Abstract
In neuronal precursors and immature neurons, the depolarizing (excitatory) effect of γ-Aminobutyric acid (GABA) signaling is associated with elevated [Cl−]i; as brain cells mature, a developmental switch occurs, leading to the decrease of [Cl−]i and to the hyperpolarizing (inhibitory) effect of GABAergic signaling. [Cl−]i is controlled by two chloride co-transporters: NKCC1, which causes Cl− to accumulate into the cells, and KCC2, which extrudes it. The ontogenetic upregulation of the latter determines the above-outlined switch; however, many other factors contribute to the correct [Cl−]i in mature neurons. The dysregulation of chloride homeostasis is involved in seizure generation and has been associated with schizophrenia, Down’s Syndrome, Autism Spectrum Disorder, and other neurodevelopmental disorders. Recently, much effort has been put into developing new drugs intended to inhibit NKCC1 activity, while no attention has been paid to the origin of [Cl−]i dysregulation. Our study examines the pathophysiology of Cl− homeostasis and focuses on the impact of oxidative stress (OS) and inflammation on the activity of Cl− co-transporters, highlighting the relevance of OS in numerous brain abnormalities and diseases. This hypothesis supports the importance of primary prevention during pregnancy. It also integrates the therapeutic framework addressed to restore normal GABAergic signaling by counteracting the alteration in chloride homeostasis in central nervous system (CNS) cells, aiming at limiting the use of drugs that potentially pose a health risk.
1. Intracellular Chloride Concentration Affects the GABAergic Signaling
Intracellular chloride concentration [Cl−]i has a critical role in osmotic equilibrium, cell volume regulation [1,2,3,4], acid-base balance [5], and cell signaling [4,6], reviewed in ref. [7,8].
Developmentally regulated [Cl−]i is the main determinant of the action of GABA at GABAA receptors. In immature neurons, high [Cl−]i results in a depolarizing response to GABA, and plays an active role in neuronal growth and the formation of synaptic connections [9,10,11,12]. On the other hand, in mature CNS neurons, a hyperpolarizing or a shunting response is generated by GABA signaling in the presence of low [Cl−]i [13]. The K+-Cl− co-transporter KCC2 mediates the efflux of Cl− from the cells [14], while the Na+, K+-2 Cl− co-transporter NKCC1 promotes Cl− uptake [15]. The decrease in [Cl−]i, which occurs during the maturation of neural cells, is a fundamental ontogenetic process under transcriptional and epigenetic control, which results in a shift of GABAergic transmission from excitation to inhibition [16]. These events occur in an asynchronous way in different brain areas following their sequential maturation, and are correlated with the regulated increase in KCC2 expression, while NKCC1 expression may decrease or remain constant [13,17]. Figure 1 shows the developmental shift in [Cl−]i, with the main cation-coupled chloride co-transporters involved and the accompanying enzymatic activities needed for their function.
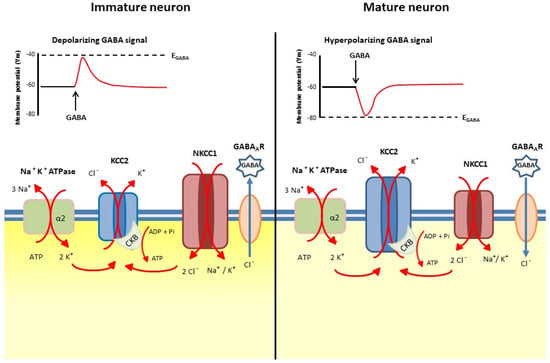
Figure 1.
The main cation-coupled chloride co-transporters which mediate [Cl−]i developmental shift. Left side: in immature neurons [Cl−]i is higher than in mature ones (right side). The intensity of the cytoplasm color refers to the intracellular chloride concentration. KCC2, K+-Cl− co-transporter 2; NKCC1, Na+, K+-2Cl− co-transporter 1; CKB, brain-type creatine kinase; Na+, K+-ATPase, sodium-potassium pump. The α2 subunit of Na⁺/K⁺-ATPase couples potassium entry into the cell with the K-Cl coordinated efflux from the cell. The size of the symbols depicting KCC2 and NKCC1 is reminiscent of the change of the co-transporter amount and activity in the developmental shift.
In the brain, KCC2 expression and its regulation are under the control of developmental cues and of brain-derived natriuretic factors (BDNF), through the extracellular signal-regulated kinase1/2(ERK1/2)-dependent upregulation of the transcription factor early growth response 4 (Egr4) [18]. It is worth mentioning that the neurotrophin BDNF plays an important role in cognitive tasks [19]. In a similar way, the insulin-like growth factor-1 (IGF-1) also positively controls KCC2 expression [20] and triiodothyronine, oxytocin, gonadic hormones and other factors affect KCC2 and NKCC1 expression and activity (reviewed in ref. [13]). In particular, the effect of estradiol on KCC2 expression insubstantia nigra pars reticulata neurons differs between males and females [21]. In rat hippocampal and neocortical neurons, oxytocin exerts its effect at birth by post-translational inhibition of NKCC1 [22]. Notably, developmental upregulation of KCC2 begins at different times for various species; for example, occurring during the second half of gestation in humans (reviewed in ref. [23]), and beginning at E18 in rats [24]. The maturation of brain regions sequentially follows the temporal pattern of neurogenesis [24], resulting in a time window for toxicant exposure during ontogenesis [25].
It is worth noting that, besides GABAA-R, other chlorine and anion channels contribute to Cl− fluxes across the membrane, the most prominent being glycine receptor (Gly-R) channels, which complement the role of GABA in chloride homeostasis, but may be involved in different processes during brain development [26]. Astrocytes may contribute to the activation of both GABAA-R and Gly-R by releasing GABA and the atypical aminosulfonic acid taurine, which activates Gly-R channels [4].
Chloride homeostasis as described here is not universal, as it differs between central and peripheral neurons (reviewed in ref. [27]). In dorsal root ganglion neurons, [Cl−]i is higher than in other cell types, since KCC2 expression is barely detectable, whilst chloride efflux is mostly mediated by KCC1 and KCC3, whose activity mostly responds to cell swelling; in addition, [Cl−]i is increased by the activity of other anion transporters [27]. Thus, in primary afferent neurons, similarly to immature CNS neurons, opening of Cl− channels usually causes membrane depolarization rather than hyperpolarization. However, the degree and the velocity of depolarization are subject to regulation and determine whether depolarization results into inhibitory or excitatory effects. For instance, intense stimulation of nocireptors and inflammatory mediators can increase [Cl−]i, and thus mediate inflammatory hyperalgesia and contribute to neuropathic pain [28]. It is interesting to note that loss-of-function of KCC3 results in Hereditary Sensorimotor Neuropathy with Agenesis of the Corpus Callosum (HSMN/ACC) and axonal swelling [29]. GABAergic neurons are also integrated in the complex cellular network constituting the enteric nervous system (ENS), which regulates, independently of the CNS, gastrointestinal functions, including motility and secretion, and relating with gut microbiota and the enteric immune system. ENS neurons have elevated intracellular Cl− concentration; therefore, activation of GABAA R results in an excitatory effect [30]. Notably, a potential anti-inflammatory role has been suggested for GABAergic signaling in ENS, in synergy with other enteric immunomodulatory mediators [31].
2. The Post-Translational Regulation of Cation-Cl− Co-Transporters
The activity of cation-Cl− co-transporters is driven by the Na+ and K+ gradients generated by the Na+, K+-ATPase. In particular, Cl− influx is driven by Na+, while Cl− efflux is mediated by K+ extrusion from the cell. The physical association of KCC2 with the α2 subunit of Na+, K+-ATPase and with the brain-type creatine kinase (CKB) at the cell membrane couples the generation of ATP by ADP phosphorylation mediated by CKB with the generation of the electrochemical gradient, which drives Cl− out of the cell. In the brain, both CKB and the α2 isoform of the α subunit of the Na+, K+-ATPase are transcriptionally co-regulated and spatially co-located with KCC2 as reviewed in [13].
The developmental shift in [Cl−]i is regulated also by several phosphorylation and dephosphorylation events on both NKCC1 and KCC2. Such post-translational modifications are differentially modulated during development and, in particular, involve the kinases PKC (α, β and γ), WNK (1–4), and SPAK/OSR1, which are downstream of WNKs reviewed in ref. [13]. It is noteworthy that the SPAK/OSR1 pathway is activated by several signals, including estradiol and osmotic stress (i.e., hypertonic conditions), and results in the phosphorylation of both NKCC1 and KCC2; the phosphorylation of the former leads to the enhancement of its activity, whereas the phosphorylation of the latter has a repressive role [32,33]. Such activity likely leads in neurons to an increase in [Cl−]i and to the modulation of GABAA-R-mediated synaptic inhibition. The role played by WNK and SPAK/OSR1 led Khale et al. [3] to suggest WNK-SPAK inhibition as a potential therapeutic target in neurological diseases not associated with cell swelling.
