
Pathophysiology of Lipid Droplets in Neuroglia



Abstract
:1. Introduction
2. Lipid Droplets in Astrocytes
2.1. Lipid Droplet Size and Sub-Cellular Localization in Astrocytes

{kind=link}
{kind=link}
| Lipid Transporter | Transporter Subtype | Function | |
|---|---|---|---|
| Astrocyte | Neuron | ||
| Fatty acid transport protein (FATP/SLC27) | FATP1/4 | Extracellular FFA uptake [7]. | Transport of neuronal de novo synthesized FFA to astrocytes [7]. |
| Fatty acid-binding protein (FABP) | FABP7 | Binding and internalization of long-chain FFA [48]. Protection from ROS toxicity through induction of LD accumulation [48]. Regulation of dendritic arbor growth, neuronal excitatory synapse formation, and synaptic transmission [49]. | / |
| Fatty acid translocase (FAT/CD36) | FAT | Increased expression upon treatment with amyloid-β [50]. Mediator of stroke-induced astrocyte activation and scar formation [51]. | Long-chain FA transport [52,53]. Uptake of saturated and unsaturated long-chain FA in glucosensing neurons of ventromedial hypothalamus [53]. |
| Apolipoprotein (Apo) | ApoE | FA transport from neurons to astrocytes [7,8]. Transport of toxic peroxidized FA from hyperactive neurons to astrocytes [8]. Release of (very-) long-chain saturated FAs from reactive astrocytes [54]. | Transport of neuronal de novo synthesized FFA to astrocytes [7] |
| ApoD | FA transport from neurons to astrocytes [7,8]. | Transport of neuronal de novo synthesized FFA to astrocytes [7]. | |
| ApoJ | Release of (very-)long-chain saturated FAs from reactive astrocytes [54]. | n.d. | |
| ATP-binding cassette transporter (ABC transporter) | ABCA1 | Export of cholesterol-transporting ApoE particles from astrocytes [55]. | Cholesterol efflux via ApoE particles from neurons [55,56] |
| Low-density lipoprotein receptor (LDL receptor) | LDL receptor | Uptake of cholesterol-transporting ApoE particles [55]. | Uptake of cholesterol-transporting ApoE particles [55]. |
| Low-density lipoprotein receptor-related protein 1 | LRP1 | n.d. | Uptake of cholesterol-containing ApoE particles [55]. |
2.2. Mobility of Lipid Droplets in Astrocytes
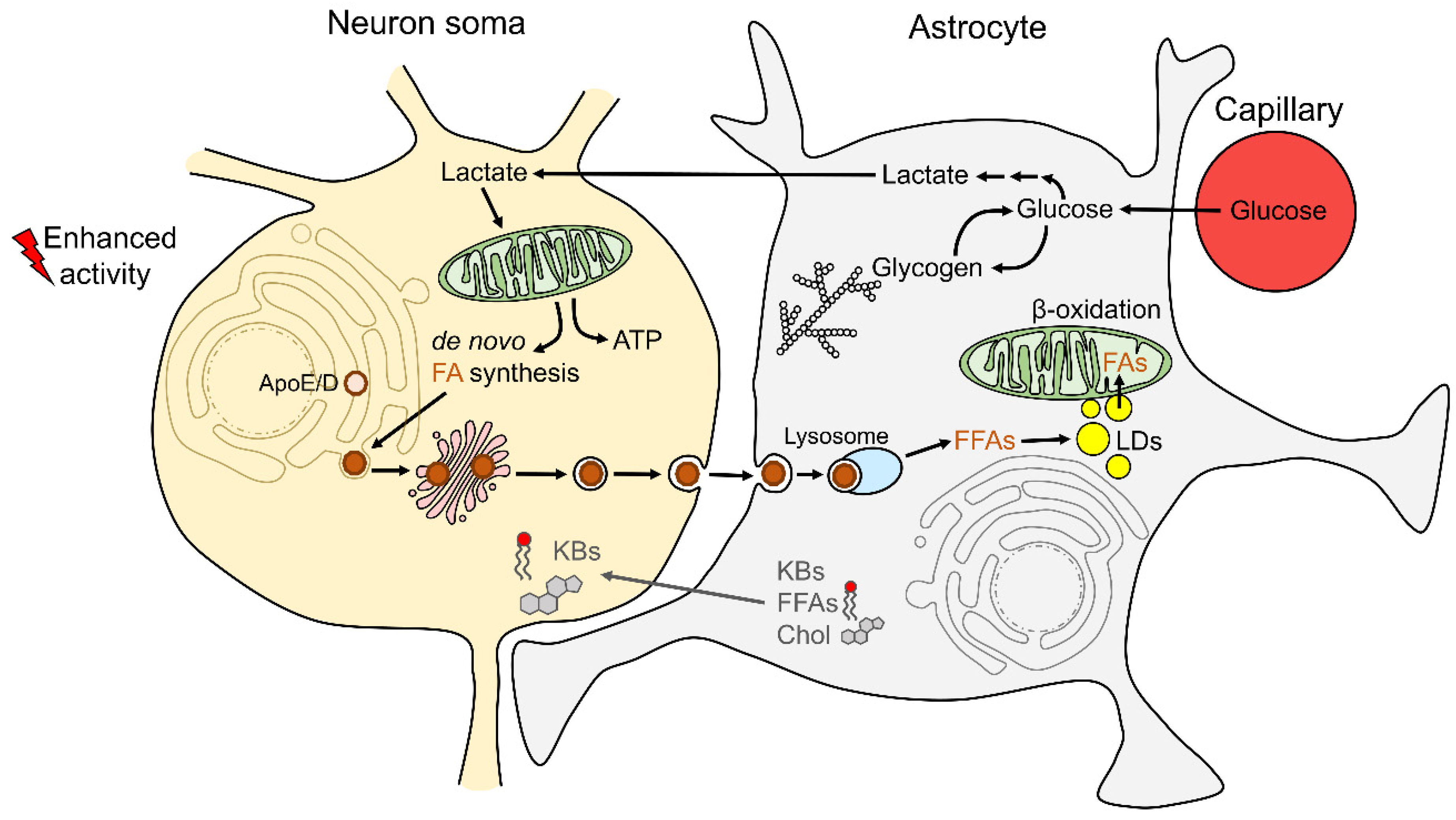
2.3. Astrocyte–Neuron Metabolic Coupling and Lipid Droplet Metabolism in Astrocytes
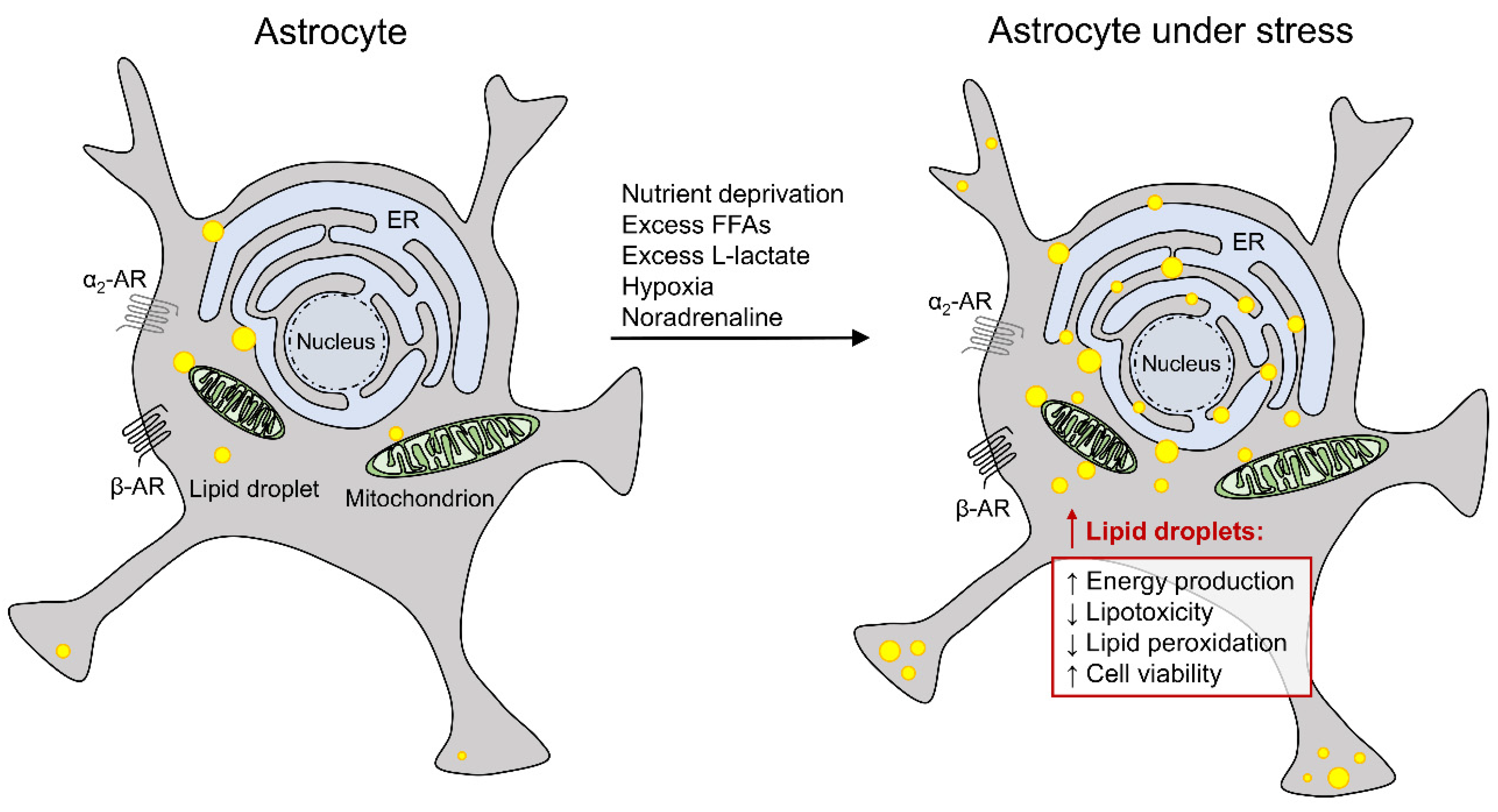
2.4. Lipid Droplets in Astrocytes and Nutrient Deprivation
2.5. Lipid Droplets in Astrocytes and Excess Extracellular Free Fatty Acids
2.6. Lipid Droplets in Astrocytes and Hypoxic Stress
2.7. Lipid Droplets in Astrocytes and Adrenergic Activation
3. Lipid Droplets in Microglia
4. Lipid Droplets in Oligodendroglia
5. Lipid Droplets in Ependymal Cells
6. Lipid Droplets in Neuroglia in Aging and Neurologic Disorders
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13c nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. BioMed Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [Green Version]
- Barber, C.N.; Raben, D.M. Lipid metabolism crosstalk in the brain: Glia and neurons. Front. Cell. Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ros induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The glia-neuron lactate shuttle and elevated ros promote lipid synthesis in neurons and lipid droplet accumulation in glia via apoe/d. Cell Metab. 2017, 26, 719–737.e716. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 2019, 177, 1522–1535.e1514. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020. [Google Scholar] [CrossRef]
- Shimabukuro, M.K.; Langhi, L.G.P.; Cordeiro, I.; Brito, J.M.; de Batista, C.M.C.; Mattson, M.P.; de Mello Coelho, V. Lipid-laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci. Rep. 2016, 6, 23795. [Google Scholar] [CrossRef] [Green Version]
- Kis, V.; Barti, B.; Lippai, M.; Sass, M. Specialized cortex glial cells accumulate lipid droplets in drosophila melanogaster. PLoS ONE 2015, 10, e0131250. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell. Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Farese, R.V., Jr.; Walther, T.C. Lipid droplets finally get a little respect. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarc, E.; Petan, T. Lipid droplets and the management of cellular stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farese, R.V., Jr. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kory, N.; Farese, R.V., Jr.; Walther, T.C. Targeting fat: Mechanisms of protein localization to lipid droplets. Trends Cell Biol. 2016, 26, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell. 2018, 44, 97–112.e117. [Google Scholar] [CrossRef]
