
Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases

 , ,
, , 
Abstract

1. Introduction
2. Oxidative Stress
3. Oxidative Stress in Cellular and Animal Models of PolyQ Diseases
3.1. Overexpression Cellular Models
3.2. Induced Pluripotent Stem Cell Models
3.3. Animal Models of PolyQ Diseases
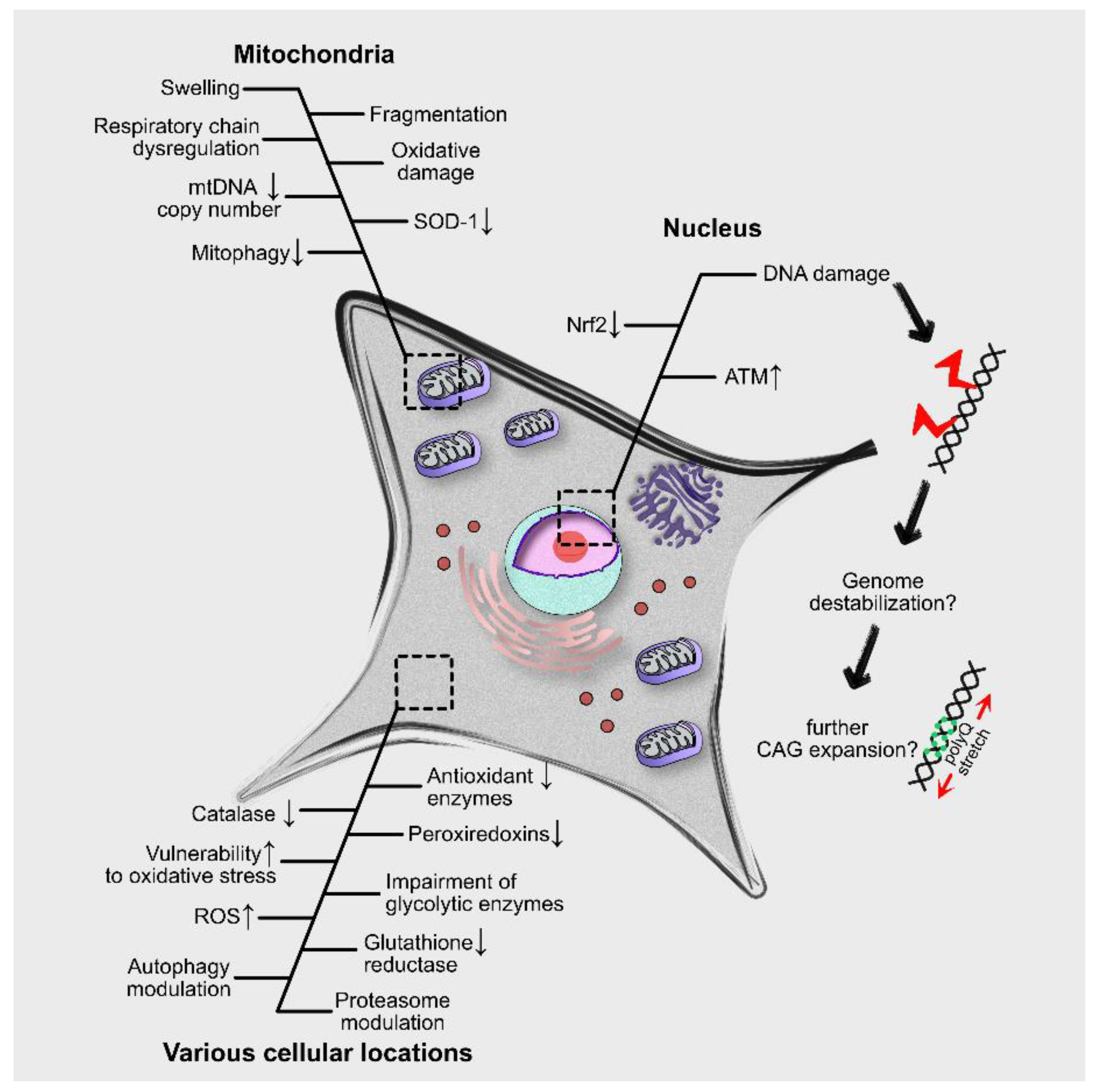
4. Molecular and Cellular Effects of Oxidative Stress in PolyQ Diseases
4.1. Proteasome Modulation
4.2. Autophagy Modulation
4.3. Mitophagy Suppression
4.4. Microglia Activation
5. Oxidative Stress, Neurodegeneration and Neuroinflammation
6. Pharmacological Strategies against PolyQ Diseases
6.1. Preclinical Studies
6.1.1. Antioxidants
6.1.2. Proteasome Activators
6.1.3. Autophagy Inducers
6.1.4. Microglia Suppressors
6.2. Clinical Trials Using Antioxidants
7. Conclusions
Funding
Conflicts of Interest
References
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [CrossRef] [PubMed]
- Haenig, C.; Atias, N.; Taylor, A.K.; Mazza, A.; Schaefer, M.H.; Russ, J.; Riechers, S.P.; Jain, S.; Coughlin, M.; Fontaine, J.F.; et al. Interactome Mapping Provides a Network of Neurodegenerative Disease Proteins and Uncovers Widespread Protein Aggregation in Affected Brains. Cell Rep. 2020, 32, 108050. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics, C. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Choudhry, S.; Mukerji, M.; Srivastava, A.K.; Jain, S.; Brahmachari, S.K. CAG repeat instability at SCA2 locus: Anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum. Mol. Genet. 2001, 10, 2437–2446. [Google Scholar] [CrossRef]
- David, G.; Durr, A.; Stevanin, G.; Cancel, G.; Abbas, N.; Benomar, A.; Belal, S.; Lebre, A.S.; Abada-Bendib, M.; Grid, D.; et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum. Mol. Genet. 1998, 7, 165–170. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T. SCA1-phosphorylation, a regulator of Ataxin-1 function and pathogenesis. Prog. Neurobiol. 2012, 99, 179–185. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Takahashi, H. Spinocerebellar Ataxia Type 17 (SCA17). Adv. Exp. Med. Biol. 2018, 1049, 219–231. [Google Scholar] [CrossRef]
- Tsuji, S. Dentatorubral-pallidoluysian atrophy. Handb. Clin. Neurol. 2012, 103, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, C.; Santos, C.; Montiel, R.; Kay, T.; Vasconcelos, J.; Maciel, P.; Lima, M. The (CAG)n tract of Machado-Joseph Disease gene (ATXN3): A comparison between DNA and mRNA in patients and controls. Eur. J. Hum. Genet. 2010, 18, 621–623. [Google Scholar] [CrossRef]
- Maiuri, T.; Hung, C.L.K.; Suart, C.; Begeja, N.; Barba-Bazan, C.; Peng, Y.; Savic, N.; Wong, T.; Truant, R. DNA Repair in Huntington’s Disease and Spinocerebellar Ataxias: Somatic Instability and Alternative Hypotheses. J. Huntingt. Dis. 2021, 10, 165–173. [Google Scholar] [CrossRef]
- Kim, M. Pathogenic polyglutamine expansion length correlates with polarity of the flanking sequences. Mol. Neurodegener. 2014, 9, 45. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, S.; Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Aggregation of polyQ-extended proteins is promoted by interaction with their natural coiled-coil partners. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhu, S.; Li, X.J.; Li, S. The Expanding Clinical Universe of Polyglutamine Disease. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2019, 25, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Todd, T.W.; Lim, J. Aggregation formation in the polyglutamine diseases: Protection at a cost? Mol. Cells 2013, 36, 185–194. [Google Scholar] [CrossRef]
- Matos, C.A.; Almeida, L.P.; Nobrega, C. Proteolytic Cleavage of Polyglutamine Disease-Causing Proteins: Revisiting the Toxic Fragment Hypothesis. Curr. Pharm. Des. 2017, 23, 753–775. [Google Scholar] [CrossRef] [PubMed]
- Michalik, A.; Van Broeckhoven, C. Pathogenesis of polyglutamine disorders: Aggregation revisited. Hum. Mol. Genet. 2003, 12 (Suppl. S2), R173–R186. [Google Scholar] [CrossRef]
- Totzeck, F.; Andrade-Navarro, M.A.; Mier, P. The Protein Structure Context of PolyQ Regions. PLoS ONE 2017, 12, e0170801. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Paulson, H.L. Polyglutamine neurodegeneration: Protein misfolding revisited. Trends Neurosci. 2008, 31, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Concept and Some Practical Aspects. Antioxidants 2020, 9, 852. [Google Scholar] [CrossRef]
