
Nrf2 in the Field of Dentistry with Special Attention to NLRP3

 ,
,
Abstract
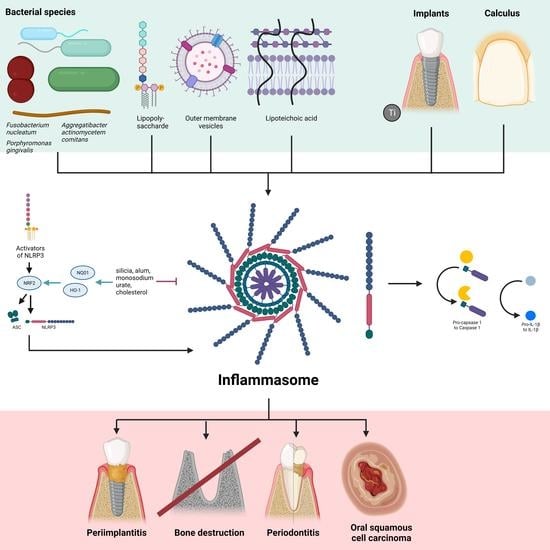
:
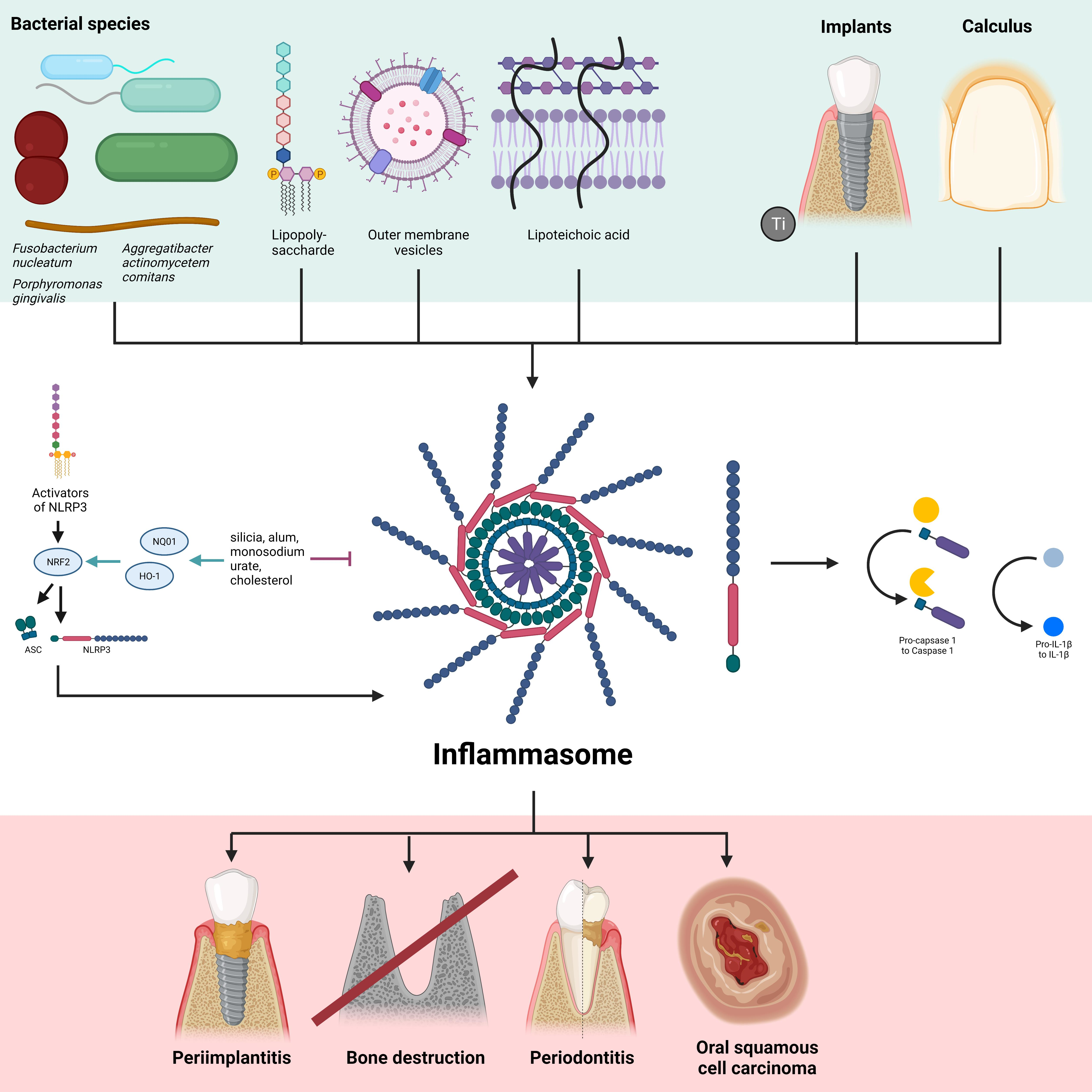
1. Introduction
2. The Oral Microbiome
3. Periodontitis
3.1. P. gingivalis
3.2. Fusobacterium Nucleatum
3.3. Aggregatibacter actinomycetemcomitans
4. Periapical Periodontitis
5. Oral Squamous Cell Carcinoma
6. Dental Implants
7. The Alveolar Bone
8. Medication-Related Osteonecrosis of the Jaw
9. Calculus
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil |
| AIM2 | absent in melanoma 2 |
| ALRs | AIM2-like receptors |
| ASC | apoptosis-associated speck-like protein containing CARD |
| ATP | adenosine triphosphate |
| BRONJ | Bisphosphonate-related osteonecrosis of the jaw |
| CASP1 | caspase-1 |
| Cdt | cytolethal distending toxin |
| CLRs | C-type lectin receptors |
| DAMPs | damage-associated molecular patterns |
| GSDMD | gasdermin D |
| HGEC | human gingival epithelial cells |
| HGFs | human gingival fibroblasts |
| HNSCC | head and neck squamous cell cancer |
| HO-1 | hemeoxygenase-1 |
| IFN | type I interferon |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LPS | lipopolysaccharide |
| LRR | leucine-rich repeat |
| LTA | lipoteichoic acid |
| Ltx | leukotoxin |
| MRONJ | medication-related osteonecrosis of the jaw |
| NF-κB | nuclear factor kappa B |
| NLRs | Nod-like receptors |
| NLRP3 | NACHT domain-, leucin-rich repeat-, and pyrin domain (PYD)-containing protein 3 |
| NOD | nucleotide-binding oligomerization domain |
| NQO1 | NAD(P)H quinone oxidoreductase-1 |
| Nrf2 | nuclear factor E2-related factor or nuclear factor (erythroid-derived 2)-like 2 |
| OA | osteoarthritis |
| OMVs | outer membrane vesicles |
| OSCC | oral squamous cell carcinoma |
| PAMPS | Pathogen-associated molecular patterns |
| PD | periodontitis |
| proCASP1 | pro-caspase-1 |
| PRRs | pattern-recognition receptors |
| PTMs | post-translational modifications |
| PYD | pyrin domain |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| RLRs | RIG-I-like receptors |
| TLRs | Toll-like receptors |
| VD3 | 1,25-dihydroxyvitamin D3 |
References
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Chen, Y.; Wang, H.Y.; Wang, R.-F. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Allen, I.C.; Bergstralh, D.T.; Brickey, W.J.; Huang, M.T.-H.; Taxman, D.J.; Duncan, J.A.; Ting, J.P.-Y. NLRP3 (NALP3, Cryopyrin) Facilitates In Vivo Caspase-1 Activation, Necrosis, and HMGB1 Release via Inflammasome-Dependent and -Independent Pathways. J. Immunol. 2009, 183, 2008–2015. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Kinio, A.; Saleh, M. Functions of NOD-Like Receptors in Human Diseases. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, I.; Miao, E.A. Pyroptotic Cell Death Defends against Intracellular Pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like Receptors in Microbial Recognition and Host Defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical Role for Cryopyrin/Nalp3 in Activation of Caspase-1 in Response to Viral Infection and Double-Stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Groß, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome Activators Induce Interleukin-1α Secretion via Distinct Pathways with Differential Requirement for the Protease Function of Caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential Expression of NLRP3 among Hematopoietic Cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutterwala, F.S.; Ogura, Y.; Szczepanik, M.; Lara-Tejero, M.; Lichtenberger, G.S.; Grant, E.P.; Bertin, J.; Coyle, A.J.; Galán, J.E.; Askenase, P.W.; et al. Critical Role for NALP3/CIAS1/Cryopyrin in Innate and Adaptive Immunity through Its Regulation of Caspase-1. Immunity 2006, 24, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Shinohara, M.L. NLRP3 Inflammasome and MS/EAE. Autoimmune Dis. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s Tyrosine Kinase Is Essential for NLRP3 Inflammasome Activation and Contributes to Ischaemic Brain Injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef]
- Nakanishi, H.; Kawashima, Y.; Kurima, K.; Chae, J.J.; Ross, A.M.; Pinto-Patarroyo, G.; Patel, S.K.; Muskett, J.A.; Ratay, J.S.; Chattaraj, P.; et al. NLRP3 Mutation and Cochlear Autoinflammation Cause Syndromic and Nonsyndromic Hearing Loss DFNA34 Responsive to Anakinra Therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E7766–E7775. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential Activation of the Inflammasome by Caspase-1 Adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Kurita-Ochiai, T.; Kobayashi, R.; Suzuki, T.; Ando, T. Regulation of the NLRP3 Inflammasome in Porphyromonas Gingivalis-Accelerated Periodontal Disease. Inflamm. Res. 2017, 66, 59–65. [Google Scholar] [CrossRef]
- Rocha, F.R.G.; Delitto, A.E.; de Souza, J.A.C.; González-Maldonado, L.A.; Wallet, S.M.; Rossa Junior, C. Relevance of Caspase-1 and Nlrp3 Inflammasome on Inflammatory Bone Resorption in A Murine Model of Periodontitis. Sci. Rep. 2020, 10, 7823. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 Forms an IL-1β-Processing Inflammasome with Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Srinivasula, S.M.; Poyet, J.-L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD Protein ASC Is an Activating Adaptor for Caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [Green Version]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.-D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e4. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Jin, T.; Zhang, X.; Zeng, Z.; Ye, B.; Wang, J.; Zhong, Y.; Xiong, X.; Gu, L. Meisoindigo Protects Against Focal Cerebral Ischemia-Reperfusion Injury by Inhibiting NLRP3 Inflammasome Activation and Regulating Microglia/Macrophage Polarization via TLR4/NF-ΚB Signaling Pathway. Front. Cell. Neurosci. 2019, 13, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An Overview of the Non-Canonical Inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Ehses, J.A.; Donath, M.Y.; Mandrup-Poulsen, T. Sustained Effects of Interleukin-1 Receptor Antagonist Treatment in Type 2 Diabetes. Diabetes Care 2009, 32, 1663–1668. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Bostanci, N.; Emingil, G.; Saygan, B.; Turkoglu, O.; Atilla, G.; Curtis, M.A.; Belibasakis, G.N. Expression and Regulation of the NALP3 Inflammasome Complex in Periodontal Diseases: NALP3 Inflammasome in Periodontal Diseases. Clin. Exp. Immunol. 2009, 157, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; Di Filippo, M.; French, L.; Beer, H.-D. The Crosstalk between Nrf2 and Inflammasomes. Int. J. Mol. Sci. 2018, 19, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, M.; Swierczynski, M.; Fichna, J.; Piechota-Polanczyk, A. The Nrf2 in the Pathophysiology of the Intestine: Molecular Mechanisms and Therapeutic Implications for Inflammatory Bowel Diseases. Pharmacol. Res. 2021, 163, 105243. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 Is Essential for Cholesterol Crystal-Induced Inflammasome Activation and Exacerbation of Atherosclerosis: Innate Immunity. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward Clinical Application of the Keap1–Nrf2 Pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE Pathway Protects Endothelial Cells from Oxidant Injury and Inhibits Inflammatory Gene Expression. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular Basis of the Keap1–Nrf2 System. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Jhang, J.-J.; Yen, G.-C. The Role of Nrf2 in NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2017, 14, 1011–1012. [Google Scholar] [CrossRef]
