
A New Insight into an Alternative Therapeutic Approach to Restore Redox Homeostasis and Functional Mitochondria in Neurodegenerative Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxidative Stress and Redox Enzymes in Neurodegenerative Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
2.3. Amyotrophic Lateral Sclerosis
3. Mitochondrial Dysfunction in Neurodegenerative Diseases
4. Neuroinflammation in Neurodegenerative Diseases
5. Importance of Mitochondrial Biogenesis and Metabolic Regulation
6. Alteration in Mitochondrial Dynamics and the Fusion/Fission Cycle in Neurodegenerative Diseases
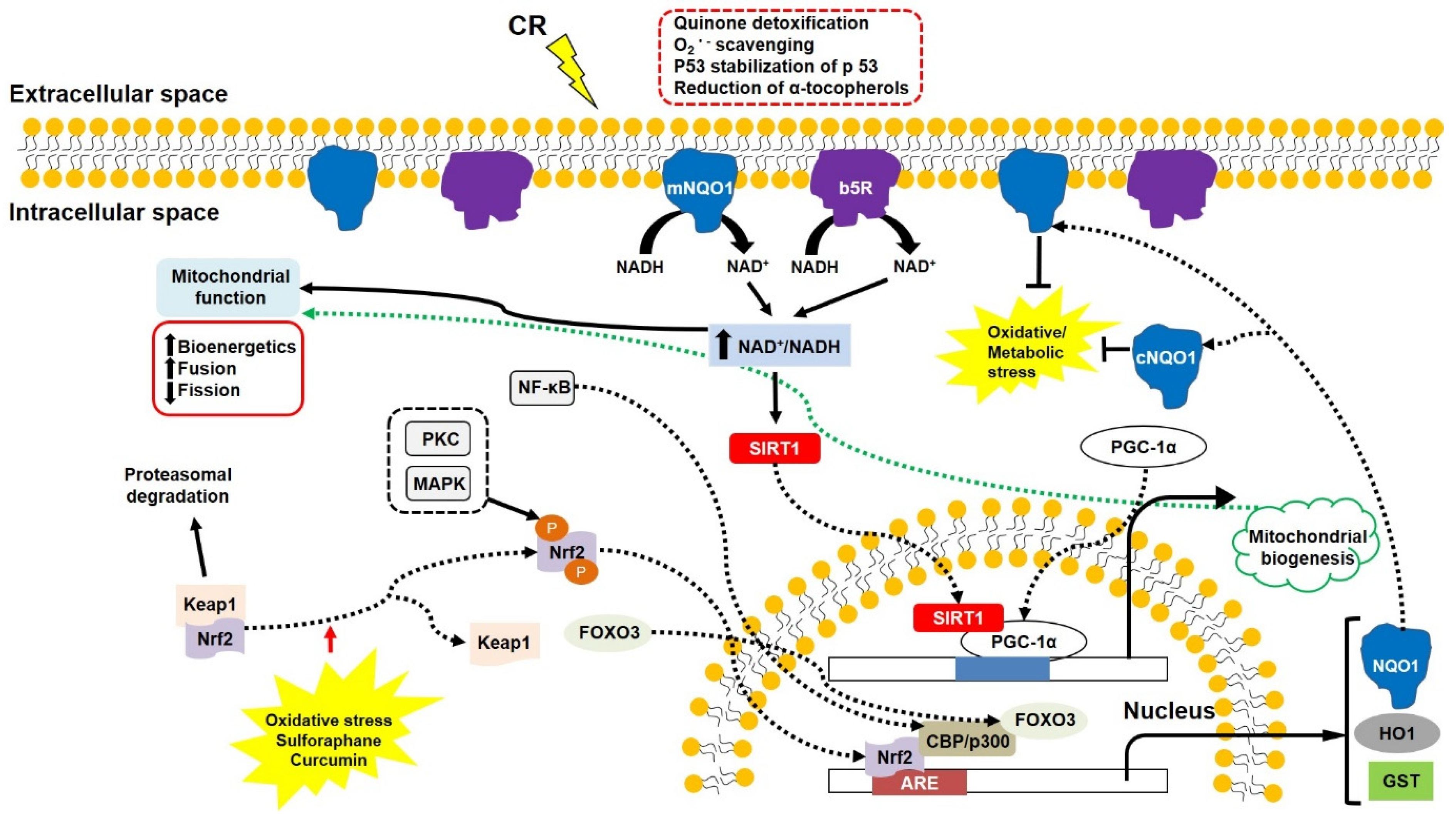
7. The New Compensatory Mechanism in Response to Mitochondrial Dysfunction
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Pomatto, L.C.D.; Davies, K.J.A. Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 2018, 124, 420–430. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, Z.; Cheng, J.; Lv, Z. A genetic program theory of aging using an RNA population model. Aging Res. Rev. 2014, 13, 46–54. [Google Scholar] [CrossRef]
- Barja, G. Towards a unified mechanistic theory of aging. Exp. Gerontol. 2019, 124, 110627. [Google Scholar] [CrossRef]
- Pirahanchi, Y.; Jessu, R.; Aeddula, N.R. Physiology, Sodium Potassium Pump. In StatPearls; Treasure Island: St. Petersburg, FL, USA, 2021. [Google Scholar]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimer’s Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.B. Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation. J. Inorg. Biochem. 2019, 194, 44–51. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Faget, K.Y. Zinc takes the center stage: Its paradoxical role in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2003, 41, 44–56. [Google Scholar] [CrossRef]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Guo, C.; Wang, P.; Zhong, M.L.; Wang, T.; Huang, X.S.; Li, J.Y.; Wang, Z.Y. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef]
- Guo, C.; Wang, T.; Zheng, W.; Shan, Z.Y.; Teng, W.P.; Wang, Z.Y. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 562–575. [Google Scholar] [CrossRef]
- Andreini, C.; Rosato, A.; Banci, L. The Relationship between Environmental Dioxygen and Iron-Sulfur Proteins Explored at the Genome Level. PLoS ONE 2017, 12, e0171279. [Google Scholar] [CrossRef]
- Beinert, H. Iron-sulfur proteins: Ancient structures, still full of surprises. J. Biol. Inorg. Chem. 2000, 5, 2–15. [Google Scholar] [CrossRef]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Ann. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Chiang, G.C.; Mao, X.; Kang, G.; Chang, E.; Pandya, S.; Vallabhajosula, S.; Isaacson, R.; Ravdin, L.D.; Alzheimer’s Disease Neuroimaging, I.; Shungu, D.C. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol. 2017, 38, 1130–1137. [Google Scholar] [CrossRef]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife 2020, 9, e56580. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Tapia, V.S.; White, C.S.; Daniels, M.J.D.; Drinkall, S.; Kennedy, P.T.; Spence, H.G.; Yu, S.; Green, J.P.; Hoyle, C.; et al. Zinc Status Alters Alzheimer’s Disease Progression through NLRP3-Dependent Inflammation. J. Neurosci. 2021, 41, 3025–3038. [Google Scholar] [CrossRef]
- Liu, Y.; Yoo, M.J.; Savonenko, A.; Stirling, W.; Price, D.L.; Borchelt, D.R.; Mamounas, L.; Lyons, W.E.; Blue, M.E.; Lee, M.K. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 13805–13814. [Google Scholar] [CrossRef]
- Pratico, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharm. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef]
- Jimenez-Jimenez, F.J.; de Bustos, F.; Molina, J.A.; Benito-Leon, J.; Tallon-Barranco, A.; Gasalla, T.; Orti-Pareja, M.; Guillamon, F.; Rubio, J.C.; Arenas, J.; et al. Cerebrospinal fluid levels of alpha-tocopherol (vitamin E) in Alzheimer’s disease. J. Neural Transm. 1997, 104, 703–710. [Google Scholar] [CrossRef]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51, 1562–1566. [Google Scholar] [CrossRef]