- Hasegawa, T.; Bando, A.; Tsuchiya, K.; Abe, S.; Okamoto, M.; Kirima, K.; Ueno, S.; Yoshizumi, M.; Houchi, H.; Tamaki, T. Enzymatic and nonenzymatic formation of reactive oxygen species from 6-anilino-5,8-quinolinequinone. Biochim. Biophys. Acta 2004, 1670, 19–27. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Leutner, S.; Eckert, A.; Muller, W.E. ROS generation, lipid peroxidation and antioxidant enzyme activities in the aging brain. J. Neural Transm. 2001, 108, 955–967. [Google Scholar] [CrossRef]
- Lu, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.; Allsop, D. Production of reactive oxygen species from aggregating proteins implicated in Alzheimer’s disease, Parkinson’s disease and other neurodegenerative diseases. Curr. Top. Med. Chem. 2001, 1, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A. Postmortem studies in Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 281–293. [Google Scholar]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Pacheco, L.S.; da Silveira, A.F.; Trott, A.; Houenou, L.J.; Algarve, T.D.; Bello, C.; Lenz, A.F.; Manica-Cattani, M.F.; da Cruz, I.B. Association between Machado-Joseph disease and oxidative stress biomarkers. Mutat. Res. 2013, 757, 99–103. [Google Scholar] [CrossRef]
- De Assis, A.M.; Saute, J.A.M.; Longoni, A.; Haas, C.B.; Torrez, V.R.; Brochier, A.W.; Souza, G.N.; Furtado, G.V.; Gheno, T.C.; Russo, A.; et al. Peripheral Oxidative Stress Biomarkers in Spinocerebellar Ataxia Type 3/Machado-Joseph Disease. Front. Neurol. 2017, 8, 485. [Google Scholar] [CrossRef]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Hakonen, A.H.; Goffart, S.; Marjavaara, S.; Paetau, A.; Cooper, H.; Mattila, K.; Lampinen, M.; Sajantila, A.; Lonnqvist, T.; Spelbrink, J.N.; et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 2008, 17, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.C.; Kuo, C.L.; Cheng, W.L.; Liu, C.S.; Hsieh, M. Decreased antioxidant enzyme activity and increased mitochondrial DNA damage in cellular models of Machado-Joseph disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar] [CrossRef]
- Bertoni, A.; Giuliano, P.; Galgani, M.; Rotoli, D.; Ulianich, L.; Adornetto, A.; Santillo, M.R.; Porcellini, A.; Avvedimento, V.E. Early and late events induced by polyQ-expanded proteins: Identification of a common pathogenic property of polYQ-expanded proteins. J. Biol. Chem. 2011, 286, 4727–4741. [Google Scholar] [CrossRef]
- Ajayi, A.; Yu, X.; Lindberg, S.; Langel, U.; Strom, A.L. Expanded ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible Spinocerebellar ataxia type 7 (SCA7) model. BMC Neurosci. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, P.; De Cristofaro, T.; Affaitati, A.; Pizzulo, G.M.; Feliciello, A.; Criscuolo, C.; De Michele, G.; Filla, A.; Avvedimento, E.V.; Varrone, S. DNA damage induced by polyglutamine-expanded proteins. Hum. Mol. Genet. 2003, 12, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lim, P.J.; Yin, C.; Rieckher, M.; Vogel, B.E.; Monteiro, M.J. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum. Mol. Genet. 2006, 15, 1025–1041. [Google Scholar] [CrossRef]
- Reina, C.P.; Zhong, X.; Pittman, R.N. Proteotoxic stress increases nuclear localization of ataxin-3. Hum. Mol. Genet. 2010, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.; Dikshit, P.; Mishra, A.; Mulherkar, S.; Nukina, N.; Jana, N.R. Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem. Biophys. Res. Commun. 2006, 342, 184–190. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Petropoulos, I.; Grimm, S.; Georgila, K.; Catalgol, B.; Friguet, B.; Grune, T.; Gonos, E.S. Protein damage, repair and proteolysis. Mol. Asp. Med. 2014, 35, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Delenclos, M.; Burgess, J.D.; Lamprokostopoulou, A.; Outeiro, T.F.; Vekrellis, K.; McLean, P.J. Cellular models of alpha-synuclein toxicity and aggregation. J. Neurochem. 2019, 150, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Samara, A.; Galbiati, M.; Luciani, P.; Deledda, C.; Messi, E.; Peri, A.; Maggi, R. Altered expression of 3-betahydroxysterol delta-24-reductase/selective Alzheimer’s disease indicator-1 gene in Huntington’s disease models. J. Endocrinol. Investig. 2014, 37, 729–737. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules 2020, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.N.; Yu, Y.V.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef]
- Laidou, S.; Alanis-Lobato, G.; Pribyl, J.; Rasko, T.; Tichy, B.; Mikulasek, K.; Tsagiopoulou, M.; Oppelt, J.; Kastrinaki, G.; Lefaki, M.; et al. Nuclear inclusions of pathogenic ataxin-1 induce oxidative stress and perturb the protein synthesis machinery. Redox Biol. 2020, 32, 101458. [Google Scholar] [CrossRef]