Chloride co-transporters may also be regulated by their internalization. PKC, by phosphorylating Ser940 of KCC2, contrasts its internalization, thus playing a positive role in KCC2 activity; in turn, PKC is activated by both the metabotropic Glutamate receptor 1 s and by the 5-hydroxytriptamine (5-HT) 2A receptor [34]. These complex interactions are mentioned here to highlight the fact that multiple neurotransmitter receptors are involved in the regulation of neuronal [Cl−]i; when in the proximity of GABAA receptors, they contribute to the modulation of the GABAergic response, which clearly appears as a key point in both physiology and pathology.
Finally, proteolytic events may modulate the chloride co-transporter amount. It was reported that peripheral nerve injury increases spinal N-methyl-d-aspartate receptor (NMDAR) activity; the consequent increase in [Ca2+]i impairs synaptic inhibition through calpain-mediated KCC2 proteolysis [35].
3. Intracellular Chloride Concentration and GABAergic Signaling in Neurological Disorders and Neurodevelopmental Diseases and the Involvement of Oxidative Stress and Inflammation
As outlined above, in CNS, both NKCC1 and KCC2 undergo a developmentally regulated alteration in their expression levels in the course of ontogenesis. However, pathological situations might alter the timing and/or the extent of the transcriptional variations of chloride co-transporters. Since GABAergic signals play a key role in orchestrating the assembly of neuronal circuits in the developing brain [36,37] and are the major inhibitory transmitters in the adult brain, it is not surprising that the dysregulation of GABAergic signaling has been associated with many neurological and neurodevelopmental disorders, such as epilepsy, schizophrenia, Down’s Syndrome (DS) and ASD [38,39]. In particular, the so-called Maternal Immune Activation (MIA), by providing a pro-inflammatory intrauterine environment, delays the developmental shift of GABAA receptors [40]. This effect, likely mediated by the pro-inflammatory cytokine IL-1β, which downregulates the transcription of KCC2, is particularly relevant because MIA is one of the more promising murine models of Autistic Spectrum Disorders (ASD) [41], but has also been linked to enhanced risk of schizophrenia in humans [42]. Besides inflammation, other environmental factors may delay the perinatal chloride shift. An example is the effect of bisphenol A on cortical neurons: bisphenol A is an endocrine disruptor, which alters chromatin, negatively affecting the expression of the Kcc2 gene, either directly or following increased MECP2 and decreased H3K9ac binding to Kcc2 regulatory regions [43]. This clearly demonstrates the impact of environmental toxicants on epigenetic machinery in neurodevelopment.
Dysregulation of NKCC1/KCC2 activity ratio is generally the key for improper [Cl−]i and alteration of GABA-induced inhibition. Epilepsy, schizophrenia and the neurodevelopmental disorders mentioned above widely differ in their etiology, severity, clinical presentation, age of onset, genetic background, and other features; therefore, it appears somehow inconsequent that this diverse set of conditions could be due to the same pathogenic mechanism: the perturbation of the inhibitory effect of GABAergic signaling.
In the next few paragraphs we will shortly review the evidence, showing that these conditions share two common features: OS and (neuro)-inflammation. In analogy with the well-known phenomenon of glutamate-induced excitotoxicity, it is reasonable to hypothesize that OS and (neuro)-inflammation may be a consequence of the intrinsic alteration of the GABAergic signaling occurring in these neurological and neurodevelopmental disorders. However, instead, we intend here to advance and support the opposite hypothesis, i.e., that OS is the cause rather than the effect of the attenuation or even the reversal of the inhibitory effect of GABA signals at GABAA receptors. We will point out that there are several mechanisms by which OS is able to reduce the expression and the activity of KCC2, thus contributing to the increase in [Cl−]i and to the global excitability of CNS cells.
The classical definition of OS was formulated by Halliwell and Guteridge in 1989 [44]: “Oxidative stress occurs due to the imbalance between the production of Reactive Oxygen Species (ROS) and the availability of antioxidants or radical scavengers”. However, by taking into account that ROS play a significant role not only as potential damaging agents but also as signaling molecules, a more comprehensive definition was put forward by Jones in 2006 [45]: “Oxidative stress may be better defined as a disruption of redox signaling and control”. ROS production occurs at many cellular sites and in manifold and varied conditions, both physiological and pathological. In particular, dysfunctional mitochondria are a relevant source of ROS [46].
In turn, inflammation is often a result of OS [47]. In particular, Advanced Glycosylation End-products (AGE)-derived from oxidized proteins, nucleic acids and lipids, may be a direct cause of inflammation; moreover, both ROS and AGE may activate inflammatory signaling cascades, sometimes leading to cell death, autophagy and inflammasome activation [48,49,50].
As pointed out by Kim et al. [51], the evidence suggests that major neuropsychiatric disorders, such as autism spectrum disorder (ASD), schizophrenia, bipolar disorder and major depressive disorder, share a number of pathogenic mechanisms based on the interaction between the environment and the genome, leading to the impairment of brain energy metabolism, mitochondrial functions and redox balance. In support of these claims, they report that alterations of the mitochondrial genome in psychiatric patients have been described [52,53], as well as several mutations and polymorphisms of mtDNA, decreased oxidative phosphorylation and increased generation of hydrogen peroxide in lymphocytes from ASD children [54]. Moreover, they point out that mitochondrial dysfunctions, in addition to generating ROS, affect mitochondrial dynamics, circadian rhythms and glutamatergic transmission. Other mechanisms of mitochondrial dysfunction in ASD are linked to dysbiosis and to environmental toxicants [52].
3.1. Epilepsy
Epilepsy is a devasting neurological disorder, affecting more than 70 million people worldwide [55]. It is characterized by recurrent seizures, due to excessive and abnormal neuronal activity in the brain cortex. About one third of epilepsy patients suffer from uncontrolled seizures despite pharmacotherapy [56]; therefore, a better insight into the pathological mechanisms is needed. Epilepsy may originate from various conditions, ranging from genetic and birth defects to brain injuries and tumors.
The importance of excitatory–inhibitory imbalance in epileptogenesis has been reviewed by Liu et al. [57], who reported the observation of upregulation of NKCC1 and/or dowregulation of KCC2 (mRNA and protein) in different animal models of chronic epilepsy. In a similar way, the epigenetic dysregulation of chloride co-transporters has been reported to occur in juvenile myoclonic epilepsy [58]. Epileptogenic brain tumors also exhibit a shift in GABAergic signaling from inhibitory to excitatory, as reviewed in ref. [57]. In support of the hypothesis of the role of the cation chloride co-transporters in the generation of seizures, a number of researches reported the attenuation of epileptic activity operated in humans [59,60] and in several animal models [57] by bumetanide, an inhibitor of NKCC. In an interesting work on human epilepsy, Kipnis et al. [61], while identifying the origin of seizure susceptibility in alterations of the chloride co-transporters, stressed the relationship between their developmental regulation operated by BDNF and gonadic hormones with sexual dimorphism observed in epileptogenesis. In addition, it has been reported that alteration of signals delivered at the synaptic metabotropic zinc receptor mZnR/GPR39, which physiologically enhance KCC2 activity, is associated with epileptic seizures [62].
Among the possible causes of chloride co-transporter unbalance, rare loss-of-function mutations of the SLC12A5 gene, encoding KCC2, were reported in patients with epilepsy [63]. Altered epigenetic regulation of genes encoding chloride co-transporters seems more plausible than rare genetic mutations in determining an improper [Cl-]i unbalance in some brain areas. The methylation state of NKCC1 and KCC2 genes has been assessed in juvenile myoclonic epilepsy patients, showing significantly lower NKCC1 DNA methylation and significantly higher KCC2 DNA methylation levels [58].
Inflammation is one of the most important mechanisms in epileptogenesis and is sustained by the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling and by OS, which justifies the proposal of anti-inflammatory and antioxidant drugs for its treatment [64].
Evidence supporting a role for ROS and for mitochondrial impairment in epilepsy has been reviewed in two recent independent papers [65,66]; in particular, Kovac et al. [65] highlighted the involvement of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases in both ROS generation and mitochondrial damage, which links ROS generation to microglia activation in a variety of brain diseases.
3.2. Schizophrenia
According to the definition of the American Psychiatric Organization, schizophrenia is a severe neurological disease, characterized by delusions, hallucinations, disorganized speech, trouble with thinking and lack of motivation (https://www.psychiatry.org/; accessed on 30 May 2021). Schizophrenia affects males 1.4 times more frequently than females [67].