- Sultana, R.; Piroddi, M.; Galli, F.; Butterfield, D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res. 2008, 33, 2540–2546. [Google Scholar] [CrossRef]
- Boyd-Kimball, D.; Poon, H.F.; Lynn, B.C.; Cai, J.; Pierce, W.M., Jr.; Klein, J.B.; Ferguson, J.; Link, C.D.; Butterfield, D.A. Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1-42): Implications for Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1239–1249. [Google Scholar] [CrossRef]
- Opii, W.O.; Joshi, G.; Head, E.; Milgram, N.W.; Muggenburg, B.A.; Klein, J.B.; Pierce, W.M.; Cotman, C.W.; Butterfield, D.A. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: Relevance to Alzheimer’s disease. Neurobiol. Aging 2008, 29, 51–70. [Google Scholar] [CrossRef]
- Sultana, R.; Reed, T.; Perluigi, M.; Coccia, R.; Pierce, W.M.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: A regional study. J. Cell Mol. Med. 2007, 11, 839–851. [Google Scholar] [CrossRef]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J. Neural. Transm. Suppl. 2001, 61, 223–235. [Google Scholar]
- Poon, H.F.; Farr, S.A.; Banks, W.A.; Pierce, W.M.; Klein, J.B.; Morley, J.E.; Butterfield, D.A. Proteomic identification of less oxidized brain proteins in aged senescence-accelerated mice following administration of antisense oligonucleotide directed at the Abeta region of amyloid precursor protein. Brain Res. Mol. Brain Res. 2005, 138, 8–16. [Google Scholar] [CrossRef]
- Thancharoen, O.; Limwattananon, C.; Waleekhachonloet, O.; Rattanachotphanit, T.; Limwattananon, P.; Limpawattana, P. Ginkgo biloba Extract (EGb761), Cholinesterase Inhibitors, and Memantine for the Treatment of Mild-to-Moderate Alzheimer’s Disease: A Network Meta-Analysis. Drugs Aging 2019, 36, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Ashoor, H.M.; Soobiah, C.; Rios, P.; Veroniki, A.A.; Hamid, J.S.; Ivory, J.D.; Khan, P.A.; Yazdi, F.; Ghassemi, M.; et al. Comparative Effectiveness and Safety of Cognitive Enhancers for Treating Alzheimer’s Disease: Systematic Review and Network Metaanalysis. J. Am. Geriatr. Soc. 2018, 66, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Brundin, P. Parkinson’s disease and alpha synuclein: Is Parkinson’s disease a prion-like disorder? Mov. Disord. 2013, 28, 31–40. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Song, N.; Xie, J.; Jiang, H. Up-regulation of divalent metal transporter 1 is involved in 1-methyl-4-phenylpyridinium (MPP(+))-induced apoptosis in MES23.5 cells. Neurobiol. Aging 2009, 30, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Bastias-Candia, S.; Zolezzi, J.M.; Inestrosa, N.C. Revisiting the Paraquat-Induced Sporadic Parkinson’s Disease-Like Model. Mol. Neurobiol. 2019, 56, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Grunblatt, E.; Mandel, S.; Youdim, M.B. Neuroprotective strategies in Parkinson’s disease using the models of 6-hydroxydopamine and MPTP. Ann. N. Y. Acad. Sci. 2000, 899, 262–273. [Google Scholar] [CrossRef]
- Torres-Vega, A.; Pliego-Rivero, B.F.; Otero-Ojeda, G.A.; Gomez-Olivan, L.M.; Vieyra-Reyes, P. Limbic system pathologies associated with deficiencies and excesses of the trace elements iron, zinc, copper, and selenium. Nutr. Rev. 2012, 70, 679–692. [Google Scholar] [CrossRef]
- Bolognin, S.; Messori, L.; Zatta, P. Metal ion physiopathology in neurodegenerative disorders. Neuromol. Med. 2009, 11, 223–238. [Google Scholar] [CrossRef]
- Gaeta, A.; Hider, R.C. The crucial role of metal ions in neurodegeneration: The basis for a promising therapeutic strategy. Br. J. Pharmacol. 2005, 146, 1041–1059. [Google Scholar] [CrossRef] [PubMed]