- Vagiona, A.C.; Andrade-Navarro, M.A.; Psomopoulos, F.; Petrakis, S. Dynamics of a Protein Interaction Network Associated to the Aggregation of polyQ-Expanded Ataxin-1. Genes 2020, 11, 1129. [Google Scholar] [CrossRef]
- Drakulic, D.; Djurovic, S.; Syed, Y.A.; Trattaro, S.; Caporale, N.; Falk, A.; Ofir, R.; Heine, V.M.; Chawner, S.; Rodriguez-Moreno, A.; et al. Copy number variants (CNVs): A powerful tool for iPSC-based modelling of ASD. Mol. Autism 2020, 11, 42. [Google Scholar] [CrossRef]
- Chuang, C.Y.; Yang, C.C.; Soong, B.W.; Yu, C.Y.; Chen, S.H.; Huang, H.P.; Kuo, H.C. Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology. Sci. Rep. 2019, 9, 1166. [Google Scholar] [CrossRef]
- Chae, J.I.; Kim, D.W.; Lee, N.; Jeon, Y.J.; Jeon, I.; Kwon, J.; Kim, J.; Soh, Y.; Lee, D.S.; Seo, K.S.; et al. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem. J. 2012, 446, 359–371. [Google Scholar] [CrossRef]
- Switonska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Marczak, L.; Stelmaszczuk, M.; Figlerowicz, M.; Figiel, M. Identification of Altered Developmental Pathways in Human Juvenile HD iPSC With 71Q and 109Q Using Transcriptome Profiling. Front. Cell. Neurosci. 2018, 12, 528. [Google Scholar] [CrossRef]
- Consortium, H.D.i. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef]
- Consortium, H.D.i. Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum. Mol. Genet. 2020, 29, 1757–1771. [Google Scholar] [CrossRef]
- Bono-Yague, J.; Gomez-Escribano, A.P.; Millan, J.M.; Vazquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Jhang, Y.L.; Cheng, P.H.; Chang, Y.F.; Mao, S.H.; Yang, H.I.; Lin, C.W.; Chen, C.M.; Yang, S.H. The Truncated C-terminal Fragment of Mutant ATXN3 Disrupts Mitochondria Dynamics in Spinocerebellar Ataxia Type 3 Models. Front. Mol. Neurosci. 2017, 10, 196. [Google Scholar] [CrossRef]
- Orr, A.L.; Li, S.; Wang, C.E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef]
- Ocampo, A.; Zambrano, A.; Barrientos, A. Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 1431–1441. [Google Scholar] [CrossRef]
- Jonson, I.; Ougland, R.; Klungland, A.; Larsen, E. Oxidative stress causes DNA triplet expansion in Huntington’s disease mouse embryonic stem cells. Stem Cell Res. 2013, 11, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Popovic, N.; Brundin, P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx 2005, 2, 447–464. [Google Scholar] [CrossRef]
- Perez-Severiano, F.; Rios, C.; Segovia, J. Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Res. 2000, 862, 234–237. [Google Scholar] [CrossRef]
- Perez-Severiano, F.; Escalante, B.; Vergara, P.; Rios, C.; Segovia, J. Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res. 2002, 951, 36–42. [Google Scholar] [CrossRef]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Torres, K.; Berrios, L.; Rosario, N.; Dufault, V.; Skatchkov, S.; Eaton, M.J.; Torres-Ramos, C.A.; Ayala-Torres, S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair 2009, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382. [Google Scholar] [CrossRef]
- Agrawal, S.; Fox, J.H. Novel proteomic changes in brain mitochondria provide insights into mitochondrial dysfunction in mouse models of Huntington’s disease. Mitochondrion 2019, 47, 318–329. [Google Scholar] [CrossRef]
- Choo, Y.S.; Mao, Z.; Johnson, G.V.; Lesort, M. Increased glutathione levels in cortical and striatal mitochondria of the R6/2 Huntington’s disease mouse model. Neurosci. Lett. 2005, 386, 63–68. [Google Scholar] [CrossRef]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Oxidative metabolism and Ca(2+) handling in striatal mitochondria from YAC128 mice, a model of Huntington’s disease. Neurochem. Int. 2017, 109, 24–33. [Google Scholar] [CrossRef]
- Brocardo, P.S.; McGinnis, E.; Christie, B.R.; Gil-Mohapel, J. Time-Course Analysis of Protein and Lipid Oxidation in the Brains of Yac128 Huntington’s Disease Transgenic Mice. Rejuvenation Res. 2016, 19, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Seo, H.; Kwak, M.; Jeon, J.; Jang, J.; Jeong, E.M.; Myeong, J.; Hwang, Y.J.; Ha, K.; Kang, M.J.; et al. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 2015, 138, 3030–3047. [Google Scholar] [CrossRef]
- de Paula Nascimento-Castro, C.; Wink, A.C.; da Fonseca, V.S.; Bianco, C.D.; Winkelmann-Duarte, E.C.; Farina, M.; Rodrigues, A.L.S.; Gil-Mohapel, J.; de Bem, A.F.; Brocardo, P.S. Antidepressant Effects of Probucol on Early-Symptomatic YAC128 Transgenic Mice for Huntington’s Disease. Neural Plast. 2018, 2018, 4056383. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Workman, J.; Hart, P.E.; Mangiarini, L.; Mahal, A.; Bates, G.; Cooper, J.M.; Schapira, A.H. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol. 2000, 47, 80–86. [Google Scholar] [CrossRef]
