
Catalase Mediates the Inhibitory Actions of PPARδ against Angiotensin II-Triggered Hypertrophy in H9c2 Cardiomyocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. Cell Culture
2.3. MTT Assay
2.4. Immunofluorescence Staining
2.5. Measurement of Intracellular Reactive Oxygen Species (ROS)
2.6. Gene Silencing with Small Interfering RNA (siRNA)
2.7. Construction of H9c2 Cells Stably Expressing Short Hairpin (sh)PPARδ
2.8. Western Blot Analysis
2.9. Real-Time PCR
2.10. Construction of the pGL3-Catalase Luciferase Reporter Plasmid and the Reporter Gene Assay
2.11. Statistical Analysis
3. Results
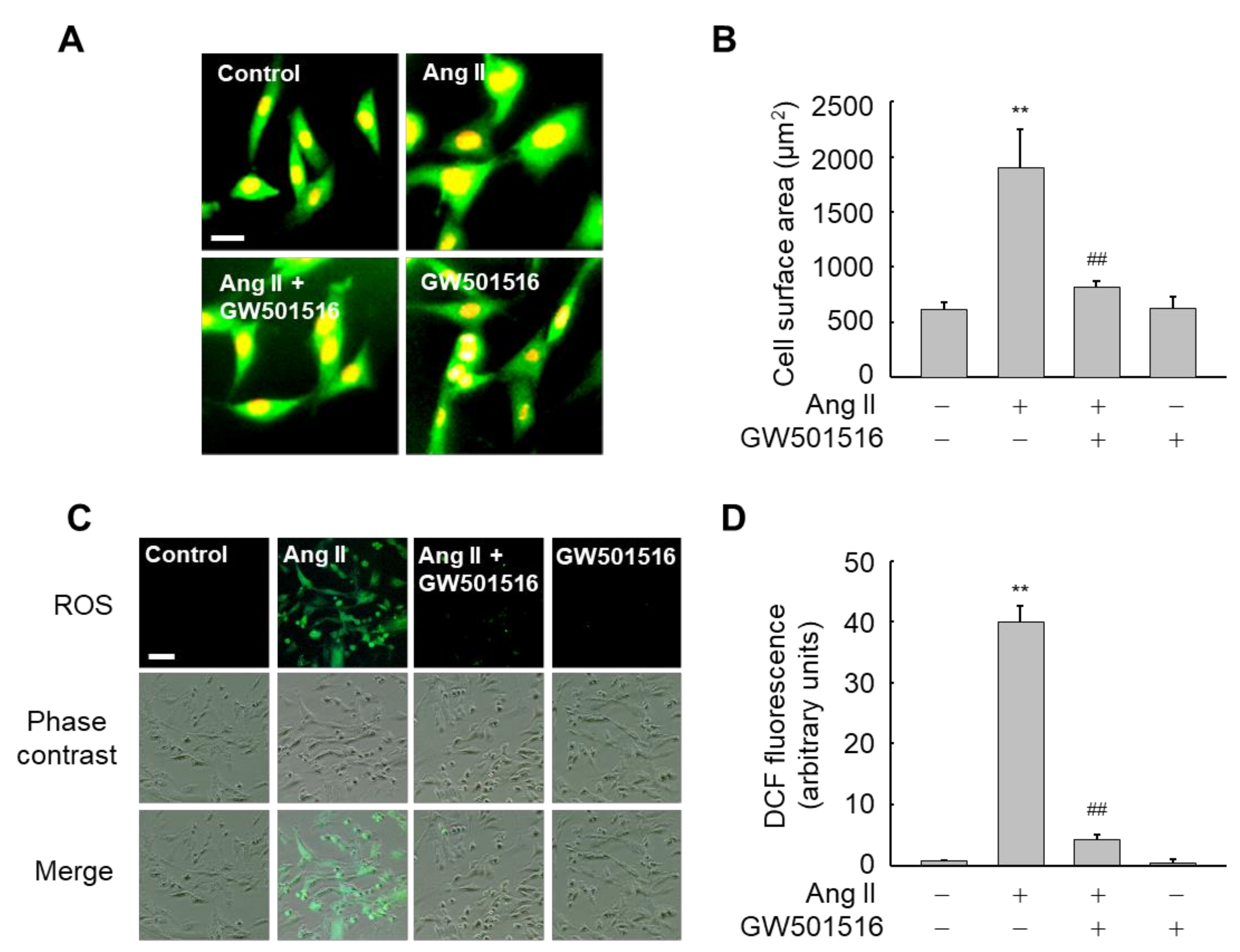
3.1. GW501516-Activated PPARδ Inhibits Ang II-Triggered Hypertrophy and ROS Generation in H9c2 Cells
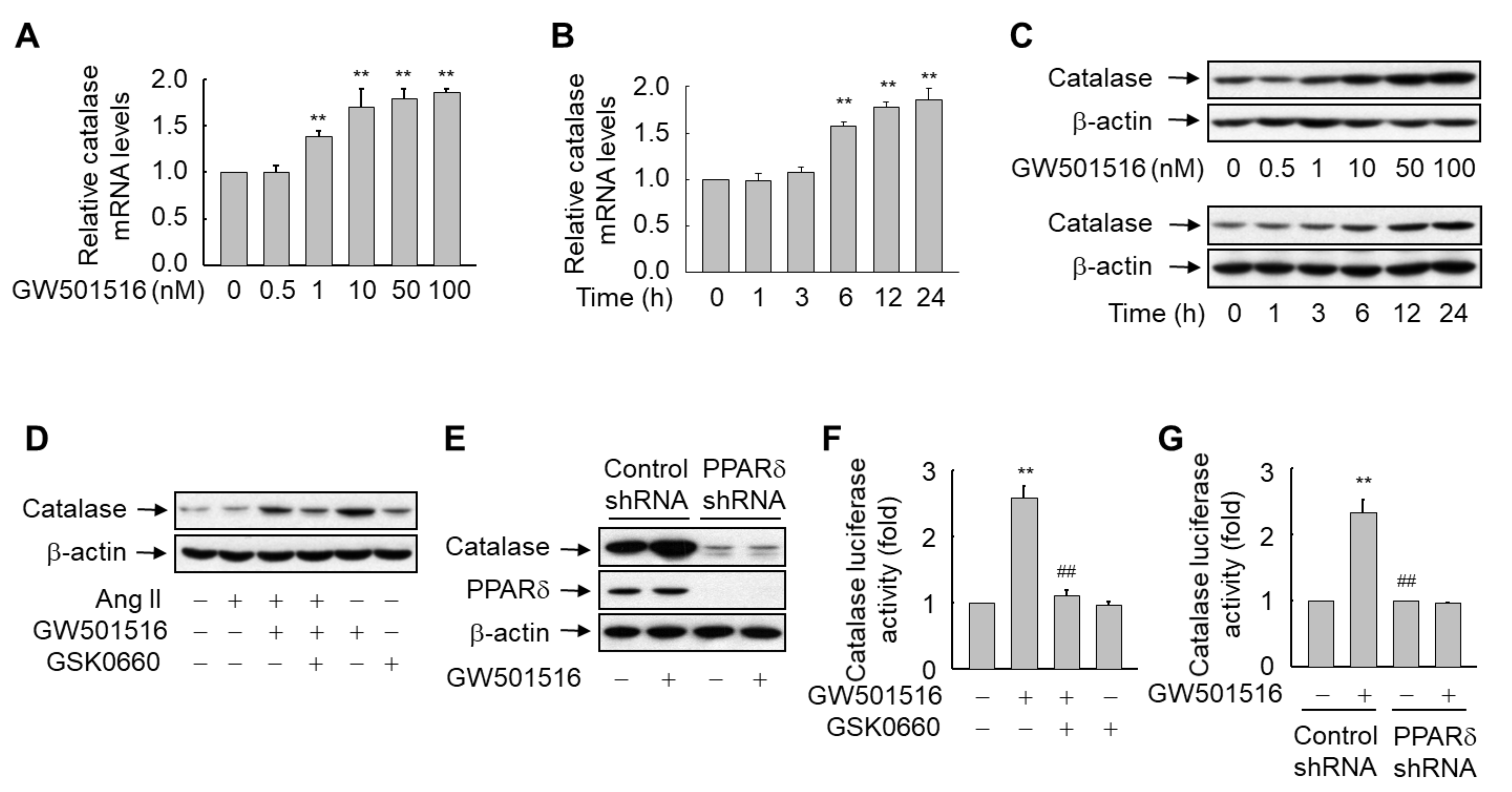
3.2. GW501516-Activated PPARδ Induces the Expression of Catalase in H9c2 Cells
3.3. GW501516-Activated PPARδ Regulates the Expression of Marker Proteins Linked to Ang II-Induced Hypertrophy of H9c2 Cardiomyocytes
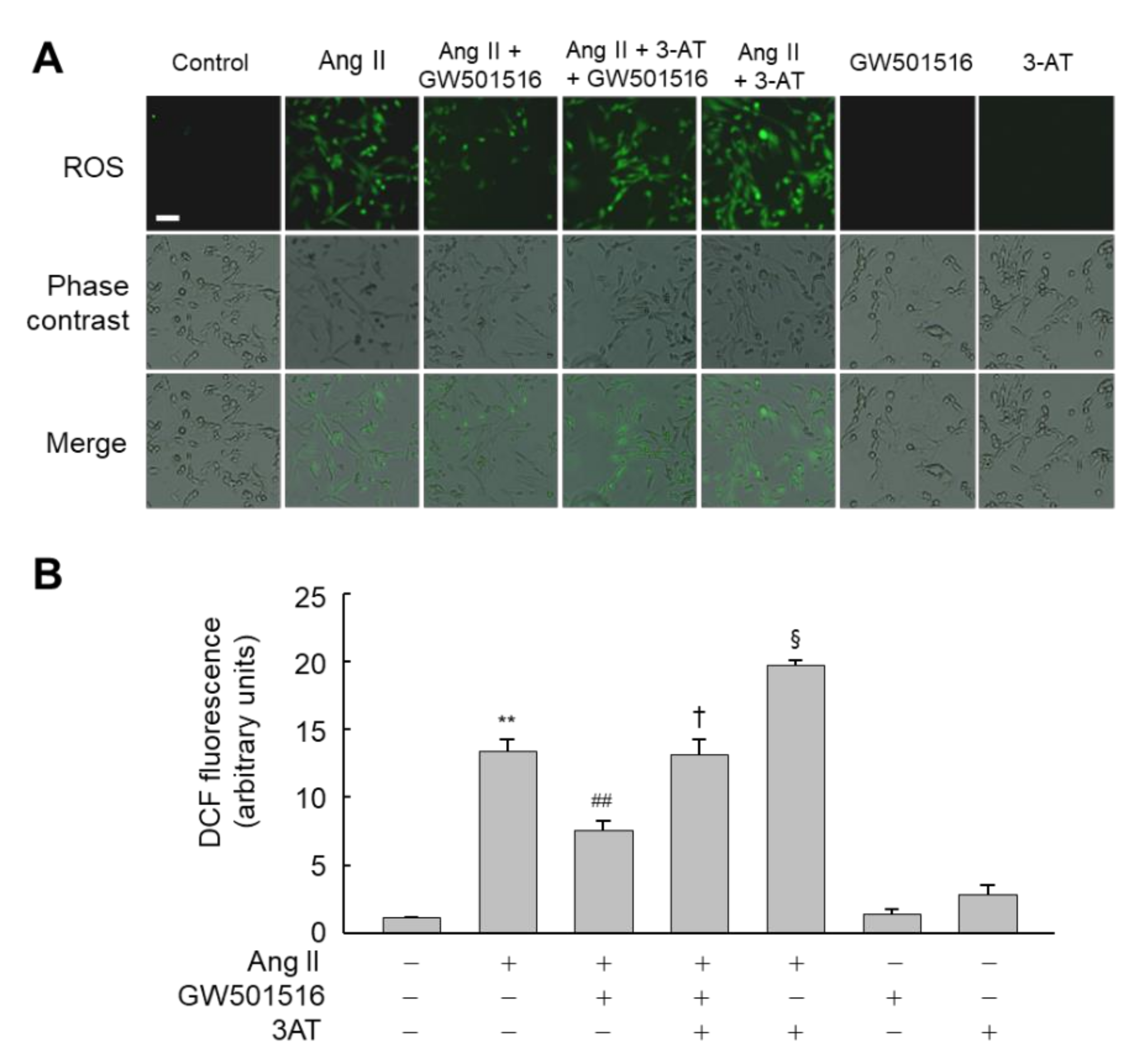
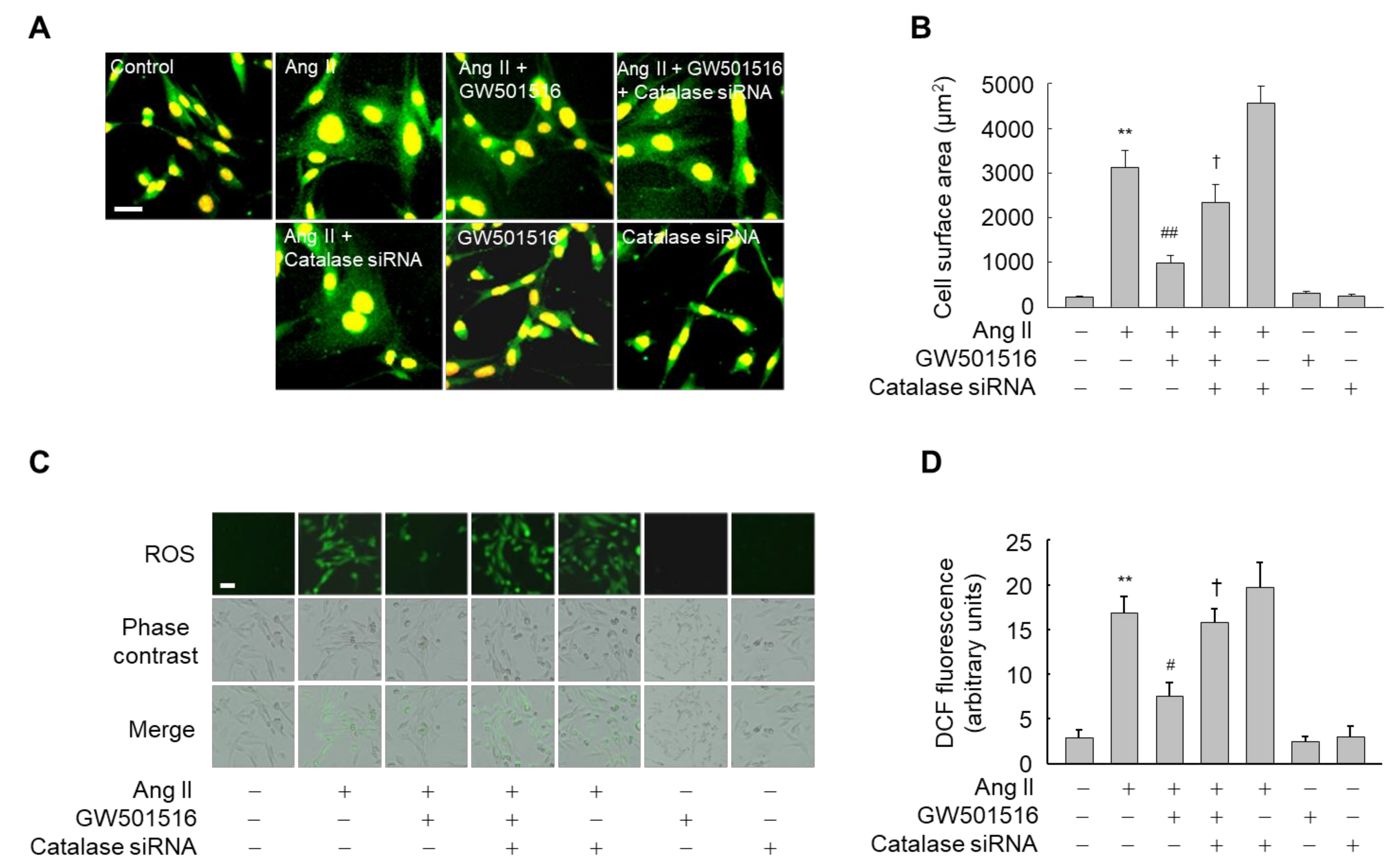
3.4. Downregulation of Catalase Abrogates the Effects of PPARδ on Hypertrophy and ROS Production
3.5. GW501516-Activated PPARδ Attenuates Ang II-Induced Hypertrophy and ROS Generation by Inhibiting p38 MAPK Signaling in H9c2 Cardiomyocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.F.; Dohmen, P.M.; Choi, Y.H.; Wahlers, T.; et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, D.R.; Amin, J.K.; Xiao, L.; Miller, T.; Viereck, J.; Oliver-Krasinski, J.; Baliga, R.; Wang, J.; Siwik, D.A.; Singh, K.; et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ. Res. 2001, 89, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Cao, F.; Tang, Q.Z.; Yan, L.; Dong, Y.G.; Zhu, L.H.; Wang, L.; Bian, Z.Y.; Li, H. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J. Nutr. Biochem. 2010, 21, 1238–1250. [Google Scholar] [CrossRef]