- Dong, Z.; Shang, H.; Chen, Y.Q.; Pan, L.-L.; Bhatia, M.; Sun, J. Sulforaphane Protects Pancreatic Acinar Cell Injury by Modulating Nrf2-Mediated Oxidative Stress and NLRP3 Inflammatory Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, J.; Pineda, M.A.; Al-Riyami, L.; Rodgers, D.T.; Huggan, J.K.; Lumb, F.E.; Khalaf, A.I.; Meakin, P.J.; Corbet, M.; Ashford, M.L.; et al. Prophylactic and Therapeutic Treatment with a Synthetic Analogue of a Parasitic Worm Product Prevents Experimental Arthritis and Inhibits IL-1β Production via NRF2-Mediated Counter-Regulation of the Inflammasome. J. Autoimmun. 2015, 60, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zeng, X.; Li, X.; Cui, C.; Hou, R.; Guo, Z.; Mehta, J.L.; Wang, X. Advances in the Molecular Mechanisms of NLRP3 Inflammasome Activators and Inactivators. Biochem. Pharmacol. 2020, 175, 113863. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Sui, J.; Fan, R.; Qu, W.; Dong, X.; Sun, D. Emodin Protects Against Acute Pancreatitis-Associated Lung Injury by Inhibiting NLPR3 Inflammasome Activation via Nrf2/HO-1 Signaling. Drug Des. Devel. Ther. 2020, Volume 14, 1971–1982. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.-H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Yu, W.-H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A Web Accessible Resource for Investigating Oral Microbe Taxonomic and Genomic Information. Database 2010, 2010, baq013. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N. Oral Microbiome Metabolism: From “Who Are They?” To “What Are They Doing?”. J. Dent. Res. 2015, 94, 1628–1637. [Google Scholar] [CrossRef]
- Kilian, M.; Chapple, I.L.C.; Hannig, M.; Marsh, P.D.; Meuric, V.; Pedersen, A.M.L.; Tonetti, M.S.; Wade, W.G.; Zaura, E. The Oral Microbiome – an Update for Oral Healthcare Professionals. Br. Dent. J. 2016, 221, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Takeshita, T. The Oral Microbiome and Human Health. J. Oral Sci. 2017, 59, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Busscher, H.J.; Rinastiti, M.; Siswomihardjo, W.; van der Mei, H.C. Biofilm Formation on Dental Restorative and Implant Materials. J. Dent. Res. 2010, 89, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, I.; De Munck, J.; Ungureanu, A.-A.; Slomka, V.; Bartic, C.; Vananroye, A.; Clasen, C.; Teughels, W.; Van Meerbeek, B.; Van Landuyt, K.L. Biofilm-Induced Changes to the Composite Surface. J. Dent. 2017, 63, 36–43. [Google Scholar] [CrossRef]
- Preethanath, R.S.; AlNahas, N.W.; Bin Huraib, S.M.; Al-Balbeesi, H.O.; Almalik, N.K.; Dalati, M.H.N.; Divakar, D.D. Microbiome of Dental Implants and Its Clinical Aspect. Microb. Pathog. 2017, 106, 20–24. [Google Scholar] [CrossRef]
- Griffen, A.L.; Beall, C.J.; Campbell, J.H.; Firestone, N.D.; Kumar, P.S.; Yang, Z.K.; Podar, M.; Leys, E.J. Distinct and Complex Bacterial Profiles in Human Periodontitis and Health Revealed by 16S Pyrosequencing. ISME J. 2012, 6, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Marsh, P.D. Dental Plaque: Biological Significance of a Biofilm and Community Life-Style. J. Clin. Periodontol. 2005, 32, 7–15. [Google Scholar] [CrossRef]
- Roberts, A.P.; Mullany, P. Oral Biofilms: A Reservoir of Transferable, Bacterial, Antimicrobial Resistance. Expert Rev. Anti Infect. Ther. 2010, 8, 1441–1450. [Google Scholar] [CrossRef]
- Kuramitsu, H.K. Virulence Factors of Mutans Streptococci: Role of Molecular Genetics. Crit. Rev. Oral Biol. Med. 1993, 4, 159–176. [Google Scholar] [CrossRef]
- Irving, J.T.; Newman, M.G.; Socransky, S.S.; Heeley, J.D. Histological Changes in Experimental Periodontal Disease in Rats Mono-Infected with a Gram-Negative Organism. Arch. Oral Biol. 1975, 20, 219–220. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L. Microbial Complexes in Subgingival Plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, R.J. Mutans Streptococci: Acquisition and Transmission. Pediatr. Dent. 2006, 28, 106–109; discussion 192–198. [Google Scholar]
- Cephas, K.D.; Kim, J.; Mathai, R.A.; Barry, K.A.; Dowd, S.E.; Meline, B.S.; Swanson, K.S. Comparative Analysis of Salivary Bacterial Microbiome Diversity in Edentulous Infants and Their Mothers or Primary Care Givers Using Pyrosequencing. PLoS ONE 2011, 6, e23503. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hao, W.; Zhou, Q.; Wang, W.; Xia, Z.; Liu, C.; Chen, X.; Qin, M.; Chen, F. Plaque Bacterial Microbiome Diversity in Children Younger than 30 Months with or without Caries Prior to Eruption of Second Primary Molars. PLoS ONE 2014, 9, e89269. [Google Scholar] [CrossRef]
- Jiang, W.; Ling, Z.; Lin, X.; Chen, Y.; Zhang, J.; Yu, J.; Xiang, C.; Chen, H. Pyrosequencing Analysis of Oral Microbiota Shifting in Various Caries States in Childhood. Microb. Ecol. 2014, 67, 962–969. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn Diagram: The Hunt for a Core Microbiome: The Hunt for a Core Microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Blum, H.E. The Human Microbiome. Adv. Med. Sci. 2017, 62, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Kilian, M. The Oral Microbiome – Friend or Foe? Eur. J. Oral Sci. 2018, 126, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Belstrøm, D.; Constancias, F.; Liu, Y.; Yang, L.; Drautz-Moses, D.I.; Schuster, S.C.; Kohli, G.S.; Jakobsen, T.H.; Holmstrup, P.; Givskov, M. Metagenomic and Metatranscriptomic Analysis of Saliva Reveals Disease-Associated Microbiota in Patients with Periodontitis and Dental Caries. Npj Biofilms Microbiomes 2017, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholizadeh, P.; Eslami, H.; Yousefi, M.; Asgharzadeh, M.; Aghazadeh, M.; Kafil, H.S. Role of Oral Microbiome on Oral Cancers, a Review. Biomed. Pharmacother. 2016, 84, 552–558. [Google Scholar] [CrossRef]
- Kinross, J.M.; Darzi, A.W.; Nicholson, J.K. Gut Microbiome-Host Interactions in Health and Disease. Genome Med. 2011, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Laugisch, O.; Johnen, A.; Maldonado, A.; Ehmke, B.; Bürgin, W.; Olsen, I.; Potempa, J.; Sculean, A.; Duning, T.; Eick, S. Periodontal Pathogens and Associated Intrathecal Antibodies in Early Stages of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 66, 105–114. [Google Scholar] [CrossRef]
- Mesa, F.; Magan-Fernandez, A.; Castellino, G.; Chianetta, R.; Nibali, L.; Rizzo, M. Periodontitis and Mechanisms of Cardiometabolic Risk: Novel Insights and Future Perspectives. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2019, 1865, 476–484. [Google Scholar] [CrossRef]
- Liu, X.-R.; Xu, Q.; Xiao, J.; Deng, Y.-M.; Tang, Z.-H.; Tang, Y.-L.; Liu, L.-S. Role of Oral Microbiota in Atherosclerosis. Clin. Chim. Acta 2020, 506, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Shimomura, Y.; Higurashi, T.; Sugi, Y.; Arimoto, J.; Umezawa, S.; Uchiyama, S.; Matsumoto, M.; Nakajima, A. Patients with Colorectal Cancer Have Identical Strains of Fusobacterium Nucleatum in Their Colorectal Cancer and Oral Cavity. Gut 2019, 68, 1335–1337. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.; Zhao, N.; Meier, R.; Koestler, D.C.; Wu, G.; del Castillo, E.; Paster, B.J.; Charpentier, K.; Izard, J.; Kelsey, K.T.; et al. Comparisons of Oral, Intestinal, and Pancreatic Bacterial Microbiomes in Patients with Pancreatic Cancer and Other Gastrointestinal Diseases. J. Oral Microbiol. 2021, 13, 1887680. [Google Scholar] [CrossRef] [PubMed]
- Hujoel, P.P.; Drangsholt, M.; Spiekerman, C.; Weiss, N.S. An Exploration of the Periodontitis–Cancer Association. Ann. Epidemiol. 2003, 13, 312–316. [Google Scholar] [CrossRef]
- Teles, F.R.F.; Alawi, F.; Castilho, R.M.; Wang, Y. Association or Causation? Exploring the Oral Microbiome and Cancer Links. J. Dent. Res. 2020, 99, 1411–1424. [Google Scholar] [CrossRef]
- Lu, M.-C.; Ji, J.-A.; Jiang, Y.-L.; Chen, Z.-Y.; Yuan, Z.-W.; You, Q.-D.; Jiang, Z.-Y. An Inhibitor of the Keap1-Nrf2 Protein-Protein Interaction Protects NCM460 Colonic Cells and Alleviates Experimental Colitis. Sci. Rep. 2016, 6, 26585. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.M.; Sefik, E.; Upadhyay, R.; Weissleder, R.; Benoist, C.; Mathis, D. Endoscopic Photoconversion Reveals Unexpectedly Broad Leukocyte Trafficking to and from the Gut. Proc. Natl. Acad. Sci. USA 2014, 111, 6696–6701. [Google Scholar] [CrossRef] [Green Version]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G.; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hu, Y.; Fang, Y.; Djukic, Z.; Yamamoto, M.; Shaheen, N.J.; Orlando, R.C.; Chen, X. Nrf2 Deficiency Impairs the Barrier Function of Mouse Oesophageal Epithelium. Gut 2014, 63, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baragetti, A.; Catapano, A.L.; Magni, P. Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease. Int. J. Mol. Sci. 2020, 21, 4459. [Google Scholar] [CrossRef]