- Aidt, F.H.; Nielsen, S.M.; Kanters, J.; Pesta, D.; Nielsen, T.T.; Norremolle, A.; Hasholt, L.; Christiansen, M.; Hagen, C.M. Dysfunctional mitochondrial respiration in the striatum of the Huntington’s disease transgenic R6/2 mouse model. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Ennis, M.D. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, RC202. [Google Scholar] [CrossRef]
- Miller, B.R.; Dorner, J.L.; Bunner, K.D.; Gaither, T.W.; Klein, E.L.; Barton, S.J.; Rebec, G.V. Up-regulation of GLT1 reverses the deficit in cortically evoked striatal ascorbate efflux in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2012, 121, 629–638. [Google Scholar] [CrossRef]
- Acuna, A.I.; Esparza, M.; Kramm, C.; Beltran, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef]
- Stucki, D.M.; Ruegsegger, C.; Steiner, S.; Radecke, J.; Murphy, M.P.; Zuber, B.; Saxena, S. Mitochondrial impairments contribute to Spinocerebellar ataxia type 1 progression and can be ameliorated by the mitochondria-targeted antioxidant MitoQ. Free Radic. Biol. Med. 2016, 97, 427–440. [Google Scholar] [CrossRef] [PubMed]
- del Cano-Espinel, M.; Acebes, J.R.; Sanchez, D.; Ganfornina, M.D. Lazarillo-related Lipocalins confer long-term protection against type I Spinocerebellar Ataxia degeneration contributing to optimize selective autophagy. Mol. Neurodegener. 2015, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Silva, S.; Neves-Carvalho, A.; Soares-Cunha, C.; Silva, J.M.; Teixeira-Castro, A.; Vieira, R.; Silva-Fernandes, A.; Maciel, P. Neuroprotective Effects of Creatine in the CMVMJD135 Mouse Model of Spinocerebellar Ataxia Type 3. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 815–826. [Google Scholar] [CrossRef]
- Snijder, P.M.; Baratashvili, M.; Grzeschik, N.A.; Leuvenink, H.G.D.; Kuijpers, L.; Huitema, S.; Schaap, O.; Giepmans, B.N.G.; Kuipers, J.; Miljkovic, J.L.; et al. Overexpression of Cystathionine gamma-Lyase Suppresses Detrimental Effects of Spinocerebellar Ataxia Type 3. Mol. Med. 2016, 21, 758–768. [Google Scholar] [CrossRef]
- Wu, Y.L.; Chang, J.C.; Lin, W.Y.; Li, C.C.; Hsieh, M.; Chen, H.W.; Wang, T.S.; Wu, W.T.; Liu, C.S.; Liu, K.L. Caffeic acid and resveratrol ameliorate cellular damage in cell and Drosophila models of spinocerebellar ataxia type 3 through upregulation of Nrf2 pathway. Free Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef]
- Wu, Y.L.; Chang, J.C.; Lin, W.Y.; Li, C.C.; Hsieh, M.; Chen, H.W.; Wang, T.S.; Liu, C.S.; Liu, K.L. Treatment with Caffeic Acid and Resveratrol Alleviates Oxidative Stress Induced Neurotoxicity in Cell and Drosophila Models of Spinocerebellar Ataxia Type3. Sci. Rep. 2017, 7, 11641. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta 2014, 1843, 86–96. [Google Scholar] [CrossRef]
- Orlowski, M.; Wilk, S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch. Biochem. Biophys. 2000, 383, 1–16. [Google Scholar] [CrossRef]
- Papaevgeniou, N.; Chondrogianni, N. UPS Activation in the Battle Against Aging and Aggregation-Related Diseases: An Extended Review. Methods Mol. Biol. 2016, 1449, 1–70. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001, 83, 301–310. [Google Scholar] [CrossRef]
- Lefaki, M.; Papaevgeniou, N.; Chondrogianni, N. Redox regulation of proteasome function. Redox Biol. 2017, 13, 452–458. [Google Scholar] [CrossRef]
- Seo, H.; Sonntag, K.C.; Isacson, O. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann. Neurol. 2004, 56, 319–328. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kukushkin, Y.; Gupta, R.; Chen, T.; Konagai, A.; Hipp, M.S.; Hayer-Hartl, M.; Hartl, F.U. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 2013, 154, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, S.; Rasko, T.; Russ, J.; Friedrich, R.P.; Stroedicke, M.; Riechers, S.P.; Muehlenberg, K.; Moller, A.; Reinhardt, A.; Vinayagam, A.; et al. Identification of human proteins that modify misfolding and proteotoxicity of pathogenic ataxin-1. PLoS Genet. 2012, 8, e1002897. [Google Scholar] [CrossRef] [PubMed]
- Bett, J.S.; Goellner, G.M.; Woodman, B.; Pratt, G.; Rechsteiner, M.; Bates, G.P. Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington’s disease mice: Exclusion of proteasome activator REGgamma as a therapeutic target. Hum. Mol. Genet. 2006, 15, 33–44. [Google Scholar] [CrossRef]