- Zatta, P.; Lucchini, R.; van Rensburg, S.J.; Taylor, A. The role of metals in neurodegenerative processes: Aluminum, manganese, and zinc. Brain Res. Bull. 2003, 62, 15–28. [Google Scholar] [CrossRef]
- Carreras, M.C.; Franco, M.C.; Peralta, J.G.; Poderoso, J.J. Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol. Asp. Med. 2004, 25, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Sarti, P.; Arese, M.; Bacchi, A.; Barone, M.C.; Forte, E.; Mastronicola, D.; Brunori, M.; Giuffre, A. Nitric oxide and mitochondrial complex IV. IUBMB Life 2003, 55, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Reeve, A.; Meagher, M.; Lax, N.; Simcox, E.; Hepplewhite, P.; Jaros, E.; Turnbull, D. The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J. Neurosci. 2013, 33, 10790–10801. [Google Scholar] [CrossRef]
- Gupta, S.P.; Patel, S.; Yadav, S.; Singh, A.K.; Singh, S.; Singh, M.P. Involvement of nitric oxide in maneb- and paraquat-induced Parkinson’s disease phenotype in mouse: Is there any link with lipid peroxidation? Neurochem. Res. 2010, 35, 1206–1213. [Google Scholar] [CrossRef]
- Selvaraj, S.; Piramanayagam, S. Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef]
- Bras, I.C.; Dominguez-Meijide, A.; Gerhardt, E.; Koss, D.; Lazaro, D.F.; Santos, P.I.; Vasili, E.; Xylaki, M.; Outeiro, T.F. Synucleinopathies: Where we are and where we need to go. J. Neurochem. 2020, 153, 433–454. [Google Scholar] [CrossRef]
- Song, D.D.; Shults, C.W.; Sisk, A.; Rockenstein, E.; Masliah, E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 2004, 186, 158–172. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhu, M.; Wilson, M.A.; Petsko, G.A.; Fink, A.L. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J. Mol. Biol. 2006, 356, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Goncalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with alpha-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef]
- Kashihara, Y.; Terao, Y.; Yoda, K.; Hirota, T.; Kubota, T.; Kimura, M.; Matsuki, S.; Hirakawa, M.; Irie, S.; Ieiri, I. Effects of magnesium oxide on pharmacokinetics of L-dopa/carbidopa and assessment of pharmacodynamic changes by a model-based simulation. Eur. J. Clin. Pharmacol. 2019, 75, 351–361. [Google Scholar] [CrossRef]
- Schneider, F.; Erisson, L.; Beygi, H.; Bradbury, M.; Cohen-Barak, O.; Grachev, I.D.; Guzy, S.; Loupe, P.S.; Levi, M.; McDonald, M.; et al. Pharmacokinetics, metabolism and safety of deuterated L-DOPA (SD-1077)/carbidopa compared to L-DOPA/carbidopa following single oral dose administration in healthy subjects. Br. J. Clin. Pharmacol. 2018, 84, 2422–2432. [Google Scholar] [CrossRef]
- Bianchi, M.L.E.; Riboldazzi, G.; Mauri, M.; Versino, M. Efficacy of safinamide on non-motor symptoms in a cohort of patients affected by idiopathic Parkinson’s disease. Neurol. Sci. 2019, 40, 275–279. [Google Scholar] [CrossRef]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- De Leo, E.; Elmonem, M.A.; Berlingerio, S.P.; Berquez, M.; Festa, B.P.; Raso, R.; Bellomo, F.; Starborg, T.; Janssen, M.J.; Abbaszadeh, Z.; et al. Cell-Based Phenotypic Drug Screening Identifies Luteolin as Candidate Therapeutic for Nephropathic Cystinosis. J. Am. Soc. Nephrol. 2020, 31, 1522–1537. [Google Scholar] [CrossRef]
- Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Co-localization of Bunina bodies and TDP-43 inclusions in lower motor neurons in amyotrophic lateral sclerosis. Neuropathology 2014, 34, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [CrossRef]
- Gebicki, J.M. Oxidative stress, free radicals and protein peroxides. Arch. Biochem. Biophys. 2016, 595, 33–39. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Aging Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Lee, C.F.; Wei, Y.H. Role of reactive oxygen species-elicited apoptosis in the pathophysiology of mitochondrial and neurodegenerative diseases associated with mitochondrial DNA mutations. J. Formos. Med. Assoc. 2009, 108, 599–611. [Google Scholar] [CrossRef]