Rare genetic mutations of cation-coupled chloride co-transporters or of elements of the WNK-SPAK/OSR1 Kinase Signaling Pathway have been identified in several neuropsychiatric and neurodevelopmental diseases reviewed in ref. [68]. Loss-of-function mutations of the SLC12A5 gene, encoding KCC2, and a gain-of-function missense variant in SLC12A2, encoding NKCC1, were described by Merner et al. [69,70]; both variants were linked to schizophrenia and the former also to ASD. Moreover, autoptic samples from schizophrenic patients showed higher transcript levels of Wnk3 and of Oxsr1, encoding OSR1 kinase [71]. Again, a pharmacological intervention aimed at restoring the correct balance of chloride co-transporters, which confirmed the involvement of this mechanism in schizophrenia and in ASD. The administration of bumetanide relieved some symptoms in schizophrenic [72] and ASD patients ([73,74,75], reviewed in ref. [76].
A Special Issue of Schizophrenia Research was devoted in 2016 to OS and inflammation in schizophrenia. In their editorial, Sawa and Sevlak [77] outlined the evidence reported by the seven manuscripts included in the issue, which cover topics such as the detection of OS and inflammatory makers in sera from schizophrenic patients and the influence of redox dysregulation and inflammation on neurotransmission [78]. Further studies were reported in a review by Koga et al. [79]. More recently, two manuscripts highlighted the interplay between OS and cellular markers of inflammation in schizophrenic patients [80,81].
3.3. Down’s Syndrome
Down’s Syndrome (DS) is a genetic disorder caused by chromosome 21 partial or full trisomy. It is to all respects a neurodevelopmental syndrome, causing, among other defects, morphogenetic brain anomalies, which in turn lead to intellectual disability and learning difficulties. At the same time, since most DS patients experience in adult life a progressive cognitive decline with neuropathological features consistent with the Alzheimer’s disease phenotype [82,83], DS is often considered a neurodegenerative disorder.
As in other brain disorders, GABAergic signaling was reportedly altered also in DS, with a reduction in GABA concentrations and an increased expression of NKCC1 in the brain of fetuses and children with DS and in the Ts65Dn mouse model of chr. 21 trisomy [84,85,86].
OS was first described in 1989 to occur in DS cells [87]. It was then recognized to be caused by mitochondrial dysfunction, which by itself has profound consequences on ATP production and deficit in total brain energy reviewed in ref. [88]. In turn, mitochondrial-originated ROS caused a progressive accumulation of oxidative damage in mtDNA, leading to early cellular aging and neurodegeneration, as reviewed in ref. [68]. Low levels of BDNF were described in the hippocampi of human fetal brains from individuals with trisomy 21 [89]. Interestingly, by utilizing a murine transgenic model of DS carrying an extra copy of DYRK1A (a candidate gene for DS), which displays both brain abnormalities and learning impairment, Guedj et al. [89] demonstrated that the overexpression of DYRK1A was responsible for the decrease in BDNF: transgenic mice fed with the DYRK1A inhibitor epigallocatechin gallate (EGCG)—a member of a natural polyphenols family from green tea—increased BDNF expression and partially rescued cognitive deficits. Many flavonoids have well-established antioxidant and free-radical-scavenging activities, but also exert their biological effects through direct actions on enzymes, receptors and signaling pathways; they are also endowed with a modulatory effect on GABAA receptors [90]. Neurodevelopmental abnormalities in DS include several impairments in neurotransmission systems; not only is the GABAergic signaling altered, but also the glutamatergic, the cholinergic and the serotonergic pathways are reviewed in ref. [85].
3.4. Autism Spectrum Disorder
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder characterized by impaired social communication skills, restricted interests and activities, repetitive patterns of behavior and sensory abnormalities, including anomalous pain sensitivity [91,92]. Intellectual disability is reported in half of the people with an ASD diagnosis [93]. About 5–46% of individuals with ASD also suffer from epilepsy [94]. The prevalence of autism is strongly unbalanced with regard to gender, being diagnosed in about four males for each female; the reason for such sexual dimorphism is still a matter of debate [95]. Autistic features are found also in people with genetic syndromes affecting neurodevelopment, including Rett and Fragile X syndromes; therefore, for the purpose of this review, we will deal with these conditions together with ASD. The genetic architecture is complex in ASD and the lack of demonstration of causal genetic abnormalities leads to defining the majority of cases as “idiopathic” [96]. The wide variability of behavioral manifestations and the frequent multisystem involvement—disorders in ASD people are not limited to the nervous system—suggest that epigenetic mechanisms impact on the neurodevelopment during the ontogenesis and in the first two years of life, which is the period of maximum neuroplasticity as reviewed in ref. [97]. Furthermore, an epigenetic model also accounts for the steep worldwide increase in ASD prevalence in the last decades, within the framework of the epidemiological transition towards a greater predominance of non-communicable diseases [98,99]. It is also of note that idiopathic forms of ASD often share disregulated gene expression/pathways with syndromic (monogenic) forms; this strongly suggests that, in idiopathic ASD forms, alterations of chromatin remodeling may mimic genetic mutations [100].
As mentioned above, in mature neurons, Gly-R contributes to the establishment of [Cl-]i, which results in the hyperpolarizing action of GABAA receptors. It is interesting to note that a number of rare mutations affecting Gly-Rs are associated with ASD [101]. At inhibitory synapses, glycine and GABAA receptors are anchored to the cytoskeleton by the scaffold protein gephyrin, which contributes to the post-synaptic clustering of GABAA receptors. Several gephyrin alterations reviewed in ref. [102], are associated with neuropsychiatric and neurodegenerative disorders, including ASD and schizophrenia. Gephyrin with the G375D missense mutation has a reduced binding affinity to GABAA and GlyRs and was found in a patients with epileptic encephalopathy [103].
Carriers of some genetic variants of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) may present with cystic fibrosis associated with ASD, probably because CFTR may be a positive regulator of NKCC1 [104]. ASD is not the only neurodevelopmental disorder where GABAergic signaling appears to be compromised. In fact, GABAergic signaling is reportedly altered also in patients carrying either of two genetic mutations that share many clinical features with ASD, namely Rett syndrome [105,106] and Fragile X syndrome [107].
Recalling the modulatory role of numerous and diverse neurotransmitter receptors localized at the GABAergic synapses, some examples where their alteration affects neurodevelopmental disorders are worth mentioning. In fact, 5-HT receptors are affected by the absence of the Fragile X mental retardation protein [108] and the abnormal methylation of 5-HT transporter gene network is associated with Attention Deficit Hyperactivity Disorder (ADHD) and with early life stress [109]. In addition, agonists of the metabotropic acethylcholine receptor reduce stereotypies in BTBR mice (a model of ASD) [110].
The first evidence that ASD was associated with OS dates back to 2004–2006 [111,112,113] and were corroborated by further studies showing that OS altered the lipid asset of erythrocyte membranes [114,115]. In addition, OS altered also the erythrocyte shape, both in ASD and in Rett patients [116,117]. Metabolomic analysis of ASD plasma and urine revealed the presence of elevated levels of AGE [118]. Several studies link OS to mitochondrial dysfunctions [119], to immune dysregulation and inflammation [120] and to gut dysbiosis, often accompanied by severe gastrointestinal problems [121,122], as reviewed in ref. [97]. Lower expression of BDNF was found in the neonatal blood of children, which were diagnosed with ASD later in life [122,123]; data from a murine model of ASD suggest that BDNF levels may be epigenetically regulated in a sexually dimorphic fashion [124]. It is worth noting that a complex relationship links BDNF expression and MeCP2, the gene whose mutation is the cause of Rett syndrome [125]. Another neurotrophic factor, IGF-1, which activates the PI3K/AKT pathway, is dysregulated in both idiopathic and syndromic ASD [19,126]. In particular, in the age range 1–4 years, the amount of BDNF in cerebrospinal fluid was significantly lower than in neurotypical children [127]; intense research is currently underway to evaluate a therapeutic use of IGF-1 in ASD [128,129].
4. How OS, Inflammation, Toxicants and Chromatin Modifiers May Affect Intracellular Chloride Concentration and GABAergic Signaling
As mentioned above, OS ensues when ROS production overwhelms the antioxidant capacities of cells or tissues; in contrast, in physiological conditions, ROS play a significant role as signaling molecules. The presence of cysteine residues in some GABAA receptor subunits, both on the extracellular and the intracellular loops, potentially makes GABAA receptors susceptible to ROS-operated modulation of ion channel gating.
Exposure of GABAA receptors to artificially elevated levels of ROS, either extracellular or intracellular, did not seem to affect the intracellular chloride concentration, since, at least in the hippocampus and in the cerebellum, ROS potentiated the inhibitory activity of GABAA receptors as reviewed in ref. [130]. However, it is worth considering that we ignore whether such experiments gave an insight into the physiological role of ROS signaling or rather simulated OS by unbalancing the redox state of the cells. Also inconclusive in this regard was the work of Toczylowska et al. [131], who studied hippocampal metabolite profiles by proton nuclear magnetic resonance spectroscopy, comparing two murine models of pharmacologically induced ASD: valproate and thalidomide. Globally, the changes in metabolites suggested disturbances in excitatory and/or inhibitory neurotransmission, in energy production and in membrane lipid composition, along with the presence of OS. While OS might justify the decrease in energy production and changes in lipid composition, the study could not discriminate whether excitatory–inhibitory unbalance was a cause or a consequence of OS.