- Baelum, V.; López, R. Periodontal Disease Epidemiology - Learned and Unlearned? Periodontol. 2000 2013, 62, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Marco del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases: Consensus Report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, F.; Saccucci, M.; Di Carlo, G.; Lucchetti, R.; Pilloni, A.; Pranno, N.; Luzzi, V.; Valesini, G.; Polimeni, A. Periodontitis and Rheumatoid Arthritis: The Same Inflammatory Mediators? Mediat. Inflamm. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yuan, Y.; Liu, H.; Li, S.; Zhang, B.; Chen, W.; An, Z.; Chen, S.; Wu, Y.; Han, B.; et al. Epidemiologic Relationship between Periodontitis and Type 2 Diabetes Mellitus. BMC Oral Health 2020, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Hoare, A.; Soto, C.; Rojas-Celis, V.; Bravo, D. Chronic Inflammation as a Link between Periodontitis and Carcinogenesis. Mediat. Inflamm. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lebeer, S.; Vanderleyden, J.; De Keersmaecker, S.C.J. Host Interactions of Probiotic Bacterial Surface Molecules: Comparison with Commensals and Pathogens. Nat. Rev. Microbiol. 2010, 8, 171–184. [Google Scholar] [CrossRef]
- Park, E.; Na, H.S.; Song, Y.-R.; Shin, S.Y.; Kim, Y.-M.; Chung, J. Activation of NLRP3 and AIM2 Inflammasomes by Porphyromonas Gingivalis Infection. Infect. Immun. 2014, 82, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Belibasakis, G.N.; Guggenheim, B.; Bostanci, N. Down-Regulation of NLRP3 Inflammasome in Gingival Fibroblasts by Subgingival Biofilms: Involvement of Porphyromonas Gingivalis. Innate Immun. 2013, 19, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Yucel-Lindberg, T.; Brunius, G. Epidermal Growth Factor Synergistically Enhances Interleukin-8 Production in Human Gingival Fibroblasts Stimulated with Interleukin-1β. Arch. Oral Biol. 2006, 51, 892–898. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The Mechanism of Osteoclast Differentiation Induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef]
- Perozini, C.; Chibebe, P.C.A.; Leao, M.V.P.; da Silva Queiroz, C.; Pallos, D. Gingival Crevicular Fluid Biochemical Markers in Periodontal Disease: A Cross-Sectional Study. Quintessence Int. Berl. Ger. 1985 2010, 41, 877–883. [Google Scholar]
- Miller, C.S.; King, C.P.; Langub, M.C.; Kryscio, R.J.; Thomas, M.V. Salivary Biomarkers of Existing Periodontal Disease: A Cross-Sectional Study. J. Am. Dent. Assoc. 1939 2006, 137, 322–329. [Google Scholar] [CrossRef]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, F.; Shu, R.; Xie, Y. The Expression of NLRP3, NLRP1 and AIM2 in the Gingival Tissue of Periodontitis Patients: RT-PCR Study and Immunohistochemistry. Arch. Oral Biol. 2015, 60, 948–958. [Google Scholar] [CrossRef]
- Isaza-Guzmán, D.M.; Medina-Piedrahíta, V.M.; Gutiérrez-Henao, C.; Tobón-Arroyave, S.I. Salivary Levels of NLRP3 Inflammasome-Related Proteins as Potential Biomarkers of Periodontal Clinical Status. J. Periodontol. 2017, 88, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Aral, K.; Berdeli, E.; Cooper, P.R.; Milward, M.R.; Kapila, Y.; Karadede Ünal, B.; Aral, C.A.; Berdeli, A. Differential Expression of Inflammasome Regulatory Transcripts in Periodontal Disease. J. Periodontol. 2020, 91, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Yilmaz, Ö. Modulation of Inflammasome Activity by Porphyromonas Gingivalis in Periodontitis and Associated Systemic Diseases. J. Oral Microbiol. 2016, 8, 30385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesan, J.; Girnary, M.S.; Jing, L.; Miao, M.Z.; Zhang, S.; Sun, L.; Morelli, T.; Schoenfisch, M.H.; Inohara, N.; Offenbacher, S.; et al. An Experimental Murine Model to Study Periodontitis. Nat. Protoc. 2018, 13, 2247–2267. [Google Scholar] [CrossRef]
- Becker, C.E.; O’Neill, L.A.J. Inflammasomes in Inflammatory Disorders: The Role of TLRs and Their Interactions with NLRs. Semin. Immunopathol. 2007, 29, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D.; Özören, N.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and Small Antiviral Compounds Activate Caspase-1 through Cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Pétrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The Inflammasome: A Danger Sensing Complex Triggering Innate Immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Johansson, A. Aggregatibacter Actinomycetemcomitans Targets NLRP3 and NLRP6 Inflammasome Expression in Human Mononuclear Leukocytes. Cytokine 2012, 59, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, F.Q.; Johnson, L.; Roberts, J.; Hung, S.-C.; Lee, J.; Atanasova, K.R.; Huang, P.-R.; Yilmaz, Ö.; Ojcius, D.M. Fusobacterium Nucleatum Infection of Gingival Epithelial Cells Leads to NLRP3 Inflammasome-Dependent Secretion of IL-1β and the Danger Signals ASC and HMGB1. Cell. Microbiol. 2016, 18, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.P.; Mueller, J.L.; Rosengren, S.; Boyle, D.L.; Schaner, P.; Cannon, S.B.; Goodyear, C.S.; Hoffman, H.M. Structural, Expression, and Evolutionary Analysis of Mouse CIAS1. Gene 2004, 338, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummer, J.A.; Broekhuizen, R.; Everett, H.; Agostini, L.; Kuijk, L.; Martinon, F.; van Bruggen, R.; Tschopp, J. Inflammasome Components NALP 1 and 3 Show Distinct but Separate Expression Profiles in Human Tissues Suggesting a Site-Specific Role in the Inflammatory Response. J. Histochem. Cytochem. 2007, 55, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Hawkins, P.N.; Lachmann, H.J.; McDermott, M.F. Interleukin-1–Receptor Antagonist in the Muckle–Wells Syndrome. N. Engl. J. Med. 2003, 348, 2583–2584. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhong, X.; Li, W.; Wang, Q. Effects of 1,25-Dihydroxyvitamin D3 on Experimental Periodontitis and AhR/NF-ΚB/NLRP3 Inflammasome Pathway in a Mouse Model. J. Appl. Oral Sci. 2019, 27, e20180713. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, P.; Wang, Q.; Ji, N.; Xia, S.; Ding, Y.; Wang, Q. Metformin Ameliorates Experimental Diabetic Periodontitis Independently of Mammalian Target of Rapamycin (MTOR) Inhibition by Reducing NIMA-Related Kinase 7 (Nek7) Expression. J. Periodontol. 2019, 90, 1032–1042. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Garstkiewicz, M.; Strittmatter, G.E.; Grossi, S.; Sand, J.; Fenini, G.; Werner, S.; French, L.E.; Beer, H.-D. Opposing Effects of Nrf2 and Nrf2-Activating Compounds on the NLRP3 Inflammasome Independent of Nrf2-Mediated Gene Expression: Innate Immunity. Eur. J. Immunol. 2017, 47, 806–817. [Google Scholar] [CrossRef] [Green Version]
- Bodet, C.; Chandad, F.; Grenier, D. Potentiel pathogénique de Porphyromonas gingivalis, Treponema denticola et Tannerella forsythia, le complexe bactérien rouge associé à la parodontite. Pathol. Biol. 2007, 55, 154–162. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Dawson, D.; Emecen-Huja, P.; Nagarajan, R.; Howard, K.; Grady, M.E.; Thompson, K.; Peyyala, R.; Al-Attar, A.; Lethbridge, K.; et al. The Periodontal War: Microbes and Immunity. Periodontol. 2000 2017, 75, 52–115. [Google Scholar] [CrossRef]
- Teles, R.; Sakellari, D.; Teles, F.; Konstantinidis, A.; Kent, R.; Socransky, S.; Haffajee, A. Relationships Among Gingival Crevicular Fluid Biomarkers, Clinical Parameters of Periodontal Disease, and the Subgingival Microbiota. J. Periodontol. 2010, 81, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Aral, K.; Milward, M.R.; Kapila, Y.; Berdeli, A.; Cooper, P.R. Inflammasomes and Their Regulation in Periodontal Disease: A Review. J. Periodontal Res. 2020, 55, 473–487. [Google Scholar] [CrossRef] [PubMed]
- de Alencar, J.B.; Zacarias, J.M.V.; Tsuneto, P.Y.; de Souza, V.H.; de Oliveira e Silva, C.; Visentainer, J.E.L.; Sell, A.M. Influence of Inflammasome NLRP3, and IL1B and IL2 Gene Polymorphisms in Periodontitis Susceptibility. PLoS ONE 2020, 15, e0227905. [Google Scholar] [CrossRef]
- Yilmaz, Ö. The Chronicles of Porphyromonas Gingivalis: The Microbium, the Human Oral Epithelium and Their Interplay. Microbiology 2008, 154, 2897–2903. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.C.; Kesavalu, L.; Walker, S.; Genco, C.A. Virulence Factors of Porphyromonas Gingivalis. Periodontol. 2000 1999, 20, 168–238. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Lambris, J.D.; Hajishengallis, G. Porphyromonas Gingivalis Disturbs Host–Commensal Homeostasis by Changing Complement Function. J. Oral Microbiol. 2017, 9, 1340085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G. Immune Evasion Strategies of Porphyromonas Gingivalis. J. Oral Biosci. 2011, 53, 233–240. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, W.; Wang, H.; Liang, S. Roles of Porphyromonas Gingivalis and Its Virulence Factors in Periodontitis. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 120, pp. 45–84. ISBN 978-0-12-821322-3. [Google Scholar]