- Lee, H.C.; Pang, C.Y.; Hsu, H.S.; Wei, Y.H. Differential accumulations of 4,977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta 1994, 1226, 37–43. [Google Scholar] [CrossRef]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef]
- Wei, Y.H.; Pang, C.Y.; You, B.J.; Lee, H.C. Tandem duplications and large-scale deletions of mitochondrial DNA are early molecular events of human aging process. Ann. N. Y. Acad. Sci. 1996, 786, 82–101. [Google Scholar] [CrossRef]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef]
- Maruszak, A.; Gaweda-Walerych, K.; Soltyszewski, I.; Zekanowski, C. Mitochondrial DNA in pathogenesis of Alzheimer’s and Parkinson’s diseases. Acta Neurobiol. Exp. 2006, 66, 153–176. [Google Scholar]
- Zullo, S.J.; Cerritos, A.; Merril, C.R. Possible relationship between conditions associated with chronic hypoxia and brain mitochondrial DNA deletions; reduction of genomic 8-hydroxyguanine levels in human brain tissues containing elevated levels of the human mitochondrial DNA4977 deletion. Arch. Biochem. Biophys. 1999, 367, 140–142. [Google Scholar] [CrossRef]
- Dannenmann, B.; Lehle, S.; Hildebrand, D.G.; Kubler, A.; Grondona, P.; Schmid, V.; Holzer, K.; Froschl, M.; Essmann, F.; Rothfuss, O.; et al. High glutathione and glutathione peroxidase-2 levels mediate cell-type-specific DNA damage protection in human induced pluripotent stem cells. Stem. Cell Rep. 2015, 4, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin and the electron transport chain. Cell. Mol. Life Sci. 2017, 74, 3883–3896. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, G.; Kowaltowski, A.J.; Barros, M.H.; Netto, L.E. Glutathione and thioredoxin peroxidases mediate susceptibility of yeast mitochondria to Ca(2+)-induced damage. Arch. Biochem. Biophys. 2004, 425, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol. Res. 2012, 34, 297–303. [Google Scholar] [CrossRef]
- Onyango, I.G.; Khan, S.M.; Bennett, J.P., Jr. Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front. Biosci. 2017, 22, 854–872. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Gudz, T.I.; Migita, C.T.; Ikeda-Saito, M.; Hassan, M.O.; Turkaly, P.J.; Hoppel, C.L. Ischemic injury to mitochondrial electron transport in the aging heart: Damage to the iron-sulfur protein subunit of electron transport complex III. Arch. Biochem. Biophys. 2001, 385, 117–128. [Google Scholar] [CrossRef]
- Greco, M.; Villani, G.; Mazzucchelli, F.; Bresolin, N.; Papa, S.; Attardi, G. Marked aging-related decline in efficiency of oxidative phosphorylation in human skin fibroblasts. FASEB J. 2003, 17, 1706–1708. [Google Scholar] [CrossRef]
- Hagen, T.M.; Yowe, D.L.; Bartholomew, J.C.; Wehr, C.M.; Do, K.L.; Park, J.Y.; Ames, B.N. Mitochondrial decay in hepatocytes from old rats: Membrane potential declines, heterogeneity and oxidants increase. Proc. Natl. Acad. Sci. USA 1997, 94, 3064–3069. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Pallardo, F.V.; Pla, R.; Pellin, A.; Juan, G.; O’Connor, J.E.; Estrela, J.M.; Miquel, J.; Vina, J. Aging of the liver: Age-associated mitochondrial damage in intact hepatocytes. Hepatology 1996, 24, 1199–1205. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, A.; Venero, J.L.; Iwasawa, R.; Hankir, M.K.; Rahman, S.; Boobis, A.; Hajji, N. PGC-1alpha controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging 2015, 7, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Role of Immunity and Inflammation in the Pathophysiology of Neurodegenerative Diseases. Neurodegener. Dis. 2015, 15, 63–69. [Google Scholar] [CrossRef]