Other evidence supports a role for OS in the alteration of the excitatory/inhibitory balance at GABAA receptors in pathological models. Hepatic encephalopathy is an example of a medical condition that may indirectly alter the redox equilibrium of CNS. It causes a motor dysfunction, originating in the substantia nigra pars reticulata (SNr), a midbrain region where the GABAergic neurons convey the final processed signals of the basal ganglia to the thalamus and superior colliculus. Bai et al. [132] reported that OS made the GABA signals lose their inhibitory effect in SNr and that such a dysfunction was accompanied by a decreased expression of the chloride co-transporter KCC2. By inhibiting the oxidative processes and reducing ROS contents, Bai et al. improved the dyskinesis caused by OS and restored normal levels of KCC2.
As discussed above [40], KCC2 expression is reduced also as a result of inflammatory stimuli, via chromatin modifications induced by pro-inflammatory cytokines. It was observed that increased levels of maternal cytokines and chemokines during gestation are associated with the development of autism in the offspring [133]. Moreover, a pro-inflammatory signature persisting after birth and can be found in ASD children [134]. Besides maternal immune stimulation, other stressful events, such as physical constraint or maternal separation of puppies, causes the increase of pro-inflammatory cytokines, as recently reviewed by Pozzi et al. [135].
Similar results are observed following exposure to toxicants. The above-mentioned bisphenol A, an endocrine disruptor [43], has been causally associated with the development of ASD [136]. In turn, perinatal exposure to Pb2+ subverts the KCC2/NKCC1 ratio, albeit through mechanisms not completely known [137]; moreover, it is worth mentioning that Pb2+ induces both oxidative stress and inflammation and causes a decrease in cerebellar BDNF [138]. Exposure of fish brain to Pb2+ demonstrated increased production of reactive oxygen species, increased lipid peroxidation, loss of protein thiol groups in synaptosomal fraction, decreased activity of Na+, K+-ATPase, partial inactivation of mitochondrial electron transport chain activity and energy depletion [139]. Scientific literature abounds with evidence that exposure to heavy metals, toxicants and endocrine disruptors may cause oxidative stress and inflammation and is associated with neurodegenerative and neurodevelopmental disorders [120,140].
Two further aspects relate the perturbation of [Cl-]i to oxidative stress and are grounds for reflection when considering the impact of GABAergic signaling in neurodevelopmental disorders. One is the above-mentioned activation of OSR1, which, by phosphorylating both NKCC1 and KCC2, enhances and represses their activities, respectively. OSR1 stands for “oxidative-stress responsive-1” and, in humans, it is encoded by the gene OXSR1. Since it is 39% identical to human SOK1 (Ste20/oxidant stress response kinase-1), a molecule that is activated by oxidative stress (OS) [141], it was hypothesized that OSR1 was also activated by OS, in the absence of any direct proof about an OS-dependent activation of OSR1. However, more recently, a global quantitative phosphoproteomics approach to identify cytoplasmic proteins altered in their phosphorylation state by an ataxia-telangiectasia-mutated kinase (ATM) detected OSR1 as a novel substrate of ATM after hydrogen peroxide exposure of cells [142]. OSR1-driven post-translational modifications of chloride co-transporters have long been seen in the context of cellular osmotic control; however, the observation that OSR1 may be activated also by oxidative stress [142] opens new perspectives to the post-translational control of chloride co-transporters. Accordingly, recent work [143] has demonstrated that two different animal models of autism—a model of Rett syndrome and the prenatal exposure to valproic acid—showed increased levels of ATM and decreased egr4 activity on the Kcc2b promoter, leading to decreased expression of Mecp2 and a delayed GABAergic developmental shift. Treatment with KU55933, an ATM inhibitor, was able to restore normal levels of KCC2, rescuing abnormal GABAergic signaling and autism-like behavior in mice. This is another example of how the autistic phenotype may be a consequence of different triggers, converging to the alteration of the same pathways. The presence of an oxidative environment may mimic specific genetic or chemical triggers by favoring decreased expression/activity of KCC2, with the consequent alteration of GABAergic signaling.
The second aspect relies on previous work on the relevant decrease of Na+, K+-ATPase activity in both erythrocytes and leukocytes of ASD children [114,144], which pointed out the importance of the membrane context embedding the Na+, K+-ATPase. Reduced membrane cholesterol, and oxidative stress-induced damage to membrane lipids, play crucial roles in decreasing the Na+, K+-ATPase activity also in schizophrenia [145]. Note that at variance with blood cells, brain Na+, K+-ATPase expresses the α2 subunit, which functionally interacts with KCC2 [146], and was reported to mediate nuclear factor kappa B (NFκB) signaling in LPS-induced immune response [147]. In addition, it should be stressed that OS often results from dysfunctional mitochondria and, in turn, impairs mitochondrial functioning, in a vicious loop reviewed in ref. [97]. As a result, cellular energy supplies are jeopardized, whilst they are required to fuel chloride efflux at the KCC2 co-transporter, which depends on the continuous supply of ATP, mediated by the membrane-embedded brain-type creatine kinase CKB.
The relationships between OS, inflammation and maintenance of [Cl−]i are shown in Figure 2.
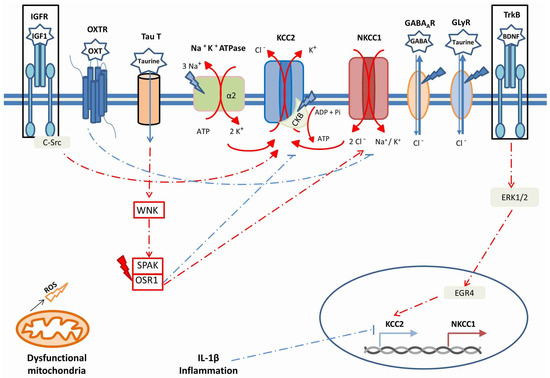
Figure 2.
The main factors modulating KCC2 and NKCC1 synthesis and activity, in relation with anomalies described in neurological and neurodevelopmental disorders. Oxidative stress/ROS is symbolized by lightning bolts; blue bolts indicate a loss-of-function effect, red bolts indicate a gain-of-function effect. The boxes surrounding IGFR and TrkB indicate that IGF-1 and BDNF signaling are both decreased in ASD and in other neurodevelopmental disorders.
5. Conclusions (and Therapeutic Suggestions)
As pointed out by several Authors, [Cl−]i is instrumental in the effect of GABA at GABAA brain receptors, which undergo a developmental shift from excitatory to inhibitory in a time-and-area-dependent fashion. Disruption of intracellular chloride homeostasis within CNS has profound consequences on the etiopathogenesis of several widespread neurological and neurodevelopmental diseases. Differential control of CNS [Cl−]i in males and females during development may provide an explanation of the gender bias in neurobiological disorders, such as ASD and schizophrenia. We outlined here the main evidence linking [Cl−]i maintenance with the occurrence of OS and inflammation and, in some way, with exposure to toxicants and chromatin modifiers, in patients with neurological and neurodevelopmental diseases. In addition, several studies even suggest that OS and inflammation might be causative in the disruption of the mechanisms maintaining the correct intracellular chloride concentration. Even if the alteration of GABAergic signals cannot be considered as the only determining aspect in the etiopathogenesis of neurodevelopmental diseases, it is indubitable that the link between GABAergic signals and brain malfunction is important. In fact, this is the rationale for pharmacological modulation of GABA function in ASD [148] and for efforts to pharmacologically control the chloride transporters in the CNS of subjects with DS and ASD, as previously mentioned for bumetanide [73,74,75,76]. Since bumetanide is a diuretic drug, prescribed to reduce symptoms of fluid retention or edema in people with congestive heart failure, liver or kidney disease, as such, is not devoid of side effects, as it inhibits both the ubiquitous isoform NKCC1 and the kidney-specific NKCC2, with consequent diuretic effects. This problem may be overcome by a newly discovered molecule, which selectively inhibits NKCC1, sparing the kidney-specific NKCC2 [149]. In any case, pharmacological targeting of chloride cotransporters might be beneficial for CNS but have negative effects for peripheral neurons, including ENS, where the activity of NKCC1 prevails. In ASD persons, pain sensitivity perturbances and gastroenteric inflammatory symptoms are quite common, which might point to altered GABAergic signaling also in the periphery. This suggests greater caution in the use of drugs targeting the GABA system.