- Buhman, K.K.; Chen, H.C.; Farese, R.V., Jr. The enzymes of neutral lipid synthesis. J. Biol. Chem. 2001, 276, 40369–40372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pol, A.; Gross, S.P.; Parton, R.G. Review: Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell. Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. Fat signals—Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism*. J. Biol. Chem. 2006, 281, 40236–40241. [Google Scholar] [CrossRef] [Green Version]
- Taschler, U.; Radner, F.P.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [Green Version]
- Etschmaier, K.; Becker, T.; Eichmann, T.O.; Schweinzer, C.; Scholler, M.; Tam-Amersdorfer, C.; Poeckl, M.; Schuligoi, R.; Kober, A.; Chirackal Manavalan, A.P.; et al. Adipose triglyceride lipase affects triacylglycerol metabolism at brain barriers. J. Neurochem. 2011, 119, 1016–1028. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [Green Version]
- Khatchadourian, A.; Bourque, S.D.; Richard, V.R.; Titorenko, V.I.; Maysinger, D. Dynamics and regulation of lipid droplet formation in lipopolysaccharide (lps)-stimulated microglia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An english translation of Alzheimer’s 1907 paper, “Uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [PubMed]
- Alzheimer, A. Uber eine eigenartige erkrankung der hirnrinde. Z. Psychiatr. Psych.-Gerichtl. Med. 1907, 18, 177–179. [Google Scholar]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Does shuttling of glycogen-derived lactate from astrocytes to neurons take place during neurotransmission and memory consolidation? J. Neurosci. Res. 2019, 97, 863–882. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef] [Green Version]
- Smolič, T.; Tavčar, P.; Horvat, A.; Černe, U.; Halužan Vasle, A.; Tratnjek, L.; Kreft, M.E.; Scholz, N.; Matis, M.; Petan, T.; et al. Astrocytes in stress accumulate lipid droplets. Glia 2021, 69, 1540–1562. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef]
- Rinholm, J.E.; Bergersen, L.H. White matter lactate—Does it matter? Neuroscience 2014, 276, 109–116. [Google Scholar] [CrossRef]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [Green Version]
- Waitt, A.E.; Reed, L.; Ransom, B.R.; Brown, A.M. Emerging roles for glycogen in the CNS. Front. Mol. Neurosci. 2017, 10, 73. [Google Scholar] [CrossRef]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Koh, H.C.; Nielsen, J.; Saltin, B.; Holmberg, H.-C.; Ortenblad, N. Pronounced limb and fibre type differences in subcellular lipid droplet content and distribution in elite skiers before and after exhaustive exercise. J. Physiol. 2017, 595, 5781–5795. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Christensen, A.E.; Nellemann, B.; Christensen, B. Lipid droplet size and location in human skeletal muscle fibers are associated with insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E721–E730. [Google Scholar] [CrossRef] [Green Version]
- Zietkowski, D.; Payne, G.S.; Nagy, E.; Mobberley, M.A.; Ryder, T.A.; de Souza, N.M. Comparison of nmr lipid profiles in mitotic arrest and apoptosis as indicators of paclitaxel resistance in cervical cell lines. Magn. Reason. Med. 2012, 68, 369–377. [Google Scholar] [CrossRef]
- Suzuki, M.; Shinohara, Y.; Ohsaki, Y.; Fujimoto, T. Lipid droplets: Size matters. J. Electron. Microsc. 2011, 60, S101–S116. [Google Scholar] [CrossRef]
- Islam, A.; Kagawa, Y.; Miyazaki, H.; Shil, S.K.; Umaru, B.A.; Yasumoto, Y.; Yamamoto, Y.; Owada, Y. Fabp7 protects astrocytes against ros toxicity via lipid droplet formation. Mol. Neurobiol. 2019, 56, 5763–5779. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Yamamoto, Y.; Sharifi, K.; Kida, H.; Kagawa, Y.; Yasumoto, Y.; Islam, A.; Miyazaki, H.; Shimamoto, C.; Maekawa, M.; et al. Astrocyte-expressed fabp7 regulates dendritic morphology and excitatory synaptic function of cortical neurons. Glia 2016, 64, 48–62. [Google Scholar] [CrossRef]
- Jones, R.S.; Minogue, A.M.; Connor, T.J.; Lynch, M.A. Amyloid-β-induced astrocytic phagocytosis is mediated by cd36, cd47 and rage. J. Neuroimmune Pharmacol. 2013, 8, 301–311. [Google Scholar] [CrossRef]
- Bao, Y.; Qin, L.; Kim, E.; Bhosle, S.; Guo, H.; Febbraio, M.; Haskew-Layton, R.E.; Ratan, R.; Cho, S. Cd36 is involved in astrocyte activation and astroglial scar formation. J. Cereb. Blood Flow Metab. 2012, 32, 1567–1577. [Google Scholar] [CrossRef]
- Le Foll, C.; Irani, B.G.; Magnan, C.; Dunn-Meynell, A.A.; Levin, B.E. Characteristics and mechanisms of hypothalamic neuronal fatty acid sensing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R655–R664. [Google Scholar] [CrossRef] [Green Version]