- Crutcher, K.A.; Gendelman, H.E.; Kipnis, J.; Perez-Polo, J.R.; Perry, V.H.; Popovich, P.G.; Weaver, L.C. Debate: “is increasing neuroinflammation beneficial for neural repair?”. J. Neuroimmune Pharmacol. 2006, 1, 195–211. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.D.; Melchior, B.; Masek, K.; Puntambekar, S.S.; Danielson, P.E.; Lo, D.D.; Sutcliffe, J.G.; Carson, M.J. Differential gene expression in LPS/IFNgamma activated microglia and macrophages: In vitro versus in vivo. J. Neurochem. 2009, 109, 117–125. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Strauss, S.; Bauer, J.; Ganter, U.; Jonas, U.; Berger, M.; Volk, B. Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer’s disease patients. Lab. Investig. 1992, 66, 223–230. [Google Scholar] [PubMed]
- Guo, J.T.; Yu, J.; Grass, D.; de Beer, F.C.; Kindy, M.S. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Willson, T.M.; Klockgether, T.; van Leuven, F.; Heneka, M.T. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2014, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Harms, A.S.; Delic, V.; Thome, A.D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A.B. Alpha-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol. Commun. 2017, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Sznejder-Pacholek, A.; Joniec-Maciejak, I.; Wawer, A.; Ciesielska, A.; Mirowska-Guzel, D. The effect of alpha-synuclein on gliosis and IL-1alpha, TNFalpha, IFNgamma, TGFbeta expression in murine brain. Pharmacol. Rep. 2017, 69, 242–251. [Google Scholar] [CrossRef]
- Earls, R.H.; Menees, K.B.; Chung, J.; Barber, J.; Gutekunst, C.A.; Hazim, M.G.; Lee, J.K. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflamm. 2019, 16, 250. [Google Scholar] [CrossRef]
- Na, S.J.; DiLella, A.G.; Lis, E.V.; Jones, K.; Levine, D.M.; Stone, D.J.; Hess, J.F. Molecular profiling of a 6-hydroxydopamine model of Parkinson’s disease. Neurochem. Res. 2010, 35, 761–772. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, J.S.; Park, J.Y.; Suh, Y.H.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. Astrogliosis in amyotrophic lateral sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics 2010, 7, 471–481. [Google Scholar] [CrossRef]
- Lasiene, J.; Yamanaka, K. Glial cells in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011, 2011, 718987. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Cozzolino, M.; Carri, M.T. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Redox (dys)Regulation. Antioxid. Redox Signal. 2018, 29, 15–36. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Canto, C.; Fan, W.; Downes, M.; Heligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.M.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.C.; Atangan, L.; Wang, M. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem. Biol. Drug Des. 2009, 74, 619–624. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Brasnyo, P.; Molnar, G.A.; Mohas, M.; Marko, L.; Laczy, B.; Cseh, J.; Mikolas, E.; Szijarto, I.A.; Merei, A.; Halmai, R.; et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br. J. Nutr. 2011, 106, 383–389. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Auwerx, J. Exploring the therapeutic space around NAD+. J. Cell Biol. 2012, 199, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H. Plasma membrane redox enzymes: New therapeutic targets for neurodegenerative diseases. Arch. Pharm. Res. 2019, 42, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Hyun, D.H.; Lee, G.H. Cytochrome b5 reductase, a plasma membrane redox enzyme, protects neuronal cells against metabolic and oxidative stress through maintaining redox state and bioenergetics. Age 2015, 37, 122. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.K.; Kim, H.K.; Mattson, M.P.; Hyun, D.H. Mitochondrial function in human neuroblastoma cells is up-regulated and protected by NQO1, a plasma membrane redox enzyme. PLoS ONE 2013, 8, e69030. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef]