The striking increase in neurological disorders occurring in the last decades represents a relevant issue for the worldwide healthcare system. In particular, major worries relate to the rise of neurodevelopmental disorders, which are usually diagnosed too late to allow intervention within two years of life and is the crucial time window for neuroplasticity [97]. The logical and experimental links shown in the present review between [Cl−]i dysfunction and occurrence of OS and inflammation highlight a novel mechanism capable of accounting for the impact of OS and mitochondrial dysfunction on neurodevelopment and the role of OS in the pathophysiology of numerous neurological diseases. As a consequence, these reflections strengthen the suggestion to place most effort into controlling OS and immune activation in pregnant mothers, infants and toddlers. In addition, since [Cl−]i dysfunction, and consequential OS and inflammation, are sometimes the result of exposure to toxicants and chromatin modifiers, a large space opens up for prevention [97,140].
The fact that dysfunctions in GABAergic signaling are a common pathogenetic trait shared by a large array of brain disorders highlights the presence of psychopathological and neurological co-occurrences in many neurodevelopmental disorders. Transition of persons with ASD to adulthood is often accompanied by high rates of psychiatric comorbidities [150]. In prefrontal cortical regions, parvalbumin-expressing GABAergic interneurons normally mature during adolescence, and are particularly affected during their development by oxidative stress, neuroinflammation and NMDAR hypofunction [78]; in fact, in patients with schizophrenia, autism and bipolar disorders, they exhibit a pattern of gene expression typical of immature cells [151]. The physiological maturation trajectory of these interneurons suggests that a transition to adult life may pose a demanding challenge to patients where OS is present.
As far as treatment is concerned, in neurological disorders in which the perturbation of GABAergic signaling is supposed, antioxidant molecules and the support of mitochondrial function seem to be the first logical line of intervention. In a different context, the combination of idebenone and tocotrienols gave promising results [152]. Furthermore, even in the case of the prescription of specific drugs targeted to brain chloride co-transporter NKCC1-i.e., bumetanide-, an integrated approach including antioxidant intervention should be considered.
In conclusion, our study proposes the modulation of GABAergic signaling by OS as a paradigmatic situation in which suggestions provided by pathophysiology can inform the clinical perspective. In an era of personalized medicine, the biological complexity of humans makes it unlikely that tailored answers can be represented by single interventions, but rather by the capability to embrace complexity and integrate interventions.
Author Contributions
Conceptualization, P.M.A., M.M. and C.P.; writing—original draft preparation, M.M.; writing—review and editing, P.M.A. and C.P.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Authors are grateful to David Muehsam for his careful reading and refinement of the manuscript.
Conflicts of Interest
Authors declare the absence of conflict of interest.
References
- Armstrong, C.M. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proc. Natl. Acad. Sci. USA 2003, 100, 6257–6262. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523. [Google Scholar] [CrossRef]
- Wilson, C.S.; Mongin, A.A. The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 2018, 689, 33–44. [Google Scholar] [CrossRef]
- Seifter, J.L.; Chang, H.-Y. Disorders of Acid-Base Balance: New Perspectives. Kidney Dis. 2016, 2, 170–186. [Google Scholar] [CrossRef] [PubMed]
- DeFazio, R.A.; Keros, S.; Quick, M.W.; Hablitz, J.J. Potassium-Coupled Chloride Cotransport Controls Intracellular Chloride in Rat Neocortical Pyramidal Neurons. J. Neurosci. 2000, 20, 8069–8076. [Google Scholar] [CrossRef] [PubMed]
- Valdivieso, G.; Santa-Coloma, T.A. The chloride anion as a signalling effector. Biol. Rev. 2019, 94, 1839–1856. [Google Scholar] [CrossRef]
- Lüscher, B.P.; Vachel, L.; Ohana, E.; Muallem, S. Cl− as a bona fide signaling ion. Am. J. Physiol. Physiol. 2020, 318, C125–C136. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Medina, I.; Chudotvorova, I. GABA Neurotransmission and Neural Cation-Chloridec Co-transporters: Actions Beyond Ion Transport. Crit. Rev. Neurobiol. 2006, 18, 105–112. [Google Scholar] [CrossRef]
- Cancedda, L.; Fiumelli, H.; Chen, K.; Poo, M.-M. Excitatory GABA Action Is Essential for Morphological Maturation of Cortical Neurons In Vivo. J. Neurosci. 2007, 27, 5224–5235. [Google Scholar] [CrossRef]
- Akerman, C.J.; Cline, H.T. Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 2007, 30, 382–389. [Google Scholar] [CrossRef]
- Watanabe, M.; Fukuda, A. Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 2015, 9, 371. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Okabe, A.; Toyoda, H.; Kilb, W.; Luhmann, H.J.; Fukuda, A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 2004, 557, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Patisaul, H.; Liedtke, W. Decoding the language of epigenetics during neural development is key for understanding development as well as developmental neurotoxicity. Epigenetics 2013, 8, 1128–1132. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA Excitatory/Inhibitory Shift in Brain Maturation and Neurological Disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Uvarov, P.; Ludwig, A.; Markkanen, M.; Rivera, C.; Airaksinen, M.S. Upregulation of the Neuron-Specific K+/Cl− Cotransporter Expression by Transcription Factor Early Growth Response 4. J. Neurosci. 2006, 26, 13463–13473. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hall, J.; Thomas, K.; Everitt, B. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat. Neurosci. 2000, 3, 533–535. [Google Scholar] [CrossRef]
- Khalil, R.B. Is insulin growth factor-1 the future for treating autism spectrum disorder and/or schizophrenia? Med. Hypotheses 2017, 99, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Galanopoulou, A.S.; Moshe, S. Role of sex hormones in the sexually dimorphic expression of KCC2 in rat substantia nigra. Exp. Neurol. 2003, 184, 1003–1009. [Google Scholar] [CrossRef]
- Khazipov, R.; Tyzio, R.; Ben-Ari, Y. Effects of oxytocin on GABA signalling in the foetal brain during delivery. Prog. Brain Res. 2008, 170, 243–257. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef]
- Li, H.; Tornberg, J.; Kaila, K.; Airaksinen, M.; Rivera, C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 2002, 16, 2358–2370. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.; Barone, S. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.; Nguyen, L.; Rigo, J.-M. Glycine receptors and brain development. Front. Cell. Neurosci. 2013, 7, 184. [Google Scholar] [CrossRef]
- Wilke, B.U.; Kummer, K.; Leitner, M.; Kress, M. Chloride—The Underrated Ion in Nociceptors. Front. Neurosci. 2020, 14, 287. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Khanna, A.; Clapham, D.E.; Woolf, C.J. Therapeutic Restoration of Spinal Inhibition via Druggable Enhancement of Potassium-Chloride Cotransporter KCC2–Mediated Chloride Extrusion in Peripheral Neuropathic Pain. JAMA Neurol. 2014, 71, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Flores, B.; Delpire, E. Osmotic Response of Dorsal Root Ganglion Neurons Expressing Wild-Type and Mutant KCC3 Transporters. Cell. Physiol. Biochem. 2020, 54, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Auteri, M.; Zizzo, M.G.; Serio, R. GABA and GABA receptors in the gastrointestinal tract: From motility to inflammation. Pharmacol. Res. 2015, 93, 11–21. [Google Scholar] [CrossRef]