- Le Foll, C.; Dunn-Meynell, A.; Musatov, S.; Magnan, C.; Levin, B.E. Fat/cd36: A major regulator of neuronal fatty acid sensing and energy homeostasis in rats and mice. Diabetes 2013, 62, 2709–2716. [Google Scholar] [CrossRef] [Green Version]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Minagawa, H.; Gong, J.-S.; Jung, C.-G.; Watanabe, A.; Lund-Katz, S.; Phillips, M.C.; Saito, H.; Michikawa, M. Mechanism underlying apolipoprotein e (apoe) isoform-dependent lipid efflux from neural cells in culture. J. Neurosci. Res. 2009, 87, 2498–2508. [Google Scholar] [CrossRef] [Green Version]
- Blanchette-Mackie, E.J.; Scow, R.O. Movement of lipolytic products to mitochondria in brown adipose tissue of young rats: An electron microscope study. J. Lipid Res. 1983, 24, 229–244. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Wolins, N.E. Packaging of fat: An evolving model of lipid droplet assembly and expansion. J. Biol. Chem. 2012, 287, 2273–2279. [Google Scholar] [CrossRef] [Green Version]
- Freyre, C.A.C.; Rauher, P.C.; Ejsing, C.S.; Klemm, R.W. Miga2 links mitochondria, the er, and lipid droplets and promotes de novo lipogenesis in adipocytes. Mol. Cell 2019, 76, 811–825.e814. [Google Scholar] [CrossRef]
- Novikoff, A.B.; Novikoff, P.M.; Rosen, O.M.; Rubin, C.S. Organelle relationships in cultured 3t3-l1 preadipocytes. J. Cell Biol. 1980, 87, 180–196. [Google Scholar] [CrossRef] [Green Version]
- Herms, A.; Bosch, M.; Reddy, B.J.; Schieber, N.L.; Fajardo, A.; Ruperez, C.; Fernandez-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. Ampk activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Goodman, J.M. The lipid droplet-a well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, J.; Ha, C.W.; Zhang, S.; Jung, J.P.; Huh, W.K.; Liu, P. Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell 2011, 2, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 2018, 27, 869–885.e866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petan, T. Lipid droplets in cancer. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Robenek, H.; Hofnagel, O.; Buers, I.; Robenek, M.J.; Troyer, D.; Severs, N.J. Adipophilin-enriched domains in the er membrane are sites of lipid droplet biogenesis. J. Cell Sci. 2006, 119, 4215–4224. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Farese, R.V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [Green Version]
- Hugenroth, M.; Bohnert, M. Come a little bit closer! Lipid droplet-er contact sites are getting crowded. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118603. [Google Scholar] [CrossRef]
- Renne, M.F.; Klug, Y.A.; Carvalho, P. Lipid droplet biogenesis: A mystery “unmixing”? Semin. Cell Dev. Biol. 2020, 108, 14–23. [Google Scholar] [CrossRef]
- Kilwein, M.D.; Welte, M.A. Lipid droplet motility and organelle contacts. Contact 2019, 2, 2515256419895688. [Google Scholar] [CrossRef] [Green Version]
- Welte, M.A. Fat on the move: Intracellular motion of lipid droplets. Biochem. Soc. Trans. 2009, 37, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Potokar, M.; Vardjan, N.; Stenovec, M.; Gabrijel, M.; Trkov, S.; Jorgačevski, J.; Kreft, M.; Zorec, R. Astrocytic vesicle mobility in health and disease. Int. J. Mol. Sci. 2013, 14, 11238–11258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potokar, M.; Kreft, M.; Pangrsic, T.; Zorec, R. Vesicle mobility studied in cultured astrocytes. Biochem. Biophys. Res. Commun. 2005, 329, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M. Tethering fat: Tethers in lipid droplet contact sites. Contact 2020, 3, 251525642090814. [Google Scholar] [CrossRef] [Green Version]
- Falchi, A.M.; Sogos, V.; Saba, F.; Piras, M.; Congiu, T.; Piludu, M. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and atp. Histochem. Cell Biol. 2013, 139, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Fink, K.; Velebit, J.; Vardjan, N.; Zorec, R.; Kreft, M. Noradrenaline-induced l-lactate production requires d-glucose entry and transit through the glycogen shunt in single-cultured rat astrocytes. J. Neurosci. Res. 2021, 99, 1084–1098. [Google Scholar] [CrossRef]