- Maynard, C.J.; Bottcher, C.; Ortega, Z.; Smith, R.; Florea, B.I.; Diaz-Hernandez, M.; Brundin, P.; Overkleeft, H.S.; Li, J.Y.; Lucas, J.J.; et al. Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc. Natl. Acad. Sci. USA 2009, 106, 13986–13991. [Google Scholar] [CrossRef]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global changes to the ubiquitin system in Huntington’s disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef]
- Kim, W.; Seo, H. Baclofen, a GABAB receptor agonist, enhances ubiquitin-proteasome system functioning and neuronal survival in Huntington’s disease model mice. Biochem. Biophys. Res. Commun. 2014, 443, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Khan, L.A.; Bauer, P.O.; Miyazaki, H.; Lindenberg, K.S.; Landwehrmeyer, B.G.; Nukina, N. Expanded polyglutamines impair synaptic transmission and ubiquitin-proteasome system in Caenorhabditis elegans. J. Neurochem. 2006, 98, 576–587. [Google Scholar] [CrossRef]
- Vilchez, D.; Morantte, I.; Liu, Z.; Douglas, P.M.; Merkwirth, C.; Rodrigues, A.P.; Manning, G.; Dillin, A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 2012, 489, 263–268. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Nollen, E.A.; Garcia, S.M.; van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Park, Y.; Hong, S.; Kim, S.J.; Kang, S. Proteasome function is inhibited by polyglutamine-expanded ataxin-1, the SCA1 gene product. Mol. Cells 2005, 19, 23–30. [Google Scholar] [PubMed]
- Persengiev, S.; Kondova, I.; Bontrop, R.E. Functional Annotation of Small Noncoding RNAs Target Genes Provides Evidence for a Deregulated Ubiquitin-Proteasome Pathway in Spinocerebellar Ataxia Type 1. J. Nucleic Acids 2012, 2012, 672536. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhai, R.G.; Zhang, F.; Hiesinger, P.R.; Cao, Y.; Haueter, C.M.; Bellen, H.J. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 2008, 452, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef]
- Wang, H.; Ying, Z.; Wang, G. Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J. Biol. Chem. 2012, 287, 28576–28585. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Koppenhafer, S.L.; Shoesmith, S.J.; Perez, M.K.; Paulson, H.L. Evidence for proteasome involvement in polyglutamine disease: Localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Genet. 1999, 8, 673–682. [Google Scholar] [CrossRef]
- Torashima, T.; Koyama, C.; Iizuka, A.; Mitsumura, K.; Takayama, K.; Yanagi, S.; Oue, M.; Yamaguchi, H.; Hirai, H. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008, 9, 393–399. [Google Scholar] [CrossRef]
- Matsumoto, M.; Yada, M.; Hatakeyama, S.; Ishimoto, H.; Tanimura, T.; Tsuji, S.; Kakizuka, A.; Kitagawa, M.; Nakayama, K.I. Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J. 2004, 23, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Li, X.J.; Li, H.; Li, S. Clearance of mutant huntingtin. Autophagy 2010, 6, 663–664. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Atwal, R.S.; Truant, R. A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 2008, 4, 91–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Kurosawa, M.; Matsumoto, G.; Kino, Y.; Okuno, M.; Kurosawa-Yamada, M.; Washizu, C.; Taniguchi, H.; Nakaso, K.; Yanagawa, T.; Warabi, E.; et al. Depletion of p62 reduces nuclear inclusions and paradoxically ameliorates disease phenotypes in Huntington’s model mice. Hum. Mol. Genet. 2015, 24, 1092–1105. [Google Scholar] [CrossRef]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef]
- Lai, S.; O’Callaghan, B.; Zoghbi, H.Y.; Orr, H.T. 14-3-3 Binding to ataxin-1(ATXN1) regulates its dephosphorylation at Ser-776 and transport to the nucleus. J. Biol. Chem. 2011, 286, 34606–34616. [Google Scholar] [CrossRef]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef]
- Herzog, L.K.; Kevei, E.; Marchante, R.; Bottcher, C.; Bindesboll, C.; Lystad, A.H.; Pfeiffer, A.; Gierisch, M.E.; Salomons, F.A.; Simonsen, A.; et al. The Machado-Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell 2020, 19, e13051. [Google Scholar] [CrossRef]
- Saitoh, Y.; Fujikake, N.; Okamoto, Y.; Popiel, H.A.; Hatanaka, Y.; Ueyama, M.; Suzuki, M.; Gaumer, S.; Murata, M.; Wada, K.; et al. p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J. Biol. Chem. 2015, 290, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Deglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011, 134, 1400–1415. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2020, 14, 612757. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Hwang, S.; Disatnik, M.H.; Mochly-Rosen, D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol. Med. 2015, 7, 1307–1326. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. Methods Mol. Biol. 2019, 2034, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNgamma+TNFalpha) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Stolzing, A.; Sethe, S.; Grune, T. Chronically active: Activation of microglial proteolysis in ageing and neurodegeneration. Redox Rep. Commun. Free Radic. Res. 2005, 10, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Dimayuga, F.O.; Wang, C.; Clark, J.M.; Dimayuga, E.R.; Dimayuga, V.M.; Bruce-Keller, A.J. SOD1 overexpression alters ROS production and reduces neurotoxic inflammatory signaling in microglial cells. J. Neuroimmunol. 2007, 182, 89–99. [Google Scholar] [CrossRef]