- Bjurstöm, H.; Wang, J.; Ericsson, I.; Bengtsson, M.; Liu, Y.; Mendu, S.K.; Issazadeh-Navikas, S.; Birnir, B. GABA, a natural immunomodulator of T lymphocytes. J. Neuroimmunol. 2008, 205, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Nugent, B.M.; Valenzuela, C.V.; Simons, T.J.; McCarthy, M.M. Kinases SPAK and OSR1 are upregulated by estradiol and activate NKCC1 in the developing hypothalamus. J. Neurosci. 2012, 32, 593–598. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inoue, K.; Furukawa, T.; Kumada, T.; Yamada, J.; Wang, T.; Inoue, R.; Fukuda, A. Taurine Inhibits K+-Cl− Cotransporter KCC2 to Regulate Embryonic Cl− Homeostasis via With-no-lysine (WNK) Protein Kinase Signaling Pathway. J. Biol. Chem. 2012, 287, 20839–20850. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Sadlaoud, K.; Boulenguez, P.; Buttigieg, D.; Liabeuf, S.; Brocard, C.; Haase, G.; Bras, H.; Vinay, L. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2012, 110, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-Y.; Chen, S.-R.; Byun, H.-S.; Chen, H.; Li, L.; Han, H.-D.; Lopez-Berestein, G.; Sood, A.K.; Pan, H.-L. N-Methyl-d-aspartate Receptor- and Calpain-mediated Proteolytic Cleavage of K+-Cl− Cotransporter-2 Impairs Spinal Chloride Homeostasis in Neuropathic Pain. J. Biol. Chem. 2012, 287, 33853–33864. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Gaiarsa, J.-L.; Tyzio, R.; Khazipov, R. GABA: A Pioneer Transmitter That Excites Immature Neurons and Generates Primitive Oscillations. Physiol. Rev. 2007, 87, 1215–1284. [Google Scholar] [CrossRef]
- Cherubini, E.; Griguoli, M.; Safiulina, V.; Lagostena, L. The Depolarizing Action of GABA Controls Early Network Activity in the Developing Hippocampus. Mol. Neurobiol. 2010, 43, 97–106. [Google Scholar] [CrossRef]
- Cellot, G.; Cherubini, E. GABAergic Signaling as Therapeutic Target for Autism Spectrum Disorders. Front. Pediatr. 2014, 2, 70. [Google Scholar] [CrossRef]
- Schulte, J.T.; Wierenga, C.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Corradini, I.; Focchi, E.; Rasile, M.; Morini, R.; Desiato, G.; Tomasoni, R.; Lizier, M.; Ghirardini, E.; Fesce, R.; Morone, D.; et al. Maternal Immune Activation Delays Excitatory-to-Inhibitory Gamma-Aminobutyric Acid Switch in Offspring. Biol. Psychiatry 2018, 83, 680–691. [Google Scholar] [CrossRef]
- Malkova, N.V.; Yu, C.Z.; Hsiao, E.Y.; Moore, M.J.; Patterson, P.H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 2012, 26, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Berglund, K.; Hanna, M.; Guo, J.U.; Kittur, J.; Torres, M.D.; Abramowitz, J.; Busciglio, J.; Gao, Y.; Birnbaumer, L.; et al. Bisphenol A delays the perinatal chloride shift in cortical neurons by epigenetic effects on theKcc2promoter. Proc. Natl. Acad. Sci. USA 2013, 110, 4315–4320. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 2nd ed.; Oxford University: Oxford, UK, 1989. [Google Scholar]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Kim, Y.; Vadodaria, K.C.; Lenkei, Z.; Kato, T.; Gage, F.H.; Marchetto, M.C.; Santos, R. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Signal. 2019, 31, 275–317. [Google Scholar] [CrossRef]
- Rose, S.; Niyazov, D.M.; Rossignol, D.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef]
- Anglin, R.E.; Mazurek, M.F.; Tarnopolsky, M.A.; Rosebush, P.I. The mitochondrial genome and psychiatric illness. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2012, 159, 749–759. [Google Scholar] [CrossRef]
- Giulivi, C.; Zhang, Y.-F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial Dysfunction in Autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef]
- Ngugi, A.K.; Kariuki, S.M.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Incidence of epilepsy: A systematic review and meta-analysis. Neurology 2011, 77, 1005–1012. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Early Identification of Refractory Epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, J.; Liang, S.; Zhang, G.; Yang, X. Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front. Neurol. 2020, 10, 1407. [Google Scholar] [CrossRef]
- Genç, F.; Kara, M.; Ünal, Y.; Küçükseymen, E.U.; Gömceli, Y.B.; Kaynar, T.; Tosun, K.; Kutlu, G. Methylation of cation–chloride cotransporters NKCC1 and KCC2 in patients with juvenile myoclonic epilepsy. Neurol. Sci. 2019, 40, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Sivakumaran, S.; Maguire, J. Bumetanide reduces seizure progression and the development of pharmacoresistant status epilepticus. Epilepsia 2015, 57, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Kourdougli, N.; Pellegrino, C.; Renko, J.-M.; Khirug, S.; Chazal, G.; Kukko-Lukjanov, T.-K.; Lauri, S.E.; Gaiarsa, J.-L.; Zhou, L.; Peret, A.; et al. Depolarizing γ-aminobutyric acid contributes to glutamatergic network rewiring in epilepsy. Ann. Neurol. 2017, 81, 251–265. [Google Scholar] [CrossRef]
- Kipnis, P.A.; Sullivan, B.J.; Kadam, S.D. Sex-Dependent Signaling Pathways Underlying Seizure Susceptibility and the Role of Chloride Cotransporters. Cells 2019, 8, 448. [Google Scholar] [CrossRef]
- Gilad, D.; Shorer, S.; Ketzef, M.; Friedman, A.; Sekler, I.; Aizenman, E.; Hershfinkel, M. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol. Dis. 2015, 81, 4–13. [Google Scholar] [CrossRef]
- Fukuda, A.; Watanabe, M. Pathogenic potential of human SLC12A5 variants causing KCC2 dysfunction. Brain Res. 2018, 1710, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Fahanik-Babaei, J.; Mohamadi-Zarch, S.-M.; Tashakori-Miyanroudi, M.; Nourabadi, D.; Nazari-Serenjeh, M.; Roghani, M.; Baluchnejadmojarad, T. Neuroprotective and anticonvulsant effects of sinomenine in kainate rat model of temporal lobe epilepsy: Involvement of oxidative stress, inflammation and pyroptosis. J. Chem. Neuroanat. 2020, 108, 101800. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Kostova, A.T.D.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species. Int. J. Mol. Sci. 2017, 18, 1935. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Smith, J.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [PubMed]
- Aleman, A.; Kahn, R.S.; Selten, J.-P. Sex Differences in the Risk of Schizophrenia. Arch. Gen. Psychiatry 2003, 60, 565–571. [Google Scholar] [CrossRef]
- Murillo-De-Ozores, A.R.; Chávez-Canales, M.; Heros, P.D.L.; Gamba, G.; Castañeda-Bueno, M. Physiological Processes Modulated by the Chloride-Sensitive WNK-SPAK/OSR1 Kinase Signaling Pathway and the Cation-Coupled Chloride Cotransporters. Front. Physiol. 2020, 11, 585907. [Google Scholar] [CrossRef]
- Merner, N.; Chandler, M.R.; Bourassa, C.V.; Liang, B.; Khanna, A.R.; Dion, P.A.; Rouleau, G.A.; Kahle, K.T. Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci. 2015, 9, 386. [Google Scholar] [CrossRef]
- Merner, N.; Mercado, A.; Khanna, A.R.; Hodgkinson, A.; Bruat, V.; Awadalla, P.; Gamba, G.; Rouleau, G.A.; Kahle, K.T. Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J. Psychiatr. Res. 2016, 77, 22–26. [Google Scholar] [CrossRef]
- Arion, D.; Lewis, D.A. Altered Expression of Regulators of the Cortical Chloride Transporters NKCC1 and KCC2 in Schizophrenia. Arch. Gen. Psychiatry 2011, 68, 21–31. [Google Scholar] [CrossRef]
- Rahmanzadeh, R.; Eftekhari, S.; Shahbazi, A.; Ardakani, M.-R.K.; Rahmanzade, R.; Mehrabi, S.; Barati, M.; Joghataei, M.T. Effect of bumetanide, a selective NKCC1 inhibitor, on hallucinations of schizophrenic patients; a double-blind randomized clinical trial. Schizophr. Res. 2017, 184, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, E.; Villeneuve, N.; Sonie, S.; Serret, S.; Rosier, A.; Roue, M.; Brosset, P.; Viellard, M.; Bernoux, D.; Rondeau, S.; et al. Erratum: Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 2017, 7, e1056–e1124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, C.-C.; Dai, Y.; Luo, Q.; Ji, Y.; Wang, K.; Deng, S.; Yu, J.; Xu, M.; Du, X.; et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Fernell, E.; Gustafsson, P.; Gillberg, C. Bumetanide for autism: Open-label trial in six children. Acta Paediatr. 2020, 110, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Lemonnier, E. Using bumetanide to treat autism appears promising but further clinical trials are needed to confirm this approach. Acta Paediatr. 2021, 110, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Sawa, A.; Sedlak, T.W. Oxidative stress and inflammation in schizophrenia. Schizophr. Res. 2016, 176, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Cabungcal, J.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A “central hub” in schizophrenia pathophysiology? Schizophr. Res. 2014, 176, 41–51. [Google Scholar] [CrossRef]