- Papparella, I.; Ceolotto, G.; Montemurro, D.; Antonello, M.; Garbisa, S.; Rossi, G.; Semplicini, A. Green tea attenuates angiotensin II-induced cardiac hypertrophy in rats by modulating reactive oxygen species production and the Src/epidermal growth factor receptor/Akt signaling pathway. J. Nutr. 2008, 138, 1596–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojic, L.A.; Huff, M.W. Peroxisome proliferator-activated receptor δ: A multifaceted metabolic player. Curr. Opin. Lipidol. 2013, 24, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Liu, J.; Yin, F.; Collins, A.R.; Lyon, C.J.; Lee, C.H.; Atkins, A.R.; Downes, M.; Barish, G.D.; Evans, R.M.; et al. KPPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4277–4282. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ham, S.A.; Kim, M.Y.; Hwang, J.S.; Lee, H.; Kang, E.S.; Yoo, T.; Woo, I.S.; Yabe-Nishimura, C.; Paek, K.S.; et al. PPARδ coordinates angiotensin II-induced senescence in vascular smooth muscle cells through PTEN-mediated inhibition of superoxide generation. J. Biol. Chem. 2011, 286, 44585–44593. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ham, S.A.; Paek, K.S.; Hwang, J.S.; Jung, S.Y.; Kim, M.Y.; Jin, H.; Kang, E.S.; Woo, I.S.; Kim, H.J.; et al. Transcriptional up-regulation of antioxidant genes by PPARδ inhibits angiotensin II-induced premature senescence in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2011, 406, 564–569. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Ham, S.A.; Yoo, T.; Lee, W.J.; Hwang, J.S.; Paek, K.S.; Lim, D.S.; Han, S.G.; Lee, C.H.; Seo, H.G. Ligand-Activated Peroxisome Proliferator-Activated Receptor δ Attenuates Vascular Oxidative Stress by Inhibiting Thrombospondin-1 Expression. J. Vasc. Res. 2018, 55, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Park, J.H.; Lee, S.; Lim, H.; Park, H.Y. PPARdelta activation inhibits angiotensin II induced cardiomyocyte hypertrophy by suppressing intracellular Ca2+ signaling pathway. J. Cell Biochem. 2009, 106, 823–834. [Google Scholar] [CrossRef]
- Cheng, K.C.; Chang, W.T.; Li, Y.; Cheng, Y.Z.; Cheng, J.T.; Chen, Z.C. GW0742 activates peroxisome proliferator-activated receptor δ to reduce free radicals and alleviate cardiac hypertrophy induced by hyperglycemia in cultured H9c2 cells. J. Cell Biochem. 2018, 119, 9532–9542. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Sadoshima, J. From Contractile Enhancement to Pathological Hypertrophy: Angiotensin II-Induced Nox2-Mediated Reactive Oxygen Species. J. Am. Coll. Cardiol. 2015, 66, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tao, Y.; Huang, Y.; Zhan, K.; Xue, M.; Wang, Y.; Ruan, D.; Liang, Y.; Huang, X.; Lin, J.; et al. Catalase ameliorates diabetes-induced cardiac injury through reduced p65/RelA- mediated transcription of BECN1. J. Cell Mol. Med. 2017, 21, 3420–3434. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ham, S.A.; Kim, M.Y.; Kim, J.H.; Paek, K.S.; Kang, E.S.; Kim, H.J.; Hwang, J.S.; Yoo, T.; Park, C.; et al. Activation of PPARδ counteracts angiotensin II-induced ROS generation by inhibiting rac1 translocation in vascular smooth muscle cells. Free Radic. Res. 2012, 46, 912–919. [Google Scholar] [CrossRef]