- Sotelo-Hitschfeld, T.; Niemeyer, M.I.; Mächler, P.; Ruminot, I.; Lerchundi, R.; Wyss, M.T.; Stobart, J.; Fernández-Moncada, I.; Valdebenito, R.; Garrido-Gerter, P.; et al. Channel-mediated lactate release by k⁺-stimulated astrocytes. J. Neurosci. 2015, 35, 4168–4178. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Gotoh, M.; Fukasawa, K.; Murakami-Murofushi, K.; Kunugi, H. Oleic acid is a potent inducer for lipid droplet accumulation through its esterification to glycerol by diacylglycerol acyltransferase in primary cortical astrocytes. Brain Res. 2019, 1725, 146484. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following nmda receptor activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The sources of reactive oxygen species and its possible role in the pathogenesis of parkinson’s disease. Parkinson’s Dis. 2018, 2018, 9163040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Gutiérrez, C.; Jiménez-Blasco, D.; Quintana-Cabrera, R. Intertwined ros and metabolic signaling at the neuron-astrocyte interface. Neurochem. Res. 2021, 46, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ros in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Belanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid processing in the brain: A key regulator of systemic metabolism. Front. Endocrinol. 2017, 8, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, V.; Frasson, D.; Kawashita, N. Several agents and pathways regulate lipolysis in adipocytes. Biochimie 2011, 93, 1631–1640. [Google Scholar] [CrossRef]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Garcia Corrales, A.V.; Haidar, M.; Bogie, J.F.J.; Hendriks, J.J.A. Fatty acid synthesis in glial cells of the cns. Int. J. Mol. Sci. 2021, 22, 8159. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Kim, H.Y. Novel metabolism of docosahexaenoic acid in neural cells. J. Biol. Chem. 2007, 282, 18661–18665. [Google Scholar] [CrossRef] [Green Version]
- Ralhan, I.; Chang, C.L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid droplets in the nervous system. J. Cell Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef] [PubMed]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. Cns synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Hall, B.; Allsop, J.; Alqarni, R.; Allen, S.P. Lipid metabolism in astrocytic structure and function. Semin. Cell Dev. Biol. 2021, 112, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8786. [Google Scholar] [CrossRef]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Reese, M.L.; Goodman, J.M. The assembly of lipid droplets and their roles in challenged cells. EMBO J. 2018, 37, e98947. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubern, A.; Barcelo-Torns, M.; Casas, J.; Barneda, D.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Lipid droplet biogenesis induced by stress involves triacylglycerol synthesis that depends on group via phospholipase a2. J. Biol. Chem. 2009, 284, 5697–5708. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. Dgat1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev. Cell 2017, 42, 9–21.e25. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, M.; Blázquez, C. Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 287–292. [Google Scholar] [CrossRef]
- Guzmán, M.; Blázquez, C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]
- Rohwedder, A.; Zhang, Q.; Rudge, S.A.; Wakelam, M.J. Lipid droplet formation in response to oleic acid in huh-7 cells is mediated by the fatty acid receptor ffar4. J. Cell Sci. 2014, 127, 3104–3115. [Google Scholar] [PubMed] [Green Version]
- Tremblay, M.E.; Zhang, I.; Bisht, K.; Savage, J.C.; Lecours, C.; Parent, M.; Titorenko, V.; Maysinger, D. Remodeling of lipid bodies by docosahexaenoic acid in activated microglial cells. J. Neuroinflamm. 2016, 13, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizato, N.; Melo, L.; Kiffer, L.; Luzete, B.; Antonio, J.; Ão, F.; Corrêa, L.H.; de Melo, H.A.B.; Santana, L.; Ito, M.; et al. Omega 3-dha and delta-tocotrienol modulate lipid droplet biogenesis and lipophagy in breast cancer cells: The impact in cancer aggressiveness. Nutrients 2019, 11, 1199. [Google Scholar] [CrossRef] [Green Version]
- Jarc, E.; Kump, A.; Malavašič, P.; Eichmann, T.O.; Zimmermann, R.; Petan, T. Lipid droplets induced by secreted phospholipase a2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 247–265. [Google Scholar] [CrossRef]
- Guštin, E.; Jarc, E.; Kump, A.; Petan, T. Lipid droplet formation in hela cervical cancer cells depends on cell density and the concentration of exogenous unsaturated fatty acids. Acta Chim. Slov. 2017, 64, 6. [Google Scholar] [CrossRef]