- Lv, K.; Wang, G.; Shen, C.; Zhang, X.; Yao, H. Role and Mechanism of the Nod-like Receptor Family Pyrin Domain-Containing 3 Inflammasome in Oral Disease. Arch. Oral Biol. 2019, 97, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Ö.; Lee, K.L. The Inflammasome and Danger Molecule Signaling: At the Crossroads of Inflammation and Pathogen Persistence in the Oral Cavity. Periodontol. 2000 2015, 69, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-KappaB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. Baltim. Md 1950 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.L.; Song, D.Z.; Yue, J.L.; Wang, T.T.; Zhou, X.D.; Zhang, P.; Zhang, L.; Huang, D.M. NLRP3 Inflammasome May Regulate Inflammatory Response of Human Periodontal Ligament Fibroblasts in an Apoptosis-Associated Speck-like Protein Containing a CARD (ASC)-Dependent Manner. Int. Endod. J. 2017, 50, 967–975. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Xue, D.; Xie, Y.-F.; Zhu, D.-W.; Dong, Y.-Y.; Wei, C.-C.; Deng, J.-Y. The Negative Feedback Regulation of MicroRNA-146a in Human Periodontal Ligament Cells after Porphyromonas Gingivalis Lipopolysaccharide Stimulation. Inflamm. Res. 2015, 64, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Okano, T.; Ashida, H.; Suzuki, S.; Shoji, M.; Nakayama, K.; Suzuki, T. Porphyromonas Gingivalis Triggers NLRP3-mediated Inflammasome Activation in Macrophages in a Bacterial Gingipains-independent Manner. Eur. J. Immunol. 2018, 48, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yu, W.; Lee, S.; Xu, Q.; Naji, A.; Le, A.D. Bisphosphonate Induces Osteonecrosis of the Jaw in Diabetic Mice via NLRP3/Caspase-1-Dependent IL-1β Mechanism: NLRP3 ACTIVATION IN BRONJ. J. Bone Miner. Res. 2015, 30, 2300–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhou, X.; Zhang, J. Induction of Heme Oxygenase-1 Attenuates Lipopolysaccharide-Induced Inflammasome Activation in Human Gingival Epithelial Cells. Int. J. Mol. Med. 2014, 34, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Wang, C.-Y.; Jin, L. Baicalin Downregulates Porphyromonas Gingivalis Lipopolysaccharide-Upregulated IL-6 and IL-8 Expression in Human Oral Keratinocytes by Negative Regulation of TLR Signaling. PLoS ONE 2012, 7, e51008. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Kyritsis, G.; Graves, D.T.; Amar, S. Interleukin-1 and Tumor Necrosis Factor Activities Partially Account for Calvarial Bone Resorption Induced by Local Injection of Lipopolysaccharide. Infect. Immun. 1999, 67, 4231–4236. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Lee, M.K.S.; Singleton, W.; Achuthan, A.; Lee, M.-C.; O’Brien-Simpson, N.M.; Cook, A.D.; Murphy, A.J.; Dashper, S.G.; Reynolds, E.C.; et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas Gingivalis and Its Outer Membrane Vesicles. Front. Cell. Infect. Microbiol. 2017, 7, 351. [Google Scholar] [CrossRef]
- Cecil, J.D.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Holden, J.A.; Singleton, W.; Perez-Gonzalez, A.; Mansell, A.; Reynolds, E.C. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front. Immunol. 2017, 8, 1017. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Xu, S.; Zhao, H.; Liu, L.; Lv, X.; Hu, F.; Wang, L.; Ji, Q. Hypoxia and Porphyromonas Gingivalis-Lipopolysaccharide Synergistically Induce NLRP3 Inflammasome Activation in Human Gingival Fibroblasts. Int. Immunopharmacol. 2021, 94, 107456. [Google Scholar] [CrossRef] [PubMed]
- Hodge, P.; Michalowicz, B. Genetic Predisposition to Periodontitis in Children and Young Adults: Genetic Predisposition to Periodontitis in Children and Young Adults. Periodontol. 2000 2001, 26, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Sun, J.; Yan, J.; Li, C.; Lv, X. Anti-Inflammatory Effects of Isorhamnetin on LPS-Stimulated Human Gingival Fibroblasts by Activating Nrf2 Signaling Pathway. Microb. Pathog. 2018, 120, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-H.; Hsu, W.-L.; Chen, T.-H.; Chou, T.-C. Activation of Nrf2/HO-1signaling Pathway Involves the Anti-Inflammatory Activity of Magnolol in Porphyromonas Gingivalis Lipopolysaccharide-Stimulated Mouse RAW 264.7 Macrophages. Int. Immunopharmacol. 2015, 29, 770–778. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, C.; Yang, P.; Chao, R.; Yue, Z.; Li, C.; Guo, J.; Li, M. Eldecalcitol Inhibits LPS-Induced NLRP3 Inflammasome-Dependent Pyroptosis in Human Gingival Fibroblasts by Activating the Nrf2/HO-1 Signaling Pathway. Drug Des. Devel. Ther. 2020, 14, 4901–4913. [Google Scholar] [CrossRef]
- Signat, B.; Roques, C.; Poulet, P.; Duffaut, D. Fusobacterium Nucleatum in Periodontal Health and Disease. Curr. Issues Mol. Biol. 2011, 13, 25–36. [Google Scholar]
- Huynh, T.; Kapur, R.V.; Kaplan, C.W.; Cacalano, N.; Kinder Haake, S.; Shi, W.; Sieling, P.; Jewett, A. The Role of Aggregation in Fusobacterium Nucleatum– Induced Immune Cell Death. J. Endod. 2011, 37, 1531–1535. [Google Scholar] [CrossRef]
- Kaplan, C.W.; Lux, R.; Huynh, T.; Jewett, A.; Shi, W.; Haake, S.K. Fusobacterium Nucleatum Apoptosis-Inducing Outer Membrane Protein. J. Dent. Res. 2005, 84, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.W. Fusobacterium Nucleatum: A Commensal-Turned Pathogen. Curr. Opin. Microbiol. 2015, 23, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Hofstad, T.; Skaug, N.; BjØrnland, T. O-Antigenic Cross-Reactivity In Fusobacterium Nucleatum. Acta Pathol. Microbiol. Scand. B 1979, 87, 371–374. [Google Scholar] [CrossRef]
- Bachrach, G.; Rosen, G.; Bellalou, M.; Naor, R.; Sela, M.N. Identification of a Fusobacterium Nucleatum 65 KDa Serine Protease. Oral Microbiol. Immunol. 2004, 19, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Inagaki, S.; Sigurdson, W.; Kuramitsu, H.K. Synergy between Tannerella Forsythia and Fusobacterium Nucleatum in Biofilm Formation. Oral Microbiol. Immunol. 2005, 20, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Tan, K.S. The Danger Signal Extracellular ATP Is an Inducer of Fusobacterium Nucleatum Biofilm Dispersal. Front. Cell. Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, P.I.; Zilm, P.S.; Rogers, A.H. Fusobacterium Nucleatum Supports the Growth of Porphyromonas Gingivalis in Oxygenated and Carbon-Dioxide-Depleted Environments. Microbiology 2002, 148, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, H.; Wang, X.; Lu, Y.; Yang, C.; Yang, P. Coinfection with Fusobacterium Nucleatum Can Enhance the Attachment and Invasion of Porphyromonas Gingivalis or Aggregatibacter Actinomycetemcomitans to Human Gingival Epithelial Cells. Arch. Oral Biol. 2015, 60, 1387–1393. [Google Scholar] [CrossRef]
- Bartold, P.M.; Gully, N.J.; Zilm, P.S.; Rogers, A.H. Identification of Components in Fusobacterium Nucleatum Chemostat-Culture Supernatants That Are Potent Inhibitors of Human Gingival Fibroblast Proliferation. J. Periodontal Res. 1991, 26, 314–322. [Google Scholar] [CrossRef]
- Jeng, J.-H.; Chan, C.-P.; Ho, Y.-S.; Lan, W.-H.; Hsieh, C.-C.; Chang, M.-C. Effects of Butyrate and Propionate on the Adhesion, Growth, Cell Cycle Kinetics, and Protein Synthesis of Cultured Human Gingival Fibroblasts. J. Periodontol. 1999, 70, 1435–1442. [Google Scholar] [CrossRef]
- Taxman, D.J.; Swanson, K.V.; Broglie, P.M.; Wen, H.; Holley-Guthrie, E.; Huang, M.T.-H.; Callaway, J.B.; Eitas, T.K.; Duncan, J.A.; Ting, J.P.Y. Porphyromonas Gingivalis Mediates Inflammasome Repression in Polymicrobial Cultures through a Novel Mechanism Involving Reduced Endocytosis. J. Biol. Chem. 2012, 287, 32791–32799. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.-C.; Huang, P.-R.; Almeida-da-Silva, C.L.C.; Atanasova, K.R.; Yilmaz, O.; Ojcius, D.M. NLRX1 Modulates Differentially NLRP3 Inflammasome Activation and NF-ΚB Signaling during Fusobacterium Nucleatum Infection. Microbes Infect. 2018, 20, 615–625. [Google Scholar] [CrossRef]
- Kong, C.; Yan, X.; Zhu, Y.; Zhu, H.; Luo, Y.; Liu, P.; Ferrandon, S.; Kalady, M.F.; Gao, R.; He, J.; et al. Fusobacterium Nucleatum Promotes the Development of Colorectal Cancer by Activating a Cytochrome P450/Epoxyoctadecenoic Acid Axis via TLR4/Keap1/NRF2 Signaling. Cancer Res. 2021, 81, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.G.; Socransky, S.S.; Savitt, E.D.; Propas, D.A.; Crawford, A. Studies of the Microbiology of Periodontosis. J. Periodontol. 1976, 47, 373–379. [Google Scholar] [CrossRef]