- Kim, M.Y.; Kang, E.S.; Ham, S.A.; Hwang, J.S.; Yoo, T.S.; Lee, H.; Paek, K.S.; Park, C.; Lee, H.T.; Kim, J.H.; et al. The PPARδ-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem. Pharmacol. 2012, 84, 1627–1634. [Google Scholar] [CrossRef]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci. Rep. 2015, 5, 15971. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.S.; Lee, W.J.; Kang, E.S.; Ham, S.A.; Yoo, T.; Paek, K.S.; Lim, D.S.; Do, J.T.; Seo, H.G. Ligand-activated peroxisome proliferator-activated receptor-δ and -γ inhibit lipopolysaccharide-primed release of high mobility group box 1 through upregulation of SIRT1. Cell Death Dis. 2014, 5, e1432. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.A.; Kim, E.; Yoo, T.; Lee, W.J.; Youn, J.H.; Choi, M.J.; Han, S.G.; Lee, C.H.; Paek, K.S.; Hwang, J.S.; et al. Ligand-activated interaction of PPARδ with c-Myc governs the tumorigenicity of breast cancer. Int. J. Cancer 2018, 143, 2985–2996. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.J.; Chan, H.W.; Osborne, J.E.; Thomas, W.G.; Hannan, R.D. Hijacking epidermal growth factor receptors by angiotensin II: New possibilities for understanding and treating cardiac hypertrophy. Cell Mol. Life Sci. 2004, 61, 2695–2703. [Google Scholar] [CrossRef]
- Savage, S.R.; McCollum, G.W.; Yang, R.; Penn, J.S. RNA-seq identifies a role for the PPARβ/δ inverse agonist GSK0660 in the regulation of TNFα-induced cytokine signaling in retinal endothelial cells. Mol. Vis. 2015, 21, 568–576. [Google Scholar]
- Malinowski, B.; Fulgheri, G.; Wicinski, M.; Grzesk, E.; Odrowaz-Sypniewska, G.; Grześk, G.; Darwish, N. Potential Markers in Cardiac Hypertrophy? EJIFCC 2012, 23, 41–46. [Google Scholar]