- Kim, A.; Garcia-Garcia, E.; Straccia, M.; Comella-Bolla, A.; Miguez, A.; Masana, M.; Alberch, J.; Canals, J.M.; Rodriguez, M.J. Reduced Fractalkine Levels Lead to Striatal Synaptic Plasticity Deficits in Huntington’s Disease. Front. Cell. Neurosci. 2020, 14, 163. [Google Scholar] [CrossRef]
- Comella Bolla, A.; Valente, T.; Miguez, A.; Brito, V.; Gines, S.; Sola, C.; Straccia, M.; Canals, J.M. CD200 is up-regulated in R6/1 transgenic mouse model of Huntington’s disease. PLoS ONE 2019, 14, e0224901. [Google Scholar] [CrossRef]
- Paldino, E.; D’Angelo, V.; Sancesario, G.; Fusco, F.R. Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: New insight on the inflammasome. Cell Death Discov. 2020, 6, 69. [Google Scholar] [CrossRef]
- Franciosi, S.; Ryu, J.K.; Shim, Y.; Hill, A.; Connolly, C.; Hayden, M.R.; McLarnon, J.G.; Leavitt, B.R. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2012, 45, 438–449. [Google Scholar] [CrossRef]
- Rub, U.; Schols, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Ingram, M.; Orr, H.; Opal, P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015, 289, 289–299. [Google Scholar] [CrossRef]
- Qu, W.; Johnson, A.; Kim, J.H.; Lukowicz, A.; Svedberg, D.; Cvetanovic, M. Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J. Neuroinflamm. 2017, 14, 107. [Google Scholar] [CrossRef]
- Rub, U.; de Vos, R.A.; Schultz, C.; Brunt, E.R.; Paulson, H.; Braak, H. Spinocerebellar ataxia type 3 (Machado-Joseph disease): Severe destruction of the lateral reticular nucleus. Brain 2002, 125, 2115–2124. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Sone, M.; Yamashita, M.; Wanker, E.E.; Okazawa, H. Glial cell lineage expression of mutant ataxin-1 and huntingtin induces developmental and late-onset neuronal pathologies in Drosophila models. PLoS ONE 2009, 4, e4262. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.; Shin, J.Y.; Roberts, M.; Wang, C.E.; Li, X.J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef]
- Olejniczak, M.; Urbanek, M.O.; Krzyzosiak, W.J. The role of the immune system in triplet repeat expansion diseases. Mediat. Inflamm. 2015, 2015, 873860. [Google Scholar] [CrossRef]
- Didonna, A.; Canto Puig, E.; Ma, Q.; Matsunaga, A.; Ho, B.; Caillier, S.J.; Shams, H.; Lee, N.; Hauser, S.L.; Tan, Q.; et al. Ataxin-1 regulates B cell function and the severity of autoimmune experimental encephalomyelitis. Proc. Natl. Acad. Sci. USA 2020, 117, 23742–23750. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimer’s Dis. 2014, 42 (Suppl. S3), S152. [Google Scholar] [CrossRef]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef]
- Krieger, S.C.; Cook, K.; De Nino, S.; Fletcher, M. The topographical model of multiple sclerosis: A dynamic visualization of disease course. Neurol. Neuroimmunol. Neuroinflam. 2016, 3, e279. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Bruck, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, J.; Witte, M.E.; Schreibelt, G.; de Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta 2011, 1812, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Bruck, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet. Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.C.; Nobre, R.J.; Pereira de Almeida, L. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, Y.; Yang, W.; Liu, D.; Ji, X.; Chi, T.; Guo, Z.; Li, L.; Zou, L. Prevention of Huntington’s Disease-Like Behavioral Deficits in R6/1 Mouse by Tolfenamic Acid Is Associated with Decreases in Mutant Huntingtin and Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 4032428. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef]
- Polyzos, A.; Holt, A.; Brown, C.; Cosme, C.; Wipf, P.; Gomez-Marin, A.; Castro, M.R.; Ayala-Pena, S.; McMurray, C.T. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum. Mol. Genet. 2016, 25, 1792–1802. [Google Scholar] [CrossRef]
- Machiela, E.; Dues, D.J.; Senchuk, M.M.; Van Raamsdonk, J.M. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol. Dis. 2016, 96, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Sonani, R.R.; Awasthi, A.; Prasad, B.; Patel, A.R.; Kumar, J.; Madamwar, D. Phycocyanin moderates aging and proteotoxicity in Caenorhabditis elegans. J. Appl. Phycol. 2016, 28, 2407–2417. [Google Scholar] [CrossRef]