- Nørskov-Lauritsen, N.; Claesson, R.; Jensen, A.B.; Åberg, C.H.; Haubek, D. Aggregatibacter Actinomycetemcomitans: Clinical Significance of a Pathobiont Subjected to Ample Changes in Classification and Nomenclature. Pathogens 2019, 8, 243. [Google Scholar] [CrossRef] [Green Version]
- Fine, D.H.; Markowitz, K.; Furgang, D.; Fairlie, K.; Ferrandiz, J.; Nasri, C.; McKiernan, M.; Gunsolley, J. Aggregatibacter Actinomycetemcomitans and Its Relationship to Initiation of Localized Aggressive Periodontitis: Longitudinal Cohort Study of Initially Healthy Adolescents. J. Clin. Microbiol. 2007, 45, 3859–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, B.A.; Novince, C.M.; Kirkwood, K.L. Aggregatibacter Actinomycetemcomitans, a Potent Immunoregulator of the Periodontal Host Defense System and Alveolar Bone Homeostasis. Mol. Oral Microbiol. 2016, 31, 207–227. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.H.; Sreenivasan, P.K.; Fives-Taylor, P.M. Evidence for Invasion of a Human Oral Cell Line by Actinobacillus Actinomycetemcomitans. Infect. Immun. 1991, 59, 2719–2726. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.T.; Murdoch, D.; Morris, A.; Holland, D.; Pappas, P.; Almela, M.; Fernández-Hidalgo, N.; Almirante, B.; Bouza, E.; Forno, D.; et al. HACEK Infective Endocarditis: Characteristics and Outcomes from a Large, Multi-National Cohort. PLoS ONE 2013, 8, e63181. [Google Scholar] [CrossRef]
- Reisinger, A.; Matzkies, L.-M.; Eller, P.; Fruhwald, F.; Krause, R. Pericardial Empyema Due to Actinomyces Israelii, Aggregatibacter Actinomycetemcomitans, and Fusobacterium Nucleatum. Pol. Arch. Intern. Med. 2019, 129, 714–715. [Google Scholar] [CrossRef] [PubMed]
- Scannapieco, F.A. Role of Oral Bacteria in Respiratory Infection. J. Periodontol. 1999, 70, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Calandrini, C.; Ribeiro, A.; Gonnelli, A.; Ota-Tsuzuki, C.; Rangel, L.; Saba-Chujfi, E.; Mayer, M. Microbial Composition of Atherosclerotic Plaques. Oral Dis. 2014, 20, e128–e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter Actinomycetemcomitans-Induced Hypercitrullination Links Periodontal Infection to Autoimmunity in Rheumatoid Arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korostoff, J.; Yamaguchi, N.; Miller, M.; Kieba, I.; Lally, E.T. Perturbation of Mitochondrial Structure and Function Plays a Central Role in Actinobacillus Actinomycetemcomitans Leukotoxin-Induced Apoptosis. Microb. Pathog. 2000, 29, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Shenker, B.J.; Ojcius, D.M.; Walker, L.P.; Zekavat, A.; Scuron, M.D.; Boesze-Battaglia, K. Aggregatibacter Actinomycetemcomitans Cytolethal Distending Toxin Activates the NLRP3 Inflammasome in Human Macrophages, Leading to the Release of Proinflammatory Cytokines. Infect. Immun. 2015, 83, 1487–1496. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Liu, J.; Pan, C.; Pan, Y. NLRP3 Inflammasome Is Required for Apoptosis of Aggregatibacter Actinomycetemcomitans-Infected Human Osteoblastic MG63 Cells. Acta Histochem. 2014, 116, 1119–1124. [Google Scholar] [CrossRef]
- Okinaga, T.; Ariyoshi, W.; Nishihara, T. Aggregatibacter Actinomycetemcomitans Invasion Induces Interleukin-1β Production Through Reactive Oxygen Species and Cathepsin B. J. Interferon Cytokine Res. 2015, 35, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Kelk, P.; Claesson, R.; Chen, C.; Sjöstedt, A.; Johansson, A. IL-1β Secretion Induced by Aggregatibacter (Actinobacillus) Actinomycetemcomitans Is Mainly Caused by the Leukotoxin. Int. J. Med. Microbiol. 2008, 298, 529–541. [Google Scholar] [CrossRef]
- Kelk, P.; Claesson, R.; Hänström, L.; Lerner, U.H.; Kalfas, S.; Johansson, A. Abundant Secretion of Bioactive Interleukin-1β by Human Macrophages Induced by Actinobacillus Actinomycetemcomitans Leukotoxin. Infect. Immun. 2005, 73, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Ando-Suguimoto, E.S.; Benakanakere, M.R.; Mayer, M.P.A.; Kinane, D.F. Distinct Signaling Pathways Between Human Macrophages and Primary Gingival Epithelial Cells by Aggregatibacter Actinomycetemcomitans. Pathogens 2020, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Stathopoulou, P.G.; Benakanakere, M.R.; Galicia, J.C.; Kinane, D.F. Epithelial Cell Pro-Inflammatory Cytokine Response Differs across Dental Plaque Bacterial Species: Cytokine Responses to Dental Plaque. J. Clin. Periodontol. 2010, 37, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, Y.; Shiba, H.; Komatsuzawa, H.; Takemoto, T.; Sakata, M.; Fujita, T.; Kawaguchi, H.; Sugai, M.; Kurihara, H. Expression of IL-1β and IL-8 by human gingival epithelial cells in response to Actinobacillus Actinomycetemcomitans. Cytokine 2001, 14, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Karamifar, K. Endodontic Periapical Lesion: An Overview on Etiology, Diagnosis and Current Treatment Modalities. Eur. Endod. J. 2020, 5, 54–67. [Google Scholar] [CrossRef]
- Sakko, M.; TjÄDerhane, L.; Rautemaa-Richardson, R. Microbiology of Root Canal Infections. Prim. Dent. J. 2016, 5, 84–89. [Google Scholar] [CrossRef]
- Cintra, L.T.A.; Estrela, C.; Azuma, M.M.; de Azevedo Queiroz, Í.O.; Kawai, T.; Gomes-Filho, J.E. Endodontic Medicine: Interrelationships among Apical Periodontitis, Systemic Disorders, and Tissue Responses of Dental Materials. Braz. Oral Res. 2018, 32 (Suppl. 1), e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran Nair, P.N. Light and Electron Microscopic Studies of Root Canal Flora and Periapical Lesions. J. Endod. 1987, 13, 29–39. [Google Scholar] [CrossRef]
- Schilder, H. Cleaning and Shaping the Root Canal. Dent. Clin. N. Am. 1974, 18, 269–296. [Google Scholar]
- European Society of Endodontology. Quality Guidelines for Endodontic Treatment: Consensus Report of the European Society of Endodontology. Int. Endod. J. 2006, 39, 921–930. [Google Scholar] [CrossRef]
- Hirsch, J.-M.; Ahlström, U.; Henrikson, P.-Å.; Heyden, G.; Peterson, L.-E. Periapical Surgery. Int. J. Oral Surg. 1979, 8, 173–185. [Google Scholar] [CrossRef]
- Barreiros, D.; Pucinelli, C.M.; de Oliveira, K.M.H.; Paula-Silva, F.W.G.; Nelson Filho, P.; da Silva, L.A.B.; Küchler, E.C.; da Silva, R.A.B. Immunohistochemical and MRNA Expression of RANK, RANKL, OPG, TLR2 and MyD88 during Apical Periodontitis Progression in Mice. J. Appl. Oral Sci. 2018, 26, e20170512. [Google Scholar] [CrossRef]
- Yin, W.; Liu, S.; Dong, M.; Liu, Q.; Shi, C.; Bai, H.; Wang, Q.; Yang, X.; Niu, W.; Wang, L. A New NLRP3 Inflammasome Inhibitor, Dioscin, Promotes Osteogenesis. Small 2020, 16, 1905977. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.P.F.A.; Pinheiro, E.T.; Gade-Neto, C.R.; Sousa, E.L.R.; Ferraz, C.C.R.; Zaia, A.A.; Teixeira, F.B.; Souza-Filho, F.J. Microbiological Examination of Infected Dental Root Canals. Oral Microbiol. Immunol. 2004, 19, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Love, R.M. Enterococcus Faecalis - a Mechanism for Its Role in Endodontic Failure: E. Faecalis Virulence. Int. Endod. J. 2001, 34, 399–405. [Google Scholar] [CrossRef]
- Yang, H.-H.; Jun, H.-K.; Jung, Y.-J.; Choi, B.-K. Enterococcus Faecalis Activates Caspase-1 Leading to Increased Interleukin-1 Beta Secretion in Macrophages. J. Endod. 2014, 40, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Tavares, B.S.; Tsosura, T.V.S.; Mattera, M.S.L.C.; Santelli, J.O.; Belardi, B.E.; Chiba, F.Y.; Cintra, L.T.A.; Silva, C.C.; Matsushita, D.H. Effects of Melatonin on Insulin Signaling and Inflammatory Pathways of Rats with Apical Periodontitis. Int. Endod. J. 2021, 54, 926–940. [Google Scholar] [CrossRef]
- Wang, L.; Jin, H.; Ye, D.; Wang, J.; Ao, X.; Dong, M.; Niu, W. Enterococcus Faecalis Lipoteichoic Acid–Induced NLRP3 Inflammasome via the Activation of the Nuclear Factor Kappa B Pathway. J. Endod. 2016, 42, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Xu, B.; Zhao, H.; Wang, Y.; Zhang, J.; Li, C.; Wu, Q.; Cao, Y.; Li, Y.; Cao, F. Dioscin Protects against Coronary Heart Disease by Reducing Oxidative Stress and Inflammation via Sirt1/Nrf2 and P38 MAPK Pathways. Mol. Med. Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, Y.; Qi, Y.; Xu, L.; Song, S.; Yin, L.; Tao, X.; Zhen, Y.; Han, X.; Ma, X.; et al. Protective Effects of Dioscin against Doxorubicin-Induced Nephrotoxicity via Adjusting FXR-Mediated Oxidative Stress and Inflammation. Toxicology 2017, 378, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Q.; Liu, Y. Immunohistochemical Localization of NALP3 Inflammasome in Experimental Periapical Lesions. Int. Endod. J. 2014, 47, 949–957. [Google Scholar] [CrossRef]