- Margoliash, E.; Novogrodsky, A. A study of the inhibition of catalase by 3-amino-1:2:4:-triazole. Biochem. J. 1958, 68, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gul, R.; Shawl, A.I.; Kim, S.H.; Kim, U.H. Cooperative interaction between reactive oxygen species and Ca2+ signals contributes to angiotensin II-induced hypertrophy in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H901–H909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Foussal, C.; Lairez, O.; Calise, D.; Pathak, A.; Guilbeau-Frugier, C.; Valet, P.; Parini, A.; Kunduzova, O. Activation of catalase by apelin prevents oxidative stress-linked cardiac hypertrophy. FEBS Lett. 2010, 584, 2363–2370. [Google Scholar] [CrossRef]
- Kang, E.S.; Hwang, J.S.; Lee, W.J.; Lee, G.H.; Choi, M.J.; Paek, K.S.; Lim, D.S.; Seo, H.G. Ligand-activated PPARδ inhibits angiotensin II-stimulated hypertrophy of vascular smooth muscle cells by targeting ROS. PLoS ONE 2019, 14, e0210482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojonazarov, B.; Luitel, H.; Sydykov, A.; Dahal, B.K.; Paul-Clark, M.J.; Bonvini, S.; Reed, A.; Schermuly, R.T.; Mitchell, J.A. The peroxisome proliferator-activated receptor β/δ agonist GW0742 has direct protective effects on right heart hypertrophy. Pulm. Circ. 2013, 3, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Ye, P.; Liu, Y.X.; Han, C.G.; Zhang, Z.Y. Peroxisome proliferator-activated receptor beta/delta activation improves angiotensin II-induced cardiac hypertrophy in vitro. Clin. Exp. Hypertens. 2008, 30, 109–119. [Google Scholar] [CrossRef]
- Planavila, A.; Rodríguez-Calvo, R.; Jové, M.; Michalik, L.; Wahli, W.; Laguna, J.C.; Vázquez-Carrera, M. Peroxisome proliferator-activated receptor beta/delta activation inhibits hypertrophy in neonatal rat cardiomyocytes. Cardiovasc. Res. 2005, 65, 832–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.D.; Vukolic, A.; Baudouy, D.; Michiels, J.F.; Wagner, N. Inducible Conditional Vascular-Specific Overexpression of Peroxisome Proliferator-Activated Receptor Beta/Delta Leads to Rapid Cardiac Hypertrophy. PPAR Res. 2016, 2016, 7631085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [CrossRef]