- Brookheart, R.T.; Michel, C.I.; Schaffer, J.E. As a matter of fat. Cell Metab. 2009, 10, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan g protein-coupled receptor gpr40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, K.; Maekawa, F.; Yada, T. Oleic acid interacts with gpr40 to induce ca2+ signaling in rat islet beta-cells: Mediation by plc and l-type ca2+ channel and link to insulin release. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670–E677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, G.; Alvarez-Curto, E.; Watterson, K.R.; Ulven, T.; Hudson, B.D. Characterizing pharmacological ligands to study the long-chain fatty acid receptors gpr40/ffa1 and gpr120/ffa4. Br. J. Pharmacol. 2015, 172, 3254–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through gpr120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Dragano, N.R.V.; Solon, C.; Ramalho, A.F.; de Moura, R.F.; Razolli, D.S.; Christiansen, E.; Azevedo, C.; Ulven, T.; Velloso, L.A. Polyunsaturated fatty acid receptors, gpr40 and gpr120, are expressed in the hypothalamus and control energy homeostasis and inflammation. J. Neuroinflamm. 2017, 14, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.Z.; He, L. The role of polyunsaturated fatty acids and gpr40 receptor in brain. Neuropharmacology 2017, 113, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lu, L.; Boneva, N.B.; Warashina, S.; Kaplamadzhiev, D.B.; Mori, Y.; Nakaya, M.A.; Kikuchi, M.; Tonchev, A.B.; Okano, H.; et al. Expression of free fatty acid receptor gpr40 in the neurogenic niche of adult monkey hippocampus. Hippocampus 2008, 18, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Falomir-Lockhart, L.J.; Cavazzutti, G.F.; Giménez, E.; Toscani, A.M. Fatty acid signaling mechanisms in neural cells: Fatty acid receptors. Front. Cell. Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Moreno, S.; Farioli-Vecchioli, S.; Ceru, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid x receptors in the adult rat cns. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Santos, M.J.; Bastias-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. Ppars in the central nervous system: Roles in neurodegeneration and neuroinflammation. Biol. Rev. Camb. Philos. Soc. 2017, 92, 2046–2069. [Google Scholar] [CrossRef]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of ppar isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Gorga, A.; Rindone, G.M.; Regueira, M.; Pellizzari, E.H.; Camberos, M.C.; Cigorraga, S.B.; Riera, M.F.; Galardo, M.N.; Meroni, S.B. Ppargamma activation regulates lipid droplet formation and lactate production in rat sertoli cells. Cell Tissue Res. 2017, 369, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Kawabori, M.; Yenari, M.A. Inflammatory responses in brain ischemia. Curr. Med. Chem. 2015, 22, 1258–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenti, G.; Benedetti, E.; D’Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Ceru, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia induces peroxisome proliferator-activated receptor alpha (pparalpha) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J. Cell. Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef]
- Zoula, S.; Rijken, P.F.; Peters, J.P.; Farion, R.; Van der Sanden, B.P.; Van der Kogel, A.J.; Decorps, M.; Remy, C. Pimonidazole binding in c6 rat brain glioma: Relation with lipid droplet detection. Br. J. Cancer 2003, 88, 1439–1444. [Google Scholar] [CrossRef] [Green Version]
- Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a metabolite and a regulator in the central nervous system. Int. J. Mol. Sci. 2016, 17, 1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosienko, V.; Teschemacher, A.; Kasparov, S. Is l-lactate a novel signaling molecule in the brain? J. Cereb. Blood Flow Metab. 2015, 35, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef]
- Morland, C.; Lauritzen, K.H.; Puchades, M.; Holm-Hansen, S.; Andersson, K.; Gjedde, A.; Attramadal, H.; Storm-Mathisen, J.; Bergersen, L.H. The lactate receptor, g-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J. Neurosci. Res. 2015, 93, 1045–1055. [Google Scholar] [CrossRef]