- Ran, S.; Liu, B.; Gu, S.; Sun, Z.; Liang, J. Analysis of the Expression of NLRP3 and AIM2 in Periapical Lesions with Apical Periodontitis and Microbial Analysis Outside the Apical Segment of Teeth. Arch. Oral Biol. 2017, 78, 39–47. [Google Scholar] [CrossRef]
- Cheng, R.; Feng, Y.; Zhang, R.; Liu, W.; Lei, L.; Hu, T. The Extent of Pyroptosis Varies in Different Stages of Apical Periodontitis. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2018, 1864, 226–237. [Google Scholar] [CrossRef]
- Ran, S.; Huang, J.; Liu, B.; Gu, S.; Jiang, W.; Liang, J. Enterococcus Faecalis Activates NLRP3 Inflammasomes Leading to Increased Interleukin-1 Beta Secretion and Pyroptosis of THP-1 Macrophages. Microb. Pathog. 2021, 154, 104761. [Google Scholar] [CrossRef]
- Guan, X.; Guan, Y.; Shi, C.; Zhu, X.; He, Y.; Wei, Z.; Yang, J.; Hou, T. Estrogen Deficiency Aggravates Apical Periodontitis by Regulating NLRP3/Caspase-1/IL-1β Axis. Am. J. Transl. Res. 2020, 12, 660–671. [Google Scholar]
- Yoo, Y.-J.; Kim, A.R.; Perinpanayagam, H.; Han, S.H.; Kum, K.-Y. Candida Albicans Virulence Factors and Pathogenicity for Endodontic Infections. Microorganisms 2020, 8, 1300. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Wang, Y.; Jiang, W.; Jia, Q.; Li, J.; Wang, W.; Wang, H.; Ding, Y.; Wang, P.; Liu, J.; et al. Nemotic Human Dental Pulp Fibroblasts Promote Human Dental Pulp Stem Cells Migration. Exp. Cell Res. 2013, 319, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Lin, Z.; He, F.; Jiang, L.; Qin, W.; Tian, Y.; Wang, R.; Huang, S. NLRP3 Is Expressed in Human Dental Pulp Cells and Tissues. J. Endod. 2012, 38, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-I.; Kang, S.-K.; Jung, H.-J.; Chun, Y.-H.; Kwon, Y.-D.; Kim, E.-C. Muramyl Dipeptide Activates Human Beta Defensin 2 and Pro-Inflammatory Mediators through Toll-like Receptors and NLRP3 Inflammasomes in Human Dental Pulp Cells. Clin. Oral Investig. 2015, 19, 1419–1428. [Google Scholar] [CrossRef]
- Jiang, W.; Lv, H.; Wang, H.; Wang, D.; Sun, S.; Jia, Q.; Wang, P.; Song, B.; Ni, L. Activation of the NLRP3/Caspase-1 Inflammasome in Human Dental Pulp Tissue and Human Dental Pulp Fibroblasts. Cell Tissue Res. 2015, 361, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Wang, P.; Ma, X.; Yin, X.; Li, J.; Wang, H.; Jiang, W.; Jia, Q.; Ni, L. Mechanisms That Lead to the Regulation of NLRP3 Inflammasome Expression and Activation in Human Dental Pulp Fibroblasts. Mol. Immunol. 2015, 66, 253–262. [Google Scholar] [CrossRef]
- Wang, D.; Sun, S.; Xue, Y.; Qiu, J.; Ye, T.; Zhang, R.; Song, B.; He, W.; Zhang, Y.; Jiang, W. MicroRNA-223 Negatively Regulates LPS-induced Inflammatory Responses by Targeting NLRP3 in Human Dental Pulp Fibroblasts. Int. Endod. J. 2021, 54, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Taïbi, F.; Metzinger-Le Meuth, V.; Massy, Z.A.; Metzinger, L. MiR-223: An Inflammatory OncomiR Enters the Cardiovascular Field. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2014, 1842, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Gupta, R.; Acharya, A.K.; Patthi, B.; Goud, V.; Reddy, S.; Garg, A.; Singla, A. Changing Trends in Oral Cancer—A Global Scenario. Nepal J. Epidemiol. 2017, 6, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, S.; Yue, K.; Wang, X.-D. The Recurrence and Survival of Oral Squamous Cell Carcinoma: A Report of 275 Cases. Chin. J. Cancer 2013, 32, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Mello, F.W.; Miguel, A.F.P.; Dutra, K.L.; Porporatti, A.L.; Warnakulasuriya, S.; Guerra, E.N.S.; Rivero, E.R.C. Prevalence of Oral Potentially Malignant Disorders: A Systematic Review and Meta-Analysis. J. Oral Pathol. Med. 2018, 47, 633–640. [Google Scholar] [CrossRef]
- Castellsagué, X.; Quintana, M.J.; Martínez, M.C.; Nieto, A.; Sánchez, M.J.; Juan, A.; Monner, A.; Carrera, M.; Agudo, A.; Quer, M.; et al. The Role of Type of Tobacco and Type of Alcoholic Beverage in Oral Carcinogenesis: Tobacco and Alcohol in Oral Carcinogenesis. Int. J. Cancer 2004, 108, 741–749. [Google Scholar] [CrossRef]
- Feller, L.; Altini, M.; Lemmer, J. Inflammation in the Context of Oral Cancer. Oral Oncol. 2013, 49, 887–892. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical Causes of Cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Hooper, S.J.; Wilson, M.J.; Crean, St.J. Exploring the Link between Microorganisms and Oral Cancer: A Systematic Review of the Literature. Head Neck 2009, 31, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Srinivasprasad, V.; Dineshshankar, J.; Sathiyajeeva, J.; Karthikeyan, M.; Sunitha, J.; Ragunathan, R. Liaison between Micro-Organisms and Oral Cancer. J. Pharm. Bioallied Sci. 2015, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, S.; Wang, H.; Tang, Y.-J.; Tang, Y.; Liang, X. Who Is Who in Oral Cancer? Exp. Cell Res. 2019, 384, 111634. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Yeh, Y.-M.; Yu, H.-Y.; Chin, C.-Y.; Hsu, C.-W.; Liu, H.; Huang, P.-J.; Hu, S.-N.; Liao, C.-T.; Chang, K.-P.; et al. Oral Microbiota Community Dynamics Associated With Oral Squamous Cell Carcinoma Staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef] [Green Version]
- Tezal, M.; Sullivan, M.A.; Hyland, A.; Marshall, J.R.; Stoler, D.; Reid, M.E.; Loree, T.R.; Rigual, N.R.; Merzianu, M.; Hauck, L.; et al. Chronic Periodontitis and the Incidence of Head and Neck Squamous Cell Carcinoma. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2406–2412. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Shen, X.; Zhou, M.; Tang, B. Periodontal Pathogens Promote Oral Squamous Cell Carcinoma by Regulating ATR and NLRP3 Inflammasome. Front. Oncol. 2021, 11, 722797. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Ahn, J.; Sampson, J.N.; Shi, J.; Yu, G.; Xiong, X.; Hayes, R.B.; Goedert, J.J. Fecal Microbiota, Fecal Metabolome, and Colorectal Cancer Interrelations. PLoS ONE 2016, 11, e0152126. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jia, Y.; Wen, L.; Mu, W.; Wu, X.; Liu, T.; Liu, X.; Fang, J.; Luan, Y.; Chen, P.; et al. Porphyromonas Gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2021, 81, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The Role of NLRP3 Inflammasome in 5-Fluorouracil Resistance of Oral Squamous Cell Carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Luo, Q.; Wang, H.; Zhang, H.; Chen, F. MicroRNA-22 Suppresses Cell Proliferation, Migration and Invasion in Oral Squamous Cell Carcinoma by Targeting NLRP3. J. Cell. Physiol. 2018, 233, 6705–6713. [Google Scholar] [CrossRef]
- Liu, B.; Lu, Y.; Chen, X.; Muthuraj, P.G.; Li, X.; Pattabiraman, M.; Zempleni, J.; Kachman, S.D.; Natarajan, S.K.; Yu, J. Protective Role of Shiitake Mushroom-Derived Exosome-Like Nanoparticles in D-Galactosamine and Lipopolysaccharide-Induced Acute Liver Injury in Mice. Nutrients 2020, 12, 477. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhou, Y.; Yu, J. Exosome-like Nanoparticles from Ginger Rhizomes Inhibited NLRP3 Inflammasome Activation. Mol. Pharm. 2019, 16, 2690–2699. [Google Scholar] [CrossRef]
- Yang, M.; Luo, Q.; Chen, X.; Chen, F. Bitter Melon Derived Extracellular Vesicles Enhance the Therapeutic Effects and Reduce the Drug Resistance of 5-Fluorouracil on Oral Squamous Cell Carcinoma. J. Nanobiotechnology 2021, 19, 259. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.T.; Doukas, S.G.; Vageli, D.P. In Vivo Short-Term Topical Application of BAY 11-7082 Prevents the Acidic Bile–Induced MRNA and MiRNA Oncogenic Phenotypes in Exposed Murine Hypopharyngeal Mucosa. Neoplasia 2018, 20, 374–386. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Casili, G.; Basilotta, R.; Lanza, M.; Filippone, A.; Raciti, G.; Puliafito, I.; Colarossi, L.; Esposito, E.; Paterniti, I. NLRP3 Inflammasome Inhibitor BAY-117082 Reduces Oral Squamous Cell Carcinoma Progression. Int. J. Mol. Sci. 2021, 22, 11108. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Zhang, L.; Ma, S.-R.; Zhao, Z.-L.; Wang, W.-M.; He, K.-F.; Zhao, Y.-F.; Zhang, W.-F.; Liu, B.; Sun, Z.-J. Clinical Significance of Keap1 and Nrf2 in Oral Squamous Cell Carcinoma. PLoS ONE 2013, 8, e83479. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Paiboonrungruan, C.; Zhang, X.; Prigge, J.R.; Schmidt, E.E.; Sun, Z.; Chen, X. Nrf2 Regulates Cellular Behaviors and Notch Signaling in Oral Squamous Cell Carcinoma Cells. Biochem. Biophys. Res. Commun. 2017, 493, 833–839. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Manoil, D. Microbial Community-Driven Etiopathogenesis of Peri-Implantitis. J. Dent. Res. 2021, 100, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Swami, V.; Vijayaraghavan, V.; Swami, V. Current Trends to Measure Implant Stability. J. Indian Prosthodont. Soc. 2016, 16, 124. [Google Scholar] [CrossRef]