- Koga, M.; Serritella, A.V.; Sawa, A.; Sedlak, T.W. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr. Res. 2016, 176, 52–71. [Google Scholar] [CrossRef]
- Upthegrove, R.; Khandaker, G.M. Cytokines, Oxidative Stress and Cellular Markers of Inflammation in Schizophrenia. Curr. Top. Behav. Neurosci. 2019, 44, 49–66. [Google Scholar] [CrossRef]
- Ma, J.; Yan, L.; Guo, T.; Yang, S.; Ni, D.; Liu, Y.; Wang, J. A pilot study of biomarkers of oxidative stress in serum and schizophrenia. Psychiatry Res. 2020, 284, 112757. [Google Scholar] [CrossRef]
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr. Alzheimer Res. 2015, 13, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zis, P.; Strydom, A. Clinical aspects and biomarkers of Alzheimer’s disease in Down syndrome. Free Radic. Biol. Med. 2017, 114, 3–9. [Google Scholar] [CrossRef]
- Contestabile, A.; Magara, S.; Cancedda, L. The GABAergic Hypothesis for Cognitive Disabilities in Down Syndrome. Front. Cell. Neurosci. 2017, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Crosti, N.; Bajer, J.; Gentile, M.; Resta, G.; Serra, A. Catalase and glutathione peroxidase activity in cells with trisomy 21. Clin. Genet. 1989, 36, 107–116. [Google Scholar] [CrossRef]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.; Vacca, R. Mitochondria as pharmacological targets in Down syndrome. Free. Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef]
- Guedj, F.; Sébrié, C.; Rivals, I.; Ledru, A.; Paly, E.; Bizot, J.C.; Smith, D.; Rubin, E.; Gillet, B.; Arbones, M.; et al. Green Tea Polyphenols Rescue of Brain Defects Induced by Overexpression of DYRK1A. PLoS ONE 2009, 4, e4606. [Google Scholar] [CrossRef]
- Hanrahan, J.R.; Chebib, M.; Johnston, G.A.R. Flavonoid modulation of GABA A receptors. Br. J. Pharmacol. 2011, 163, 234–245. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Elsabbagh, M.; Divan, G.; Koh, Y.-J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global Prevalence of Autism and Other Pervasive Developmental Disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef]
- Keller, R.; Basta, R.; Salerno, L.; Elia, M. Autism, epilepsy, and synaptopathies: A not rare association. Neurol. Sci. 2017, 38, 1353–1361. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Li, C.; Zhang, Z.; Teng, H.; Wang, Y.; Zhao, T.; Shi, L.; Zhang, K.; Xia, K.; et al. Genetic evidence of gender difference in autism spectrum disorder supports the female-protective effect. Transl. Psychiatry 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Casanova, E.L.; Frye, R.E.; Baeza-Velasco, C.; LaSalle, J.M.; Hagerman, R.J.; Scherer, S.; Natowicz, M.R. Editorial: Secondary vs. Idiopathic Autism. Front. Psychiatry 2020, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Panisi, C.; Guerini, F.R.; Abruzzo, P.M.; Balzola, F.; Biava, P.M.; Bolotta, A.; Brunero, M.; Burgio, E.; Chiara, A.; Clerici, M.; et al. Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. J. Pers. Med. 2021, 11, 70. [Google Scholar] [CrossRef]
- Burgio, E. Environment and Fetal Programming: The origins of some current “pandemics”. J. Pediatr. Neonat. Individual. Med. 2015, 4, 2. [Google Scholar] [CrossRef]
- Street, M.E.; Angelini, S.; Bernasconi, S.; Burgio, E.; Cassio, A.; Catellani, C.; Cirillo, F.; Deodati, A.; Fabbrizi, E.; Fanos, V.; et al. Current Knowledge on Endocrine Disrupting Chemicals (EDCs) from Animal Biology to Humans, from Pregnancy to Adulthood: Highlights from a National Italian Meeting. Int. J. Mol. Sci. 2018, 19, 1647. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Zoghbi, H. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef]
- Pilorge, M.; Fassier, C.; Le Corronc, H.; Potey, A.; Bai, J.; De Gois, S.; Delaby, E.; Assouline, B.; Guinchat, V.; Devillard, F.; et al. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry 2015, 21, 936–945. [Google Scholar] [CrossRef]
- Pizzarelli, R.; Griguoli, M.; Zacchi, P.; Petrini, E.M.; Barberis, A.; Cattaneo, A.; Cherubini, E. Tuning GABAergic Inhibition: Gephyrin Molecular Organization and Functions. Neuroscience 2019, 439, 125–136. [Google Scholar] [CrossRef]
- Dejanovic, B.; Djémié, T.; Grünewald, N.; Suls, A.; Kress, V.; Hetsch, F.; Craiu, D.; Zemel, M.; Gormley, P.; Lal, D.; et al. Simultaneous impairment of neuronal and metabolic function of mutated gephyrin in a patient with epileptic encephalopathy. EMBO Mol. Med. 2015, 7, 1580–1594. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Kreiser, N.L.; Urbano, M.R.; Burket, J.A.; Pickle, J.C. Autism presenting in the context of a genetic variant of CFTR and early HSV exposure confounded by chronic pain, altered gut microbiota and paternal abandonment; limitations of current pharmacotherapy and barriers to personalized treatment recommendations. Pers. Med. Psychiatry 2017, 3, 24–29. [Google Scholar] [CrossRef][Green Version]
- Chao, H.-T.; Chen, H.; Samaco, R.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.; Gong, S.; Lu, H.-C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Lozovaya, N.; Nardou, R.; Tyzio, R.; Chiesa, M.; Pons-Bennaceur, A.; Eftekhari, S.; Bui, T.-T.; Billon-Grand, M.; Rasero, J.; Bonifazi, P.; et al. Early alterations in a mouse model of Rett syndrome: The GABA developmental shift is abolished at birth. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Qi, L.; Yang, Z.; Yang, T.; Zhang, Y.; Xu, H.; Zhao, H. Impaired GABA Neural Circuits Are Critical for Fragile X Syndrome. Neural Plast. 2018, 2018, 8423420. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.T.; Spatuzza, M.; D’Antoni, S.; Bonaccorso, C.M.; Trovato, C.; Musumeci, S.A.; Leopoldo, M.; Lacivita, E.; Catania, M.V.; Ciranna, L. Activation of 5-HT7 Serotonin Receptors Reverses Metabotropic Glutamate Receptor-Mediated Synaptic Plasticity in Wild-Type and Fmr1 Knockout Mice, a Model of Fragile X Syndrome. Biol. Psychiatry 2012, 72, 924–933. [Google Scholar] [CrossRef] [PubMed]
- De Lima, R.M.S.; Barth, B.; Arcego, D.M.; Filho, E.J.D.M.; Clappison, A.; Patel, S.; Wang, Z.; Pokhvisneva, I.; Sassi, R.B.; Hall, G.B.C.; et al. Amygdala 5-HTT Gene Network Moderates the Effects of Postnatal Adversity on Attention Problems: Anatomo-Functional Correlation and Epigenetic Changes. Front. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Amodeo, D.A.; Eyi, J.; Sweeney, J.A.; Ragozzino, M.E. Oxotremorine treatment reduces repetitive behaviors in BTBR T+ tf/J mice. Front. Synaptic Neurosci. 2014, 6, 17. [Google Scholar] [CrossRef]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2006, 141, 947–956. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; et al. Oxidative Stress and Erythrocyte Membrane Alterations in Children with Autism: Correlation with Clinical Features. PLoS ONE 2013, 8, e66418. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, G.; Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Marzetti, C.; Spyratou, E.; Georgakilas, A.G.; Marini, M.; Abruzzo, P.M.; Bolotta, A.; et al. High predictive values of RBC membrane-based diagnostics by biophotonics in an integrated approach for Autism Spectrum Disorders. Sci. Rep. 2017, 7, 9854. [Google Scholar] [CrossRef]
- Bolotta, A.; Battistelli, M.; Falcieri, E.; Ghezzo, A.; Manara, M.C.; Manfredini, S.; Marini, M.; Posar, A.; Visconti, P.; Abruzzo, P.M. Oxidative Stress in Autistic Children Alters Erythrocyte Shape in the Absence of Quantitative Protein Alterations and of Loss of Membrane Phospholipid Asymmetry. Oxidative Med. Cell. Longev. 2018, 2018, 643060. [Google Scholar] [CrossRef] [PubMed]
- Ciccoli, L.; De Felice, C.; Paccagnini, E.; Leoncini, S.; Pecorelli, A.; Signorini, C.; Belmonte, G.; Valacchi, G.; Rossi, M.; Hayek, J. Morphological changes and oxidative damage in Rett Syndrome erythrocytes. Biochim. Biophys. Acta 2012, 1820, 511–520. [Google Scholar] [CrossRef]