- Horvat, A.; Zorec, R.; Vardjan, N. Lactate as an astroglial signal augmenting aerobic glycolysis and lipid metabolism. Front. Physiol. 2021, 12, 1660. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ye, X.; Xie, M.; Ye, J. Induction of triglyceride accumulation and mitochondrial maintenance in muscle cells by lactate. Sci. Rep. 2016, 6, 33732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan g-protein-coupled receptor, gpr81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.; Tunaru, S.; Tang, C.; Müller, M.; Gille, A.; Sassmann, A.; Hanson, J.; Offermanns, S. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through gpr81. Cell Metab. 2010, 11, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Sanchez-Abarca, L.I.; Tabernero, A.; Medina, J.M. Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 2001, 36, 321–329. [Google Scholar] [CrossRef]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Ma, X.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Zhao, Y. In chronic hypoxia, glucose availability and hypoxic severity dictate the balance between hif-1 and hif-2 in astrocytes. FASEB J. 2019, 33, 11123–11136. [Google Scholar] [CrossRef] [Green Version]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a hif1alpha-ppargamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via srebp1c activation in hepg2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by hif-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-inducible factors and the regulation of lipid metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Chmurzyńska, A. The multigene family of fatty acid-binding proteins (fabps): Function, structure and polymorphism. J. Appl. Genet. 2006, 47, 39–48. [Google Scholar] [CrossRef]
- Toprak, U.; Hegedus, D.; Doğan, C.; Güney, G. A journey into the world of insect lipid metabolism. Arch. Insect Biochem. Physiol. 2020, 104, e21682. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.G.; Laranjeira, A.; Van Huffel, L.; Gartner, A.; Vilain, S.; Bastianen, J.; Van Veldhoven, P.P.; Dotti, C.G. Glial beta-oxidation regulates drosophila energy metabolism. Sci. Rep. 2015, 5, 7805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.; Zeppenfeld, D.; McConnell, E.; Pena, S.; Nedergaard, M. Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 2012, 37, 2496–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A.; Cruz, N.F. Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J. Neurochem. 2016, 138, 14–52. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, M.B.; Rasineni, K.; Weller, S.G.; Schulze, R.J.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. Β-adrenergic induction of lipolysis in hepatocytes is inhibited by ethanol exposure. J. Biol. Chem. 2017, 292, 11815–11828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardjan, N.; Chowdhury, H.H.; Horvat, A.; Velebit, J.; Malnar, M.; Muhič, M.; Kreft, M.; Krivec, Š.G.; Bobnar, S.T.; Miš, K.; et al. Enhancement of astroglial aerobic glycolysis by extracellular lactate-mediated increase in camp. Front. Mol. Neurosci. 2018, 11, 148. [Google Scholar] [CrossRef]
- Pardo, L.; Valor, L.M.; Eraso-Pichot, A.; Barco, A.; Golbano, A.; Hardingham, G.E.; Masgrau, R.; Galea, E. Creb regulates distinct adaptive transcriptional programs in astrocytes and neurons. Sci. Rep. 2017, 7, 6390. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in neurological diseases: A road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Yao, M.; Tabuchi, H.; Nagashima, Y.; Baba, M.; Nakaigawa, N.; Ishiguro, H.; Hamada, K.; Inayama, Y.; Kishida, T.; Hattori, K.; et al. Gene expression analysis of renal carcinoma: Adipose differentiation-related protein as a potential diagnostic and prognostic biomarker for clear-cell renal carcinoma. J. Pathol. 2005, 205, 377–387. [Google Scholar] [CrossRef]
- Bozza, P.T.; Viola, J.P. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fat. Acids 2010, 82, 243–250. [Google Scholar] [CrossRef]
- Loving, B.A.; Tang, M.; Neal, M.C.; Gorkhali, S.; Murphy, R.; Eckel, R.H.; Bruce, K.D. Lipoprotein lipase regulates microglial lipid droplet accumulation. Cells 2021, 10, 198. [Google Scholar] [CrossRef]
- Gong, H.; Dong, W.; Rostad, S.W.; Marcovina, S.M.; Albers, J.J.; Brunzell, J.D.; Vuletic, S. Lipoprotein lipase (lpl) is associated with neurite pathology and its levels are markedly reduced in the dentate gyrus of Alzheimer’s disease brains. J. Histochem. Cytochem. 2013, 61, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14949–14954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klosinski, L.P.; Yao, J.; Yin, F.; Fonteh, A.N.; Harrington, M.G.; Christensen, T.A.; Trushina, E.; Brinton, R.D. White matter lipids as a ketogenic fuel supply in aging female brain: Implications for Alzheimer’s disease. EBioMedicine 2015, 2, 1888–1904. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Worthington, W.C., Jr.; Cathcart, R.S., 3rd. Ependymal cilia: Distribution and activity in the adult human brain. Science 1963, 139, 221–222. [Google Scholar] [CrossRef]
- Del Bigio, M.R. Ependymal cells: Biology and pathology. Acta Neuropathol. 2010, 119, 55–73. [Google Scholar] [CrossRef]