- Carlsson, L.; Röstlund, T.; Albrektsson, B.; Albrektsson, T.; Brånemark, P.-I. Osseointegration of Titanium Implants. Acta Orthop. Scand. 1986, 57, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Amengual-Peñafiel, L.; Brañes-Aroca, M.; Marchesani-Carrasco, F.; Jara-Sepúlveda, M.C.; Parada-Pozas, L.; Cartes-Velásquez, R. Coupling between Osseointegration and Mechanotransduction to Maintain Foreign Body Equilibrium in the Long-Term: A Comprehensive Overview. J. Clin. Med. 2019, 8, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trindade, R.; Albrektsson, T.; Galli, S.; Prgomet, Z.; Tengvall, P.; Wennerberg, A. Osseointegration and Foreign Body Reaction: Titanium Implants Activate the Immune System and Suppress Bone Resorption during the First 4 Weeks after Implantation. Clin. Implant Dent. Relat. Res. 2018, 20, 82–91. [Google Scholar] [CrossRef]
- Zhang, F.; Finkelstein, J. The Relationship between Single Nucleotide Polymorphisms and Dental Implant Loss: A Scoping Review. Clin. Cosmet. Investig. Dent. 2019, Volume 11, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Chappuis, V.; Avila-Ortiz, G.; Araújo, M.G.; Monje, A. Medication-related Dental Implant Failure: Systematic Review and Meta-analysis. Clin. Oral Implant. Res. 2018, 29, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Srinivasan, M.; McKenna, G.; Müller, F. Effect of Advanced Age and/or Systemic Medical Conditions on Dental Implant Survival: A Systematic Review and Meta-analysis. Clin. Oral Implant. Res. 2018, 29, 311–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadadi, A.; Mezied, M. Evidence-Based Analysis of the Effect of Smoking on Osseointegrated Implant Outcome. Natl. J. Maxillofac. Surg. 2021, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Albrektsson, T.; Becker, W.; Coli, P.; Jemt, T.; Mölne, J.; Sennerby, L. Bone Loss around Oral and Orthopedic Implants: An Immunologically Based Condition. Clin. Implant Dent. Relat. Res. 2019, 21, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Eger, M.; Hiram-Bab, S.; Liron, T.; Sterer, N.; Carmi, Y.; Kohavi, D.; Gabet, Y. Mechanism and Prevention of Titanium Particle-Induced Inflammation and Osteolysis. Front. Immunol. 2018, 9, 2963. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.S. Systemic Risk Factors for the Development of Periimplant Diseases. Implant Dent. 2019, 28, 115–119. [Google Scholar] [CrossRef]
- Khammissa, R.A.G.; Feller, L.; Meyerov, R.; Lemmer, J. Peri-Implant Mucositis and Peri-Implantitis: Clinical and Histopathological Characteristics and Treatment. SADJ J. S. Afr. Dent. Assoc. Tydskr. Van Suid-Afr. Tandheelkd. Ver. 2012, 67, 122, 124–126. [Google Scholar]
- Rakic, M.; Galindo-Moreno, P.; Monje, A.; Radovanovic, S.; Wang, H.-L.; Cochran, D.; Sculean, A.; Canullo, L. How Frequent Does Peri-Implantitis Occur? A Systematic Review and Meta-Analysis. Clin. Oral Investig. 2018, 22, 1805–1816. [Google Scholar] [CrossRef]
- Kordbacheh Changi, K.; Finkelstein, J.; Papapanou, P.N. Peri-implantitis Prevalence, Incidence Rate, and Risk Factors: A Study of Electronic Health Records at a U.S. Dental School. Clin. Oral Implant. Res. 2019, 30, 306–314. [Google Scholar] [CrossRef]
- French, D.; Ofec, R.; Levin, L. Long Term Clinical Performance of 10 871 Dental Implants with up to 22 Years of Follow-up: A Cohort Study in 4247 Patients. Clin. Implant Dent. Relat. Res. 2021, 23, 289–297. [Google Scholar] [CrossRef]
- Nowzari, H.; Phamduong, S.; Botero, J.E.; Villacres, M.C.; Rich, S.K. The Profile of Inflammatory Cytokines in Gingival Crevicular Fluid around Healthy Osseointegrated Implants: Profile of Inflammatory Cytokines. Clin. Implant Dent. Relat. Res. 2012, 14, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Duarte, P.M.; Serrão, C.R.; Miranda, T.S.; Zanatta, L.C.S.; Bastos, M.F.; Faveri, M.; Figueiredo, L.C.; Feres, M. Could Cytokine Levels in the Peri-Implant Crevicular Fluid Be Used to Distinguish between Healthy Implants and Implants with Peri-Implantitis? A Systematic Review. J. Periodontal Res. 2016, 51, 689–698. [Google Scholar] [CrossRef]
- Jin, Q.; Teng, F.; Cheng, Z. Association between Common Polymorphisms in IL-1 and TNFα and Risk of Peri-Implant Disease: A Meta-Analysis. PLoS ONE 2021, 16, e0258138. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.; Anwar, M.; Gupta, C.; Chand, P.; Singh, S. Association of Interleukin-1 Gene Polymorphism and Early Crestal Bone Loss around Submerged Dental Implants: A Systematic Review and Meta-Analysis. J. Indian Prosthodont. Soc. 2021, 21, 116. [Google Scholar] [CrossRef]
- de Morais, L.S.; Serra, G.G.; Albuquerque Palermo, E.F.; Andrade, L.R.; Müller, C.A.; Meyers, M.A.; Elias, C.N. Systemic Levels of Metallic Ions Released from Orthodontic Mini-Implants. Am. J. Orthod. Dentofac. Orthop. 2009, 135, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Q.; Pang, Z.; Gong, M.; Tang, L. Titanium Ions Promote Exogenous Calcium-Dependent Calcium Influx in Activated Jurkat T Cells: A Possible Mechanism to Explain Its Immunostimulatory Properties. Mediat. Inflamm. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tang, L.; Thu, Y.M.; Chen, D. Titanium Ions Play a Synergistic Role in the Activation of NLRP3 Inflammasome in Jurkat T Cells. Inflammation 2020, 43, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, F.; Becker, K.; Rahn, S.; Hegewald, A.; Pfeffer, K.; Henrich, B. Real-Time PCR Analysis of Fungal Organisms and Bacterial Species at Peri-Implantitis Sites. Int. J. Implant Dent. 2015, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Wellington, M.; Koselny, K.; Sutterwala, F.S.; Krysan, D.J. Candida Albicans Triggers NLRP3-Mediated Pyroptosis in Macrophages. Eukaryot. Cell 2014, 13, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ling, J.; Jiang, Q. Inflammasomes in Alveolar Bone Loss. Front. Immunol. 2021, 12, 691013. [Google Scholar] [CrossRef]
- Qu, C.; Bonar, S.L.; Hickman-Brecks, C.L.; Abu-Amer, S.; McGeough, M.D.; Peña, C.A.; Broderick, L.; Yang, C.; Grimston, S.K.; Kading, J.; et al. NLRP3 Mediates Osteolysis through Inflammation-dependent and -independent Mechanisms. FASEB J. 2015, 29, 1269–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Alippe, Y.; Wang, C.; Ricci, B.; Xiao, J.; Qu, C.; Zou, W.; Novack, D.V.; Abu-Amer, Y.; Civitelli, R.; Mbalaviele, G. Bone Matrix Components Activate the NLRP3 Inflammasome and Promote Osteoclast Differentiation. Sci. Rep. 2017, 7, 6630. [Google Scholar] [CrossRef]
- Jiang, N.; An, J.; Yang, K.; Liu, J.; Guan, C.; Ma, C.; Tang, X. NLRP3 Inflammasome: A New Target for Prevention and Control of Osteoporosis? Front. Endocrinol. 2021, 12, 752546. [Google Scholar] [CrossRef] [PubMed]
- Cline-Smith, A.; Axelbaum, A.; Shashkova, E.; Chakraborty, M.; Sanford, J.; Panesar, P.; Peterson, M.; Cox, L.; Baldan, A.; Veis, D.; et al. Ovariectomy Activates Chronic Low-Grade Inflammation Mediated by Memory T Cells, Which Promotes Osteoporosis in Mice. J. Bone Miner. Res. 2020, 35, 1174–1187. [Google Scholar] [CrossRef] [PubMed]
- Percegoni, N.; Ferreira, A.; Rodrigues, C.; Rosenthal, D.; Castelo Branco, M.; Rumjanek, V.; Carvalho, D. Profile of Serum IL-1β and IL-10 Shortly after Ovariectomy and Estradiol Replacement in Rats. Horm. Metab. Res. 2009, 41, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Song, J.H.; Oh, S.H.; Kim, J.W.; Lee, M.N.; Piao, X.; Yang, J.W.; Kim, O.S.; Kim, T.S.; Kim, S.H.; et al. Targeting NLRP3 Inflammasome Reduces Age-Related Experimental Alveolar Bone Loss. J. Dent. Res. 2020, 99, 1287–1295. [Google Scholar] [CrossRef]
- Li, H.; Zhong, X.; Chen, Z.; Li, W. Suppression of NLRP3 Inflammasome Improves Alveolar Bone Defect Healing in Diabetic Rats. J. Orthop. Surg. 2019, 14, 167. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, H.; Wei, J.; Lin, S.; Zong, Z.; Gong, F.; Huang, X.; Sun, J.; Li, P.; Lin, H.; et al. Inhibition of Nrf2/HO-1 Signaling Leads to Increased Activation of the NLRP3 Inflammasome in Osteoarthritis. Arthritis Res. Ther. 2019, 21, 300. [Google Scholar] [CrossRef] [Green Version]