- Anwar, A.; Abruzzo, P.M.; Pasha, S.; Rajpoot, K.; Bolotta, A.; Ghezzo, A.; Marini, M.; Posar, A.; Visconti, P.; Thornalley, P.J.; et al. Advanced glycation endproducts, dityrosine and arginine transporter dysfunction in autism—A source of biomarkers for clinical diagnosis. Mol. Autism 2018, 9, 3. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2011, 17, 290–314. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2011, 17, 389–401. [Google Scholar] [CrossRef]
- Frye, R.E.; Rose, S.; Slattery, J.; Macfabe, D.F. Gastrointestinal dysfunction in autism spectrum disorder: The role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 2015, 26, 27458. [Google Scholar] [CrossRef]
- Mezzelani, A.; Landini, M.; Facchiano, F.; Raggi, M.E.; Villa, L.; Molteni, M.; De Santis, B.; Brera, C.; Caroli, A.M.; Milanesi, L.; et al. Environment, dysbiosis, immunity and sex-specific susceptibility: A translational hypothesis for regressive autism pathogenesis. Nutr. Neurosci. 2014, 18, 145–161. [Google Scholar] [CrossRef]
- Liu, Y.-K.; Gao, H.; Jin, S.-B.; Tu, W.-J.; Chen, Y.-J. Association of neonatal blood levels of brain-derived neurotrophic factor with development of autism spectrum disorder: A systematic review and meta-analysis. World J. Pediatr. 2021, 17, 164–170. [Google Scholar] [CrossRef]
- Konopko, M.A.; Densmore, A.L.; Krueger, B.K. Sexually Dimorphic Epigenetic Regulation of Brain-Derived Neurotrophic Factor in Fetal Brain in the Valproic Acid Model of Autism Spectrum Disorder. Dev. Neurosci. 2017, 39, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, S.; Del Bigio, M.R.; Rastegar, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Front. Cell Dev. Biol. 2020, 8, 763. [Google Scholar] [CrossRef]
- Riikonen, R. Insulin-Like Growth Factors in the Pathogenesis of Neurological Diseases in Children. Int. J. Mol. Sci. 2017, 18, 2056. [Google Scholar] [CrossRef] [PubMed]
- Steinman, G.; Mankuta, D. Molecular biology of autism’s etiology—An alternative mechanism. Med. Hypotheses 2019, 130, 109272. [Google Scholar] [CrossRef] [PubMed]
- Costales, J.; Kolevzon, A. The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci. Biobehav. Rev. 2016, 63, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Linker, S.B.; Mendes, A.P.D.; Marchetto, M.C. IGF-1 treatment causes unique transcriptional response in neurons from individuals with idiopathic autism. Mol. Autism 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- González, A.N.B.; Pazos, M.I.L.; Calvo, D.J. Reactive Oxygen Species in the Regulation of the GABA Mediated Inhibitory Neurotransmission. Neuroscience 2019, 439, 137–145. [Google Scholar] [CrossRef]
- Toczylowska, B.; Zieminska, E.; Senator, P.; Lazarewicz, J.W. Hippocampal Metabolite Profiles in Two Rat Models of Autism: NMR-Based Metabolomics Studies. Mol. Neurobiol. 2020, 57, 3089–3105. [Google Scholar] [CrossRef]
- Bai, Y.; Bai, Y.; Wang, S.; Wu, F.; Wang, D.H.; Chen, J.; Huang, J.; Li, H.; Li, Y.; Wu, S.; et al. Targeted upregulation of uncoupling protein 2 within the basal ganglia output structure ameliorates dyskinesia after severe liver failure. Free Radic. Biol. Med. 2018, 124, 40–50. [Google Scholar] [CrossRef]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2016, 22, 273–279. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Rasile, M.; Corradini, I.; Matteoli, M. Environmental regulation of the chloride transporter KCC2: Switching inflammation off to switch the GABA on? Transl. Psychiatry 2020, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.P.; Schluter, M.D.; Steer, R.A.; Guo, L.; Ming, X. Bisphenol A Exposure in Children With Autism Spectrum Disorders. Autism Res. 2015, 8, 272–283. [Google Scholar] [CrossRef]
- Neuwirth, L.S.; Phillips, G.R.; El Idrissi, A. Perinatal Pb2+ exposure alters the expression of genes related to the neurodevelopmental GABA-shift in postnatal rats. J. Biomed. Sci. 2018, 25, 45. [Google Scholar] [CrossRef]
- Nam, S.M.; Choi, S.-H.; Cho, H.-J.; Seo, J.S.; Choi, M.; Nahm, S.-S.; Chang, B.-J.; Nah, S.-Y. Ginseng Gintonin Attenuates Lead-Induced Rat Cerebellar Impairments during Gestation and Lactation. Biomolecules 2020, 10, 385. [Google Scholar] [CrossRef]
- Maiti, A.; Saha, N.C.; Paul, G. Effect of Lead on Oxidative Stress, Na+K+ATPase Activity and Mitochondrial Electron Transport Chain Activity of the Brain of Clarias batrachus L. Bull. Environ. Contam. Toxicol. 2010, 84, 672–676. [Google Scholar] [CrossRef]
- Gialloreti, L.E.; Mazzone, L.; Benvenuto, A.; Fasano, A.; Alcon, A.G.; Kraneveld, A.; Moavero, R.; Raz, R.; Riccio, M.P.; Siracusano, M.; et al. Risk and Protective Environmental Factors Associated with Autism Spectrum Disorder: Evidence-Based Principles and Recommendations. J. Clin. Med. 2019, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Tamari, M.; Daigo, Y.; Nakamura, Y. Isolation and characterization of a novel serine threonine kinase gene on chromosome 3p22-21.3. J. Hum. Genet. 1999, 44, 116–120. [Google Scholar] [CrossRef]
- Kozlov, S.V.; Waardenberg, A.; Engholm-Keller, K.; Arthur, J.W.; Graham, M.; Lavin, M. Reactive Oxygen Species (ROS)-Activated ATM-Dependent Phosphorylation of Cytoplasmic Substrates Identified by Large-Scale Phosphoproteomics Screen. Mol. Cell. Proteom. 2016, 15, 1032–1047. [Google Scholar] [CrossRef]
- Pizzamiglio, L.; Focchi, E.; Cambria, C.; Ponzoni, L.; Ferrara, S.; Bifari, F.; Desiato, G.; Landsberger, N.; Murru, L.; Passafaro, M.; et al. The DNA repair protein ATM as a target in autism spectrum disorder. JCI Insight 2021, 6, e133654. [Google Scholar] [CrossRef] [PubMed]
- Bolotta, A.; Visconti, P.; Fedrizzi, G.; Ghezzo, A.; Marini, M.; Manunta, P.; Messaggio, E.; Posar, A.; Vignini, A.; Abruzzo, P.M. Na+, K+-ATPase activity in children with autism spectrum disorder: Searching for the reason(s) of its decrease in blood cells. Autism Res. 2018, 11, 1388–1403. [Google Scholar] [CrossRef]
- Dasgupta, A.; Roy, S.; Banerjee, U.; Chowdhury, P.; Mukhopadhyay, A.; Saha, G.; Singh, O. Role of membrane cholesterol and lipid peroxidation in regulating the Na+/K+-ATPase activity in schizophrenia. Indian J. Psychiatry 2016, 58, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Onimaru, H.; Yamada, J.; Inoue, K.; Ueno, S.; Onaka, T.; Toyoda, H.; Arata, A.; Ishikawa, T.-O.; Taketo, M.M.; et al. Malfunction of Respiratory-Related Neuronal Activity in Na+, K+-ATPase α2 Subunit-Deficient Mice Is Attributable to Abnormal Cl− Homeostasis in Brainstem Neurons. J. Neurosci. 2004, 24, 10693–10701. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.A.; Isaksen, T.J.; Heuck, A.; Scavone, C.; Lykke-Hartmann, K. The α2 Na+/K+-ATPase isoform mediates LPS-induced neuroinflammation. Sci. Rep. 2020, 10, 14180. [Google Scholar] [CrossRef] [PubMed]
- Brondino, N.; Fusar-Poli, L.; Panisi, C.; Damiani, S.; Barale, F.; Politi, P. Pharmacological Modulation of GABA Function in Autism Spectrum Disorders: A Systematic Review of Human Studies. J. Autism Dev. Disord. 2015, 46, 825–839. [Google Scholar] [CrossRef]
- Savardi, A.; Borgogno, M.; Narducci, R.; La Sala, G.; Ortega, J.A.; Summa, M.; Armirotti, A.; Bertorelli, R.; Contestabile, A.; De Vivo, M.; et al. Discovery of a Small Molecule Drug Candidate for Selective NKCC1 Inhibition in Brain Disorders. Chem 2020, 6, 2073–2096. [Google Scholar] [CrossRef]
- Keller, R.; Chieregato, S.; Bari, S.; Castaldo, R.; Rutto, F.; Chiocchetti, A.; Dianzani, U. Autism in Adulthood: Clinical and Demographic Characteristics of a Cohort of Five Hundred Persons with Autism Analyzed by a Novel Multistep Network Model. Brain Sci. 2020, 10, 416. [Google Scholar] [CrossRef]
- Gandal, M.J.; Nesbitt, A.M.; McCurdy, R.M.; Alter, M.D. Measuring the Maturity of the Fast-Spiking Interneuron Transcriptional Program in Autism, Schizophrenia, and Bipolar Disorder. PLoS ONE 2012, 7, e41215. [Google Scholar] [CrossRef]
- Bolotta, A.; Pini, A.; Abruzzo, P.M.; Ghezzo, A.; Modesti, A.; Gamberi, T.; Ferreri, C.; Bugamelli, F.; Fortuna, F.; Vertuani, S.; et al. Effects of tocotrienol supplementation in Friedreich’s ataxia: A model of oxidative stress pathology. Exp. Biol. Med. 2019, 245, 201–212. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).