- Olstad, E.W.; Ringers, C.; Hansen, J.N.; Wens, A.; Brandt, C.; Wachten, D.; Yaksi, E.; Jurisch-Yaksi, N. Ciliary beating compartmentalizes cerebrospinal fluid flow in the brain and regulates ventricular development. Curr. Biol. 2019, 29, 229–241.e226. [Google Scholar] [CrossRef] [Green Version]
- Petrik, D.; Myoga, M.H.; Grade, S.; Gerkau, N.J.; Pusch, M.; Rose, C.R.; Grothe, B.; Götz, M. Epithelial sodium channel regulates adult neural stem cell proliferation in a flow-dependent manner. Cell Stem Cell 2018, 22, 865–878.e868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamoto, K.; Wichterle, H.; Gonzalez-Perez, O.; Cholfin, J.A.; Yamada, M.; Spassky, N.; Murcia, N.S.; Garcia-Verdugo, J.M.; Marin, O.; Rubenstein, J.L.; et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 2006, 311, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Capilla-Gonzalez, V.; Cebrian-Silla, A.; Guerrero-Cazares, H.; Garcia-Verdugo, J.M.; Quiñones-Hinojosa, A. Age-related changes in astrocytic and ependymal cells of the subventricular zone. Glia 2014, 62, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Bouab, M.; Paliouras, G.N.; Aumont, A.; Forest-Bérard, K.; Fernandes, K.J. Aging of the subventricular zone neural stem cell niche: Evidence for quiescence-associated changes between early and mid-adulthood. Neuroscience 2011, 173, 135–149. [Google Scholar] [CrossRef]
- Rawish, E.; Nickel, L.; Schuster, F.; Stölting, I.; Frydrychowicz, A.; Saar, K.; Hübner, N.; Othman, A.; Kuerschner, L.; Raasch, W. Telmisartan prevents development of obesity and normalizes hypothalamic lipid droplets. J. Endocrinol. 2020, 244, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Lamberz, C.; Piotrowitz, K.; Offermann, N.; But, D.; Scheller, A.; Al-Amoudi, A.; Kuerschner, L. Tanycytes and a differential fatty acid metabolism in the hypothalamus. Glia 2017, 65, 231–249. [Google Scholar] [CrossRef]
- Gajera, C.R.; Emich, H.; Lioubinski, O.; Christ, A.; Beckervordersandforth-Bonk, R.; Yoshikawa, K.; Bachmann, S.; Christensen, E.I.; Götz, M.; Kempermann, G.; et al. Lrp2 in ependymal cells regulates bmp signaling in the adult neurogenic niche. J. Cell. Sci. 2010, 123, 1922–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Chiba, Y.; Fujihara, R.; Kubo, H.; Sakamoto, H.; Ueno, M. Immunohistochemical analysis of transporters related to clearance of amyloid-β peptides through blood-cerebrospinal fluid barrier in human brain. Histochem. Cell Biol. 2015, 144, 597–611. [Google Scholar] [CrossRef]
- Enos, N.; Takenaka, H.; Scott, S.; Salfity, H.V.N.; Kirk, M.; Egar, M.W.; Sarria, D.A.; Slayback-Barry, D.; Belecky-Adams, T.; Chernoff, E.A.G. Meningeal foam cells and ependymal cells in axolotl spinal cord regeneration. Front. Immunol. 2019, 10, 2558. [Google Scholar] [CrossRef]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein e4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, V.; Maciel, P.; Costa, V. Leading the way in the nervous system: Lipid droplets as new players in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158820. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. Tar DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.B.; Lee, V.M.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of tdp43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, S.A.; Barres, B.A. Glia as primary drivers of neuropathology in tdp-43 proteinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 4439–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. Tdp-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Velebit, J.; Horvat, A.; Smolič, T.; Prpar Mihevc, S.; Rogelj, B.; Zorec, R.; Vardjan, N. Astrocytes with tdp-43 inclusions exhibit reduced noradrenergic camp and ca2+ signaling and dysregulated cell metabolism. Sci. Rep. 2020, 10, 6003. [Google Scholar] [CrossRef] [Green Version]
- Mou, Y.; Dong, Y.; Chen, Z.; Denton, K.R.; Duff, M.O.; Blackstone, C.; Zhang, S.-C.; Li, X.-J. Impaired lipid metabolism in astrocytes underlies degeneration of cortical projection neurons in hereditary spastic paraplegia. Acta Neuropathol. Commun. 2020, 8, 214. [Google Scholar] [CrossRef]
- Renvoisé, B.; Malone, B.; Falgairolle, M.; Munasinghe, J.; Stadler, J.; Sibilla, C.; Park, S.H.; Blackstone, C. Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum. Mol. Genet. 2016, 25, 5111–5125. [Google Scholar]
- Papadopoulos, C.; Orso, G.; Mancuso, G.; Herholz, M.; Gumeni, S.; Tadepalle, N.; Jüngst, C.; Tzschichholz, A.; Schauss, A.; Höning, S.; et al. Spastin binds to lipid droplets and affects lipid metabolism. PloS Genet. 2015, 11, e1005149. [Google Scholar] [CrossRef] [Green Version]