- Bicca Obetine Baptista, F.; Arantes, L.P.; Machado, M.L.; da Silva, A.F.; Marafiga Cordeiro, L.; da Silveira, T.L.; Soares, F.A.A. Diphenyl diselenide protects a Caenorhabditis elegans model for Huntington’s disease by activation of the antioxidant pathway and a decrease in protein aggregation. Met. Integr. Biometal Sci. 2020, 12, 1142–1158. [Google Scholar] [CrossRef]
- Boasquivis, P.F.; Silva, G.M.M.; Paiva, F.A.; Cavalcanti, R.M.; Nunez, C.V.; de Paula Oliveira, R. Guarana (Paullinia cupana) Extract Protects Caenorhabditis elegans Models for Alzheimer Disease and Huntington Disease through Activation of Antioxidant and Protein Degradation Pathways. Oxid. Med. Cell. Longev. 2018, 2018, 9241308. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, H.; Roxo, M.; Rohrig, T.; Richling, E.; Wang, X.; Wink, M. Anti-Aging and Antioxidant Potential of Paullinia cupana var. sorbilis: Findings in Caenorhabditis elegans Indicate a New Utilization for Roasted Seeds of Guarana. Medicines 2017, 4, 61. [Google Scholar] [CrossRef]
- Pohl, F.; Teixeira-Castro, A.; Costa, M.D.; Lindsay, V.; Fiuza-Fernandes, J.; Goua, M.; Bermano, G.; Russell, W.; Maciel, P.; Kong Thoo Lin, P. GST-4-Dependent Suppression of Neurodegeneration in C. elegans Models of Parkinson’s and Machado-Joseph Disease by Rapeseed Pomace Extract Supplementation. Front. Neurosci. 2019, 13, 1091. [Google Scholar] [CrossRef]
- Vilasboas-Campos, D.; Costa, M.D.; Teixeira-Castro, A.; Rios, R.; Silva, F.G.; Bessa, C.; Dias, A.C.P.; Maciel, P. Neurotherapeutic effect of Hyptis spp. leaf extracts in Caenorhabditis elegans models of tauopathy and polyglutamine disease: Role of the glutathione redox cycle. Free Radic. Biol. Med. 2021, 162, 202–215. [Google Scholar] [CrossRef]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pfleger, C.M.; Friedman, L.; Vittorino, R.; Zhao, W.; Qian, X.; Conley, L.; Ho, L.; Pasinetti, G.M. Potential Application of Grape Derived Polyphenols in Huntington’s Disease. Transl. Neurosci. 2010, 1, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Chongtham, A.; Agrawal, N. Curcumin modulates cell death and is protective in Huntington’s disease model. Sci. Rep. 2016, 6, 18736. [Google Scholar] [CrossRef]
- Underwood, B.R.; Imarisio, S.; Fleming, A.; Rose, C.; Krishna, G.; Heard, P.; Quick, M.; Korolchuk, V.I.; Renna, M.; Sarkar, S.; et al. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 2010, 19, 3413–3429. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Bauer, P.O.; Kurosawa, M.; Goswami, A.; Washizu, C.; Machida, Y.; Tosaki, A.; Yamada, M.; Knopfel, T.; Nakamura, T.; et al. Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum. Mol. Genet. 2008, 17, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Browne, S.E.; Choi, D.K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef]
- Billes, V.; Kovacs, T.; Hotzi, B.; Manzeger, A.; Tagscherer, K.; Komlos, M.; Tarnoci, A.; Padar, Z.; Erdos, A.; Bjelik, A.; et al. AUTEN-67 (Autophagy Enhancer-67) Hampers the Progression of Neurodegenerative Symptoms in a Drosophila model of Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 133–147. [Google Scholar] [CrossRef]
- Fardghassemi, Y.; Maios, C.; Parker, J.A. Small Molecule Rescue of ATXN3 Toxicity in C. elegans via TFEB/HLH-30. Neurotherapeutics 2021, 18, 1151–1165. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133, 93–104. [Google Scholar] [CrossRef]
- Marcelo, A.; Brito, F.; Carmo-Silva, S.; Matos, C.A.; Alves-Cruzeiro, J.; Vasconcelos-Ferreira, A.; Koppenol, R.; Mendonca, L.; de Almeida, L.P.; Nobrega, C. Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Secomandi, E.; Castiglioni, A.; Melone, M.A.B.; Isidoro, C. Resveratrol protects neuronal-like cells expressing mutant Huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem. Int. 2018, 117, 174–187. [Google Scholar] [CrossRef]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Paldino, E.; Giampa, C.; Bernardi, G.; Fusco, F.R. PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington’s Disease. PLoS ONE 2015, 10, e0134482. [Google Scholar] [CrossRef]
- Paldino, E.; D’Angelo, V.; Laurenti, D.; Angeloni, C.; Sancesario, G.; Fusco, F.R. Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor, in the R6/2 Mouse Model of Huntington’s Disease. Cells 2020, 9, 2286. [Google Scholar] [CrossRef]
