference Number | Working Concentration |
|---|---|---|
| Rabbit Anti-GCLC | ab 53179 (Abcam) | 1:1000 |
| Rabbit Anti-GCLM | ab 124827 (Abcam) | 1:1000 |
| Rabbit Anti-SLC25A11 | ab 155196 (Abcam) | 1:1000 |
| F (ab′) 2–HRP goat Anti-rabbit | 31461 (Invitrogen) | 1:10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantó, A.; Martínez-González, J.; Almansa, I.; López-Pedrajas, R.; Hernández-Rabaza, V.; Olivar, T.; Miranda, M. Time-Course Changes in Oxidative Stress and Inflammation in the Retinas of rds Mice: A Retinitis Pigmentosa Model. Antioxidants 2022, 11, 1950. https://doi.org/10.3390/antiox11101950
Cantó A, Martínez-González J, Almansa I, López-Pedrajas R, Hernández-Rabaza V, Olivar T, Miranda M. Time-Course Changes in Oxidative Stress and Inflammation in the Retinas of rds Mice: A Retinitis Pigmentosa Model. Antioxidants. 2022; 11(10):1950. https://doi.org/10.3390/antiox11101950
Chicago/Turabian StyleCantó, Antolín, Javier Martínez-González, Inmaculada Almansa, Rosa López-Pedrajas, Vicente Hernández-Rabaza, Teresa Olivar, and María Miranda. 2022. "Time-Course Changes in Oxidative Stress and Inflammation in the Retinas of rds Mice: A Retinitis Pigmentosa Model" Antioxidants 11, no. 10: 1950. https://doi.org/10.3390/antiox11101950
APA StyleCantó, A., Martínez-González, J., Almansa, I., López-Pedrajas, R., Hernández-Rabaza, V., Olivar, T., & Miranda, M. (2022). Time-Course Changes in Oxidative Stress and Inflammation in the Retinas of rds Mice: A Retinitis Pigmentosa Model. Antioxidants, 11(10), 1950. https://doi.org/10.3390/antiox11101950