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ottenberg, G.; Sferrazza, G.F.; Hubbs, C.; Fallahi, M.; Rumbaugh, G.; Brantley, A.F.; Lasmezas, C.I. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 2015, 138, 992–1008. [Google Scholar] [CrossRef]
- Willows, R.; Navaratnam, N.; Lima, A.; Read, J.; Carling, D. Effect of different gamma-subunit isoforms on the regulation of AMPK. Biochem. J. 2017, 474, 1741–1754. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Whitfield, J.; Janzen, N.R.; Belhaj, M.R.; Galic, S.; Murray-Segal, L.; Smiles, W.J.; Ling, N.X.Y.; Dite, T.A.; Scott, J.W.; et al. Genetic loss of AMPK-glycogen binding destabilises AMPK and disrupts metabolism. Mol. Metab. 2020, 41, 101048. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.A.; Pinkosky, S.L.; Filippov, S.; Hanselman, J.C.; Cramer, C.T.; Newton, R.S. AMP-activated protein kinase: An emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J. Lipid. Res. 2012, 53, 2490–2514. [Google Scholar] [CrossRef]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 2019, 8, e47866. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Ruegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef]
- Chae, S.; Ahn, B.Y.; Byun, K.; Cho, Y.M.; Yu, M.H.; Lee, B.; Hwang, D.; Park, K.S. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci. Signal 2013, 6, rs4. [Google Scholar] [CrossRef]
- Li, P.; Fan, W.; Xu, J.; Lu, M.; Yamamoto, H.; Auwerx, J.; Sears, D.D.; Talukdar, S.; Oh, D.; Chen, A.; et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 2011, 147, 815–826. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnaune-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Trefts, E.; Shaw, R.J. AMPK: Restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Zhang, X.; Wang, G.; Zhang, W.; Cai, Z.; Pan, B.S.; Gu, H.; Xu, C.; Jin, G.; Xu, X.; et al. Inositol serves as a natural inhibitor of mitochondrial fission by directly targeting AMPK. Mol. Cell 2021, 81, 3803–3819.e3807. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Magrane, J.; Hervias, I.; Henning, M.S.; Damiano, M.; Kawamata, H.; Manfredi, G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 2009, 18, 4552–4564. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef]
- Lee, J.J.; Sanchez-Martinez, A.; Martinez Zarate, A.; Beninca, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Tanaka, K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy 2010, 6, 952–954. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug. Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; E, S.Y.; Lamming, D.W.; et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef]
- Moreno, D.; Knecht, E.; Viollet, B.; Sanz, P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008, 582, 2650–2654. [Google Scholar] [CrossRef]
- Rocchi, S.; Picard, F.; Vamecq, J.; Gelman, L.; Potier, N.; Zeyer, D.; Dubuquoy, L.; Bac, P.; Champy, M.F.; Plunket, K.D.; et al. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol. Cell 2001, 8, 737–747. [Google Scholar] [CrossRef]
- Garza-Lombo, C.; Schroder, A.; Reyes-Reyes, E.M.; Franco, R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 2018, 8, 102–110. [Google Scholar] [CrossRef]
- Simula, L.; Nazio, F.; Campello, S. The mitochondrial dynamics in cancer and immune-surveillance. Semin. Cancer Biol. 2017, 47, 29–42. [Google Scholar] [CrossRef]
- Stoker, M.L.; Newport, E.; Hulit, J.C.; West, A.P.; Morten, K.J. Impact of pharmacological agents on mitochondrial function: A growing opportunity? Biochem. Soc. Trans. 2019, 47, 1757–1772. [Google Scholar] [CrossRef]
- Kitami, T.; Logan, D.J.; Negri, J.; Hasaka, T.; Tolliday, N.J.; Carpenter, A.E.; Spiegelman, B.M.; Mootha, V.K. A chemical screen probing the relationship between mitochondrial content and cell size. PLoS ONE 2012, 7, e33755. [Google Scholar] [CrossRef] [PubMed]