- Falk, J.; Rohde, M.; Bekhite, M.M.; Neugebauer, S.; Hemmerich, P.; Kiehntopf, M.; Deufel, T.; Hübner, C.A.; Beetz, C. Functional mutation analysis provides evidence for a role of reep1 in lipid droplet biology. Hum. Mutat. 2014, 35, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Inloes, J.M.; Hsu, K.-L.; Dix, M.M.; Viader, A.; Masuda, K.; Takei, T.; Wood, M.R.; Cravatt, B.F. The hereditary spastic paraplegia-related enzyme ddhd2 is a principal brain triglyceride lipase. Proc. Natl. Acad. Sci. USA 2014, 111, 14924–14929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inloes, J.M.; Kiosses, W.B.; Wang, H.; Walther, T.C.; Farese, R.V., Jr.; Cravatt, B.F. Functional contribution of the spastic paraplegia-related triglyceride hydrolase ddhd2 to the formation and content of lipid droplets. Biochemistry 2018, 57, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Lew, M. Overview of parkinson’s disease. Pharmacotherapy 2007, 27, 155s–160s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the parkinson’s disease protein α-synuclein*. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outeiro, T.F.; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef] [Green Version]
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell type-specific lipid storage changes in parkinson’s disease patient brains are recapitulated by experimental glycolipid disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Dufresne, M.; Joppé, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabé-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease. Cell Stem Cell 2015, 17, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Corder, E.; Saunders, A.; Strittmatter, W.; Schmechel, D.; Gaskell, P.; Small, G.; Roses, A.; Haines, J.; Pericak-Vance, M. Gene dose of apolipoprotein e type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. Apoe4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. Trem2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 2020, 105, 837–854.e839. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2021; online ahead of print. [Google Scholar] [CrossRef]
- Gasparovic, C.; Rosenberg, G.A.; Wallace, J.A.; Estrada, E.Y.; Roberts, K.; Pastuszyn, A.; Ahmed, W.; Graham, G.D. Magnetic resonance lipid signals in rat brain after experimental stroke correlate with neutral lipid accumulation. Neurosci. Lett. 2001, 301, 87–90. [Google Scholar] [CrossRef]
- Geng, F.; Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3, 443. [Google Scholar] [CrossRef] [Green Version]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of soat1 suppresses glioblastoma growth via blocking srebp-1-mediated lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. Srebp maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid droplets maintain energy homeostasis and glioblastoma growth via autophagic release of stored fatty acids. iScience 2020, 23, 101569. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An lxr agonist promotes glioblastoma cell death through inhibition of an egfr/akt/srebp-1/ldlr-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hager, L.; Li, L.; Pun, H.; Liu, L.; Hossain, M.A.; Maguire, G.F.; Naples, M.; Baker, C.; Magomedova, L.; Tam, J.; et al. Lecithin:Cholesterol acyltransferase deficiency protects against cholesterol-induced hepatic endoplasmic reticulum stress in mice. J. Biol. Chem. 2012, 287, 20755–20768. [Google Scholar] [CrossRef] [Green Version]
- Lai, E.; Bikopoulos, G.; Wheeler, M.B.; Rozakis-Adcock, M.; Volchuk, A. Differential activation of er stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E540–E550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedi, X.; Ming, Y.; Yongping, W.; Yi, Y.; Xiaoxiang, Z. Free cholesterol overloading induced smooth muscle cells death and activated both er- and mitochondrial-dependent death pathway. Atherosclerosis 2009, 207, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smolič, T.; Zorec, R.; Vardjan, N. Pathophysiology of Lipid Droplets in Neuroglia. Antioxidants 2022, 11, 22. https://doi.org/10.3390/antiox11010022
Smolič T, Zorec R, Vardjan N. Pathophysiology of Lipid Droplets in Neuroglia. Antioxidants. 2022; 11(1):22. https://doi.org/10.3390/antiox11010022
Chicago/Turabian StyleSmolič, Tina, Robert Zorec, and Nina Vardjan. 2022. "Pathophysiology of Lipid Droplets in Neuroglia" Antioxidants 11, no. 1: 22. https://doi.org/10.3390/antiox11010022
APA StyleSmolič, T., Zorec, R., & Vardjan, N. (2022). Pathophysiology of Lipid Droplets in Neuroglia. Antioxidants, 11(1), 22. https://doi.org/10.3390/antiox11010022