- Paldino, E.; Balducci, C.; La Vitola, P.; Artioli, L.; D’Angelo, V.; Giampa, C.; Artuso, V.; Forloni, G.; Fusco, F.R. Neuroprotective Effects of Doxycycline in the R6/2 Mouse Model of Huntington’s Disease. Mol. Neurobiol. 2020, 57, 1889–1903. [Google Scholar] [CrossRef]
- Giampa, C.; Alvino, A.; Magatti, M.; Silini, A.R.; Cardinale, A.; Paldino, E.; Fusco, F.R.; Parolini, O. Conditioned medium from amniotic cells protects striatal degeneration and ameliorates motor deficits in the R6/2 mouse model of Huntington’s disease. J. Cell. Mol. Med. 2019, 23, 1581–1592. [Google Scholar] [CrossRef]
- Peyser, C.E.; Folstein, M.; Chase, G.A.; Starkstein, S.; Brandt, J.; Cockrell, J.R.; Bylsma, F.; Coyle, J.T.; McHugh, P.R.; Folstein, S.E. Trial of d-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry 1995, 152, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Vaddadi, K.S.; Soosai, E.; Chiu, E.; Dingjan, P. A randomised, placebo-controlled, double blind study of treatment of Huntington’s disease with unsaturated fatty acids. Neuroreport 2002, 13, 29–33. [Google Scholar] [CrossRef]
- Puri, B.K.; Bydder, G.M.; Counsell, S.J.; Corridan, B.J.; Richardson, A.J.; Hajnal, J.V.; Appel, C.; McKee, H.M.; Vaddadi, K.S.; Horrobin, D.F. MRI and neuropsychological improvement in Huntington disease following ethyl-EPA treatment. Neuroreport 2002, 13, 123–126. [Google Scholar] [CrossRef]
- Neuhaus, O.; Stuve, O.; Archelos, J.J.; Hartung, H.P. Putative mechanisms of action of statins in multiple sclerosis—Comparison to interferon-beta and glatiramer acetate. J. Neurol. Sci. 2005, 233, 173–177. [Google Scholar] [CrossRef]
- Markovic-Plese, S.; Singh, A.K.; Singh, I. Therapeutic potential of statins in multiple sclerosis: Immune modulation, neuroprotection and neurorepair. Future Neurol. 2008, 3, 153. [Google Scholar] [CrossRef]
- Neuhaus, O.; Stuve, O.; Zamvil, S.S.; Hartung, H.P. Are statins a treatment option for multiple sclerosis? Lancet. Neurol. 2004, 3, 369–371. [Google Scholar] [CrossRef]
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 1997, 29, 315–331. [Google Scholar] [CrossRef]
- Shay, K.P.; Michels, A.J.; Li, W.; Kong, A.N.; Hagen, T.M. Cap-independent Nrf2 translation is part of a lipoic acid-stimulated detoxification stress response. Biochim. Biophys. Acta 2012, 1823, 1102–1109. [Google Scholar] [CrossRef]
- Spain, R.; Powers, K.; Murchison, C.; Heriza, E.; Winges, K.; Yadav, V.; Cameron, M.; Kim, E.; Horak, F.; Simon, J.; et al. Lipoic acid in secondary progressive MS: A randomized controlled pilot trial. Neurol. Neuroimmunol. Neuroinflam. 2017, 4, e374. [Google Scholar] [CrossRef]
- Gruber, R.C.; Chretien, N.; Dufault, M.R.; Proto, J.; Zhang, M.; LaMorte, M.; Havari, E.; Samad, T.A.; Turner, T.; Chomyk, A.; et al. Central Effects of BTK Inhibition in Neuroinflammation (808). Neurology 2020, 94, 808. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PolyQ Disease | Causative Gene | Transmission | Normal/Mutant Repeat Tract Length |
|---|---|---|---|
| Spinocerebellar ataxia type 1 (SCA1) | ATXN1 | Autosomal Dominant | 6–35/39–83 |
| Spinocerebellar ataxia type 2 (SCA2) | ATXN2 | Autosomal Dominant | 13–31/>32 |
| Spinocerebellar ataxia type 3 (SCA3) | ATXN3 | Autosomal Dominant | 12–41/52–91 |
| Spinocerebellar ataxia type 6 (SCA6) | CACNAIA | Autosomal Dominant | 4–18/20–33 |
| Spinocerebellar ataxia type 7 (SCA7) | ATXN7 | Autosomal Dominant | 7–27/37–460 |
| Spinocerebellar ataxia type 17 (SCA17) | TBP | Autosomal Dominant | 25–42/46–55 |
| Dentatorubral-pallidoluysian atrophy (DRPLA) | ATN1 | Autosomal Dominant | 6–35/48–93 |
| spinal and bulbar muscular atrophy (SBMA) | AR | X-linkedDominant | 11–36/38–62 |
| Huntington’s disease (HD) | HTT | Autosomal Dominant | 6–35/40–121 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkekas, I.; Gioran, A.; Boziki, M.K.; Grigoriadis, N.; Chondrogianni, N.; Petrakis, S. Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases. Antioxidants 2021, 10, 1450. https://doi.org/10.3390/antiox10091450
Gkekas I, Gioran A, Boziki MK, Grigoriadis N, Chondrogianni N, Petrakis S. Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases. Antioxidants. 2021; 10(9):1450. https://doi.org/10.3390/antiox10091450
Chicago/Turabian StyleGkekas, Ioannis, Anna Gioran, Marina Kleopatra Boziki, Nikolaos Grigoriadis, Niki Chondrogianni, and Spyros Petrakis. 2021. "Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases" Antioxidants 10, no. 9: 1450. https://doi.org/10.3390/antiox10091450
APA StyleGkekas, I., Gioran, A., Boziki, M. K., Grigoriadis, N., Chondrogianni, N., & Petrakis, S. (2021). Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases. Antioxidants, 10(9), 1450. https://doi.org/10.3390/antiox10091450