- Piechota, J.; Szczesny, R.; Wolanin, K.; Chlebowski, A.; Bartnik, E. Nuclear and mitochondrial genome responses in HeLa cells treated with inhibitors of mitochondrial DNA expression. Acta Biochim. Pol. 2006, 53, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Heller, S.; Loffler, B.; Schafer, I.; Seibel, M.; Villani, G.; Seibel, P. Generation of Rho Zero Cells: Visualization and Quantification of the mtDNA Depletion Process. Int. J. Mol. Sci. 2015, 16, 9850–9865. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H.; Hunt, N.D.; Emerson, S.S.; Hernandez, J.O.; Mattson, M.P.; de Cabo, R. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J. Neurochem. 2007, 100, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L.; Navas, P.; Low, H.; Sun, I.L.; de Cabo, R. Sirtuin activation: A role for plasma membrane in the cell growth puzzle. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 368–370. [Google Scholar] [CrossRef][Green Version]
- Lenaz, G.; Paolucci, U.; Fato, R.; D’Aurelio, M.; Parenti Castelli, G.; Sgarbi, G.; Biagini, G.; Ragni, L.; Salardi, S.; Cacciari, E. Enhanced activity of the plasma membrane oxidoreductase in circulating lymphocytes from insulin-dependent diabetes mellitus patients. Biochem. Biophys. Res. Commun. 2002, 290, 1589–1592. [Google Scholar] [CrossRef]
- Rodriguez-Aguilera, J.C.; Lopez-Lluch, G.; Santos-Ocana, C.; Villalba, J.M.; Gomez-Diaz, C.; Navas, P. Plasma membrane redox system protects cells against oxidative stress. Redox. Rep. 2000, 5, 148–150. [Google Scholar] [CrossRef][Green Version]
- Villalba, J.M.; Navas, P. Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid. Redox. Signal 2000, 2, 213–230. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Smith, J. Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol. 2002, 12, 404–406. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Torres-Lista, V.; Parrado-Fernandez, C.; Alvarez-Monton, I.; Frontinan-Rubio, J.; Duran-Prado, M.; Peinado, J.R.; Johansson, B.; Alcain, F.J.; Gimenez-Llort, L. Neophobia, NQO1 and SIRT1 as premorbid and prodromal indicators of AD in 3xTg-AD mice. Behav. Brain Res. 2014, 271, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Yi, Q.; Wang, W. Association between NQO1 C609T polymorphism and bladder cancer susceptibility: A systemic review and meta-analysis. Tumour. Biol. 2013, 34, 2551–2556. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Medina-Carmona, E.; Timson, D.J. Natural Small Molecules as Stabilizers and Activators of Cancer-Associated NQO1 Polymorphisms. Curr. Drug Targets 2016, 17, 1506–1514. [Google Scholar] [CrossRef]
- Bian, J.T.; Zhao, H.L.; Zhang, Z.X.; Bi, X.H.; Zhang, J.W. Association of NAD(P)H:quinone oxidoreductase 1 polymorphism and Alzheimer’s disease in Chinese. J. Mol. Neurosci. 2008, 34, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P. Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer disease. J. Alzheimers Dis. 2002, 4, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Nakanishi, M.; Hamato, F.; Sasaki, K.; Takahashi, S. Concentrations of alpha-tocopherol and its quinone derivative in cerebrospinal fluid from patients with vascular dementia of the Binswanger type and Alzheimer type dementia. Neurosci. Lett. 1994, 174, 73–76. [Google Scholar] [CrossRef]
- Hyun, D.H.; Mughal, M.R.; Yang, H.; Lee, J.H.; Ko, E.J.; Hunt, N.D.; de Cabo, R.; Mattson, M.P. The plasma membrane redox system is impaired by amyloid beta-peptide and in the hippocampus and cerebral cortex of 3xTgAD mice. Exp. Neurol. 2010, 225, 423–429. [Google Scholar] [CrossRef]
- De Cabo, R.; Cabello, R.; Rios, M.; Lopez-Lluch, G.; Ingram, D.K.; Lane, M.A.; Navas, P. Calorie restriction attenuates age-related alterations in the plasma membrane antioxidant system in rat liver. Exp. Gerontol. 2004, 39, 297–304. [Google Scholar] [CrossRef]