- Lagarto, J.L.; Nickdel, M.B.; Kelly, D.J.; Price, A.; Nanchahal, J.; Dunsby, C.; French, P.; Itoh, Y. Autofluorescence Lifetime Reports Cartilage Damage in Osteoarthritis. Sci. Rep. 2020, 10, 2154. [Google Scholar] [CrossRef]
- McAllister, M.J.; Chemaly, M.; Eakin, A.J.; Gibson, D.S.; McGilligan, V.E. NLRP3 as a Potentially Novel Biomarker for the Management of Osteoarthritis. Osteoarthr. Cartil. 2018, 26, 612–619. [Google Scholar] [CrossRef]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory Mediators in Osteoarthritis: A Critical Review of the State-of-the-Art, Current Prospects, and Future Challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Detzen, L.; Cheat, B.; Besbes, A.; Hassan, B.; Marchi, V.; Baroukh, B.; Lesieur, J.; Sadoine, J.; Torrens, C.; Rochefort, G.; et al. NLRP3 Is Involved in Long Bone Edification and the Maturation of Osteogenic Cells. J. Cell. Physiol. 2021, 236, 4455–4469. [Google Scholar] [CrossRef] [PubMed]
- Marx, R.E. Pamidronate (Aredia) and Zoledronate (Zometa) Induced Avascular Necrosis of the Jaws: A Growing Epidemic. J. Oral Maxillofac. Surg. 2003, 61, 1115–1117. [Google Scholar] [CrossRef]
- Anesi, A.; Generali, L.; Sandoni, L.; Pozzi, S.; Grande, A. From Osteoclast Differentiation to Osteonecrosis of the Jaw: Molecular and Clinical Insights. Int. J. Mol. Sci. 2019, 20, 4925. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Munz, A.; Reinert, S.; Hoefert, S. Gingival Fibroblasts and Medication-Related Osteonecrosis of the Jaw: Results by Real-Time and Wound Healing in Vitro Assays. J. Cranio-Maxillofac. Surg. 2019, 47, 1464–1474. [Google Scholar] [CrossRef]
- Ruggiero, S.L.; Dodson, T.B.; Fantasia, J.; Goodday, R.; Aghaloo, T.; Mehrotra, B.; O’Ryan, F. American Association of Oral and Maxillofacial Surgeons Position Paper on Medication-Related Osteonecrosis of the Jaw—2014 Update. J. Oral Maxillofac. Surg. 2014, 72, 1938–1956. [Google Scholar] [CrossRef]
- Tsurushima, H.; Kokuryo, S.; Sakaguchi, O.; Tanaka, J.; Tominaga, K. Bacterial Promotion of Bisphosphonate-Induced Osteonecrosis in Wistar Rats. Int. J. Oral Maxillofac. Surg. 2013, 42, 1481–1487. [Google Scholar] [CrossRef]
- Tanaka, J.; Kokuryo, S.; Yoshiga, D.; Tsurushima, H.; Sakaguchi, O.; Habu, M.; Nishihara, T.; Yoshioka, I.; Tominaga, K. An Osteonecrosis Model Induced by Oral Bisphosphonate in Ovariectomised Rats. Oral Dis. 2015, 21, 969–976. [Google Scholar] [CrossRef]
- Sakaguchi, O.; Kokuryo, S.; Tsurushima, H.; Tanaka, J.; Habu, M.; Uehara, M.; Nishihara, T.; Tominaga, K. Lipopolysaccharide Aggravates Bisphosphonate-Induced Osteonecrosis in Rats. Int. J. Oral Maxillofac. Surg. 2015, 44, 528–534. [Google Scholar] [CrossRef]
- Hallmer, F.; Bjørnland, T.; Andersson, G.; Becktor, J.P.; Kristoffersen, A.K.; Enersen, M. Bacterial Diversity in Medication-Related Osteonecrosis of the Jaw. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2017, 123, 436–444. [Google Scholar] [CrossRef]
- Kizub, D.A.; Miao, J.; Schubert, M.M.; Paterson, A.H.G.; Clemons, M.; Dees, E.C.; Ingle, J.N.; Falkson, C.I.; Barlow, W.E.; Hortobagyi, G.N.; et al. Risk Factors for Bisphosphonate-Associated Osteonecrosis of the Jaw in the Prospective Randomized Trial of Adjuvant Bisphosphonates for Early-Stage Breast Cancer (SWOG 0307). Support. Care Cancer 2021, 29, 2509–2517. [Google Scholar] [CrossRef]
- Kaneko, J.; Okinaga, T.; Hikiji, H.; Ariyoshi, W.; Yoshiga, D.; Habu, M.; Tominaga, K.; Nishihara, T. Zoledronic Acid Exacerbates Inflammation through M1 Macrophage Polarization. Inflamm. Regen. 2018, 38, 16. [Google Scholar] [CrossRef]
- Lee, S.S.; Kim, S.M.; Kim, Y.S.; Lee, S.K. Extensive Protein Expression Changes Induced by Pamidronate in RAW 264.7 Cells as Determined by IP-HPLC. PeerJ 2020, 8, e9202. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.E.; Shanley, D. Formation and Inhibition of Dental Calculus. J. Periodontol. 1969, 40, 643–646. [Google Scholar] [CrossRef]
- Sato, Y.; Iikubo, M.; Nishioka, T.; Yoda, N.; Kusunoki, T.; Nakagawa, A.; Sasaki, K.; Tominaga, T. The Effectiveness of an Actuator-Driven Pulsed Water Jet for the Removal of Artificial Dental Calculus: A Preliminary Study. BMC Oral Health 2020, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.M.; Sottosanti, J.S. A Re-evaluation of Scaling and Root Planing. J. Periodontol. 2021, 92, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, M.; Weijden, G. Risk Factors for Periodontitis: Risk Factors for Periodontitis. Int. J. Dent. Hyg. 2006, 4, 2–7. [Google Scholar] [CrossRef]
- Jepsen, S.; Deschner, J.; Braun, A.; Schwarz, F.; Eberhard, J. Calculus Removal and the Prevention of Its Formation: Calculus Removal and Prevention. Periodontol. 2000 2011, 55, 167–188. [Google Scholar] [CrossRef]
- Mombelli, A. Microbial Colonization of the Periodontal Pocket and Its Significance for Periodontal Therapy. Periodontol. 2000 2018, 76, 85–96. [Google Scholar] [CrossRef]
- Bastendorf, K.-D.; Strafela-Bastendorf, N.; Lussi, A. Mechanical Removal of the Biofilm: Is the Curette Still the Gold Standard? In Monographs in Oral Science; Eick, S., Ed.; S. Karger: Basel, Switzerland, 2021; Volume 29, pp. 105–118. ISBN 978-3-318-06851-1. [Google Scholar]
- Montenegro Raudales, J.L.; Yoshimura, A.; Sm, Z.; Kaneko, T.; Ozaki, Y.; Ukai, T.; Miyazaki, T.; Latz, E.; Hara, Y. Dental Calculus Stimulates Interleukin-1β Secretion by Activating NLRP3 Inflammasome in Human and Mouse Phagocytes. PLoS ONE 2016, 11, e0162865. [Google Scholar] [CrossRef] [PubMed]
- Ziauddin, S.M.; Yoshimura, A.; Montenegro Raudales, J.L.; Ozaki, Y.; Higuchi, K.; Ukai, T.; Kaneko, T.; Miyazaki, T.; Latz, E.; Hara, Y. Crystalline Structure of Pulverized Dental Calculus Induces Cell Death in Oral Epithelial Cells. J. Periodontal Res. 2018, 53, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Xu, S.; Hao, Y.; Peterson, B.; Li, B.; Yang, K.; Lv, X.; Zhou, Q.; Ji, Q. Biological Effects on Tooth Root Surface Topographies Induced by Various Mechanical Treatments. Colloids Surf. B Biointerfaces 2020, 188, 110748. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, X.; Tang, K.; Ye, T.; Duan, C.; Lv, P.; Yan, L.; Wu, X.; Chen, Z.; Liu, J.; et al. Sulforaphane Elicts Dual Therapeutic Effects on Renal Inflammatory Injury and Crystal Deposition in Calcium Oxalate Nephrocalcinosis. Theranostics 2020, 10, 7319–7334. [Google Scholar] [CrossRef]
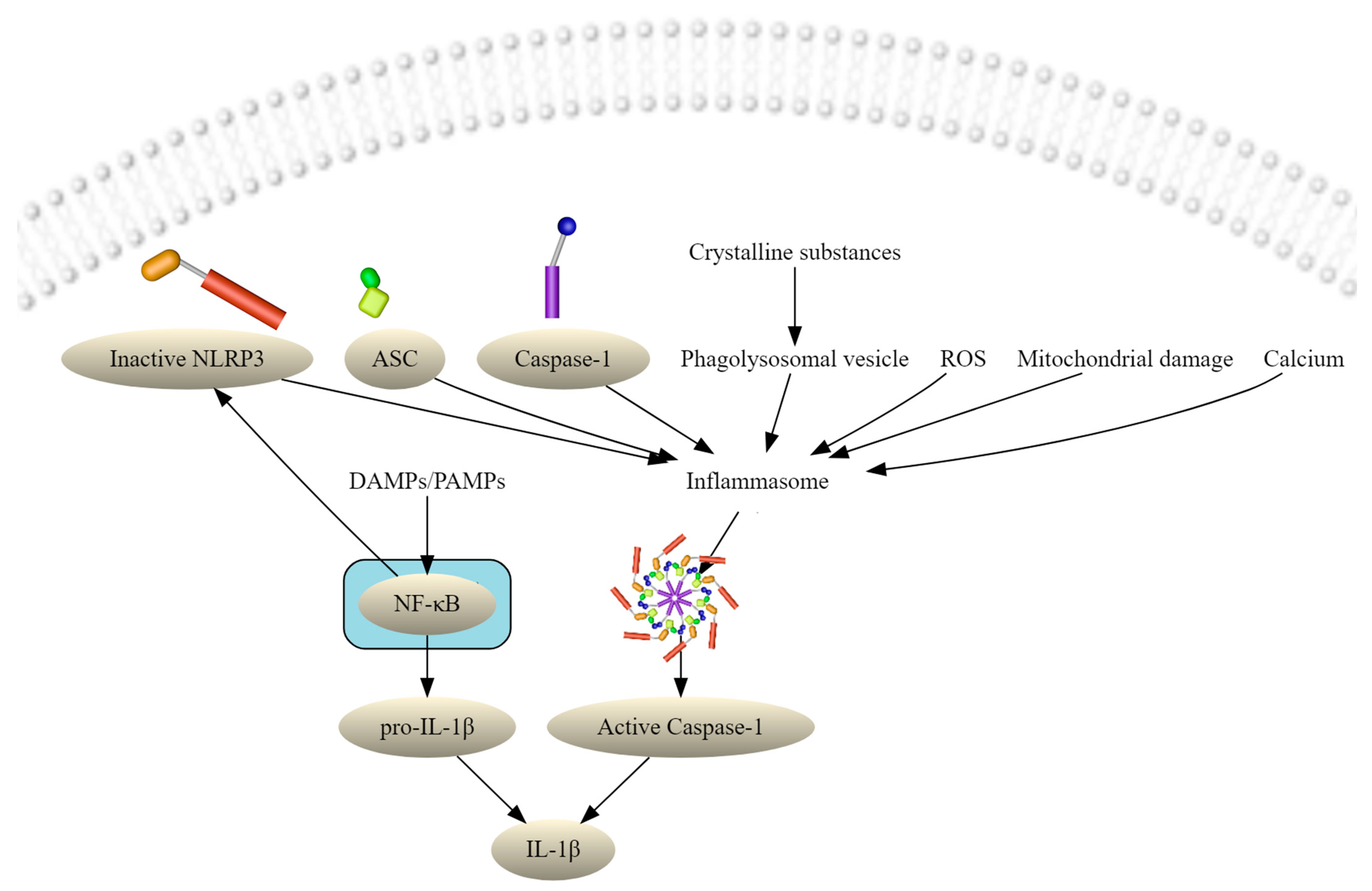
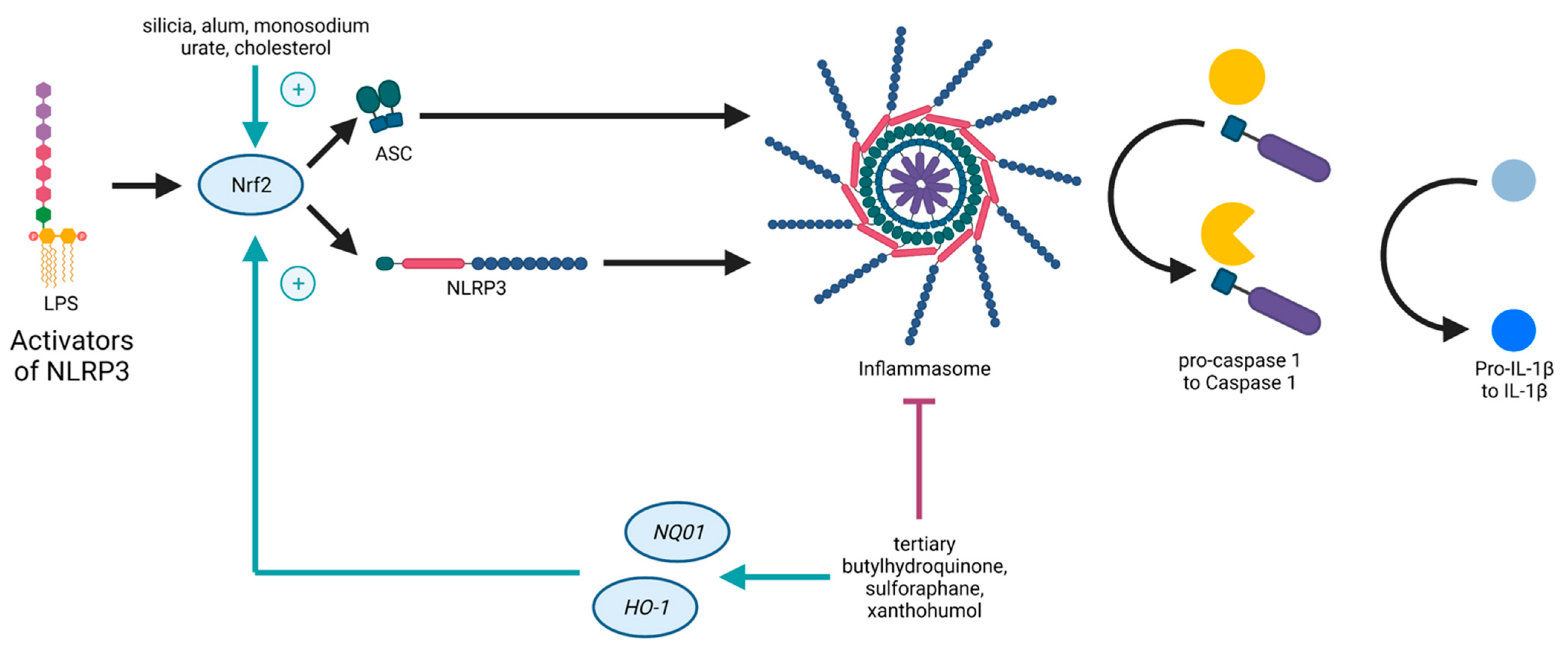
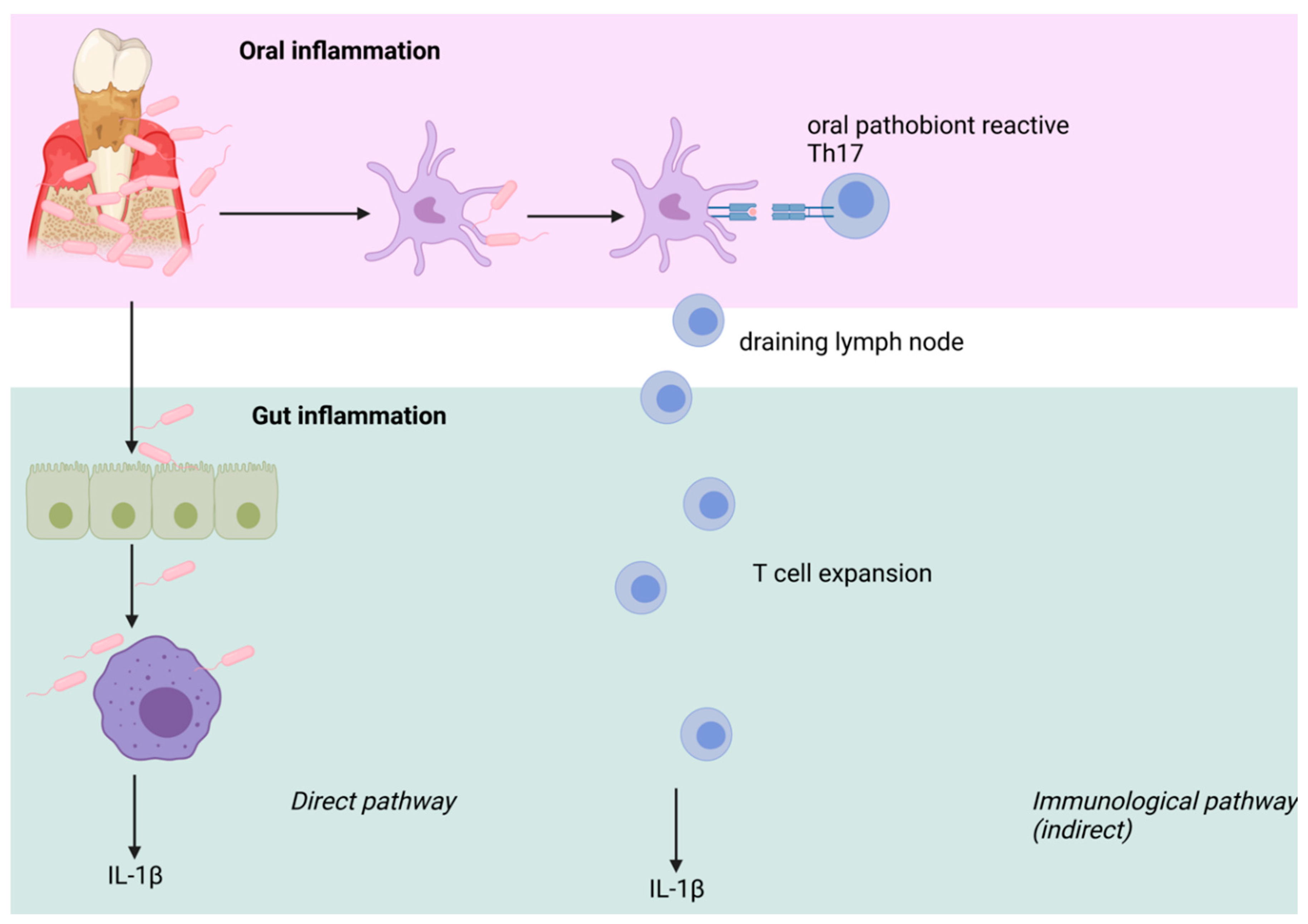

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease/Symptoms | Cause/Association | Mechanism |
|---|---|---|
| Periodontitis | pathogenic bacteria | Activation of NLRP3 and RANKL Nrf2 suppresses subscription of NLRP3-associated genes |
| Colorectal cancer | P. gingivalis | Activation of NLRP3 |
| F. nucleatum | Metastasis by TLR4/Keap1/Nrf2 axis | |
| Periapical periodontis | E. faecalis | Lipoteichoic acid stimulating NF-κB, resulting in a decrease in osteoblasts |
| Bone destruction | P. gingivalis | Outer membrane vesicles, resulting in an increase in osteoclasts Lipopolysaccharide stimulating IL-1β |
| F. nucleatum | via NF-κB, resulting in an increase in osteoclasts | |
| Calculus | Stimulating osteoclasts | |
| Pyroptosis | C. albicans | Affecting macrophages and |
| P. gingivalis | Affecting dental pulp cells | |
| Implants | Via Ti-Ions affecting macrophages | |
| Periimplants | Via Ti-Ions affecting macrophages | |
| Oral squamous cell carcinoma | P. gingivalis | Via cytokines |
| F. nucleatum | Via cytokines |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schieffer, L.; Manzl, C.; Schatz, C.; Haybaeck, J.; Crismani, A. Nrf2 in the Field of Dentistry with Special Attention to NLRP3. Antioxidants 2022, 11, 149. https://doi.org/10.3390/antiox11010149
Schieffer L, Manzl C, Schatz C, Haybaeck J, Crismani A. Nrf2 in the Field of Dentistry with Special Attention to NLRP3. Antioxidants. 2022; 11(1):149. https://doi.org/10.3390/antiox11010149
Chicago/Turabian StyleSchieffer, Lisa, Claudia Manzl, Christoph Schatz, Johannes Haybaeck, and Adriano Crismani. 2022. "Nrf2 in the Field of Dentistry with Special Attention to NLRP3" Antioxidants 11, no. 1: 149. https://doi.org/10.3390/antiox11010149
APA StyleSchieffer, L., Manzl, C., Schatz, C., Haybaeck, J., & Crismani, A. (2022). Nrf2 in the Field of Dentistry with Special Attention to NLRP3. Antioxidants, 11(1), 149. https://doi.org/10.3390/antiox11010149