- Hyun, D.H.; Emerson, S.S.; Jo, D.G.; Mattson, M.P.; de Cabo, R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc. Natl. Acad. Sci. USA 2006, 103, 19908–19912. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Sun, Y.; Diaz-Ruiz, A.; Ali, A.; Gutierrez, V.; Palacios, H.H.; Curtis, J.; Siendones, E.; Ariza, J.; Abulwerdi, G.A.; et al. Cytochrome b5 reductase and the control of lipid metabolism and healthspan. NPJ Aging Mech. Dis. 2016, 2, 16006. [Google Scholar] [CrossRef]
- Gong, X.; Kole, L.; Iskander, K.; Jaiswal, A.K. NRH:quinone oxidoreductase 2 and NAD(P)H:quinone oxidoreductase 1 protect tumor suppressor p53 against 20s proteasomal degradation leading to stabilization and activation of p53. Cancer Res. 2007, 67, 5380–5388. [Google Scholar] [CrossRef]
- Jaber, S.; Polster, B.M. Idebenone and neuroprotection: Antioxidant, pro-oxidant, or electron carrier? J. Bioenerg. Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Aging Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Li, X.; Li, L.; Zhang, H. Theaflavins alleviate sevoflurane-induced neurocytotoxicity via Nrf2 signaling pathway. Int. J. Neurosci. 2020, 130, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Masutani, H.; Kim, Y.C.; Nishikawa, M.; Ohira, A.; Yodoi, J. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, A.Y.; Simonyi, A.; Jensen, M.D.; Shelat, P.B.; Rottinghaus, G.E.; MacDonald, R.S.; Miller, D.K.; Lubahn, D.E.; Weisman, G.A.; et al. Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J. Neurosci. Res. 2005, 82, 138–148. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Messina, E.; Scafidi, V.; Di Carlo, M. Curcumin induces apoptosis in human neuroblastoma cells via inhibition of AKT and Foxo3a nuclear translocation. Free Radic. Res. 2014, 48, 1397–1408. [Google Scholar] [CrossRef]
- Oi, Y.; Kawada, T.; Shishido, C.; Wada, K.; Kominato, Y.; Nishimura, S.; Ariga, T.; Iwai, K. Allyl-containing sulfides in garlic increase uncoupling protein content in brown adipose tissue, and noradrenaline and adrenaline secretion in rats. J. Nutr. 1999, 129, 336–342. [Google Scholar] [CrossRef]
- Chen, C.; Pung, D.; Leong, V.; Hebbar, V.; Shen, G.; Nair, S.; Li, W.; Kong, A.N. Induction of detoxifying enzymes by garlic organosulfur compounds through transcription factor Nrf2: Effect of chemical structure and stress signals. Free Radic. Biol. Med. 2004, 37, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Son, T.G.; Jo, D.G.; Kim, D.C.; Hyun, D.H. Cytotoxicity of lipid-soluble ginseng extracts is attenuated by plasma membrane redox enzyme NQO1 through maintaining redox homeostasis and delaying apoptosis in human neuroblastoma cells. Arch. Pharm. Res. 2016, 39, 1339–1348. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyun, D.-H.; Lee, J. A New Insight into an Alternative Therapeutic Approach to Restore Redox Homeostasis and Functional Mitochondria in Neurodegenerative Diseases. Antioxidants 2022, 11, 7. https://doi.org/10.3390/antiox11010007
Hyun D-H, Lee J. A New Insight into an Alternative Therapeutic Approach to Restore Redox Homeostasis and Functional Mitochondria in Neurodegenerative Diseases. Antioxidants. 2022; 11(1):7. https://doi.org/10.3390/antiox11010007
Chicago/Turabian StyleHyun, Dong-Hoon, and Jaewang Lee. 2022. "A New Insight into an Alternative Therapeutic Approach to Restore Redox Homeostasis and Functional Mitochondria in Neurodegenerative Diseases" Antioxidants 11, no. 1: 7. https://doi.org/10.3390/antiox11010007
APA StyleHyun, D.-H., & Lee, J. (2022). A New Insight into an Alternative Therapeutic Approach to Restore Redox Homeostasis and Functional Mitochondria in Neurodegenerative Diseases. Antioxidants, 11(1), 7. https://doi.org/10.3390/antiox11010007