- Newsholme, S.J.; Dunsford, W.S.; Brodie, T.; Brennan, C.; Brown, M.; Geiger, L.E. Mouse carcinogenicity study with GW501516, a PPAR delta agonist. 48th Annual Meeting of the Society of Toxicology. Toxicologist 2009, 108, 896. [Google Scholar]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, I.; Wang, H.X.; Feng, X.; Alenina, N.; Bader, M.; Prabhakar, B.S.; Li, P.F. Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 2008, 283, 5996–6004. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.Q.; Wang, K.; Lv, D.Y.; Li, P.F. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 2008, 283, 29730–29739. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Luo, D.; Rando, T.A. The regulation of catalase gene expression in mouse muscle cells is dependent on the CCAAT-binding factor NF-Y. Biochem. Biophys. Res. Commun. 2003, 303, 609–618. [Google Scholar] [CrossRef]
- Nenoi, M.; Ichimura, S.; Mita, K.; Yukawa, O.; Cartwright, I.L. Regulation of the catalase gene promoter by Sp1, CCAAT-recognizing factors, and a WT1/Egr-related factor in hydrogen peroxide-resistant HP100 cells. Cancer Res. 2001, 61, 5885–5894. [Google Scholar]
- Taniguchi, M.; Hashimoto, M.; Hori, N.; Sato, K. CCAAT/enhancer binding protein-beta (C/EBP-beta), a pivotal regulator of the TATA-less promoter in the rat catalase gene. FEBS Lett. 2005, 579, 5785–5790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnun, G.D.; Domann, F.E.; Moore, S.A.; Robbins, M.E. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 2002, 16, 2793–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, Y.; Matsuda, M.; Miyata, Y.; Fukuhara, A.; Komuro, R.; Shimabukuro, M.; Shimomura, I. Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a response element distinct from that of mouse. Endocr. J. 2010, 57, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.H.; Lee, S.R.; Kim, M.K.; Shin, C.Y.; Lee, D.H.; Chung, J.H. Activation of Peroxisome Proliferator-Activated Receptor Alpha Improves Aged and UV-Irradiated Skin by Catalase Induction. PLoS ONE 2016, 11, e0162628. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.S.; Hur, J.; Lee, W.J.; Won, J.P.; Lee, H.G.; Lim, D.-S.; Kim, E.; Seo, H.G. Catalase Mediates the Inhibitory Actions of PPARδ against Angiotensin II-Triggered Hypertrophy in H9c2 Cardiomyocytes. Antioxidants 2021, 10, 1223. https://doi.org/10.3390/antiox10081223
Hwang JS, Hur J, Lee WJ, Won JP, Lee HG, Lim D-S, Kim E, Seo HG. Catalase Mediates the Inhibitory Actions of PPARδ against Angiotensin II-Triggered Hypertrophy in H9c2 Cardiomyocytes. Antioxidants. 2021; 10(8):1223. https://doi.org/10.3390/antiox10081223
Chicago/Turabian StyleHwang, Jung Seok, Jinwoo Hur, Won Jin Lee, Jun Pil Won, Hyuk Gyoon Lee, Dae-Seog Lim, Eunsu Kim, and Han Geuk Seo. 2021. "Catalase Mediates the Inhibitory Actions of PPARδ against Angiotensin II-Triggered Hypertrophy in H9c2 Cardiomyocytes" Antioxidants 10, no. 8: 1223. https://doi.org/10.3390/antiox10081223
APA StyleHwang, J. S., Hur, J., Lee, W. J., Won, J. P., Lee, H. G., Lim, D.-S., Kim, E., & Seo, H. G. (2021). Catalase Mediates the Inhibitory Actions of PPARδ against Angiotensin II-Triggered Hypertrophy in H9c2 Cardiomyocytes. Antioxidants, 10(8), 1223. https://doi.org/10.3390/antiox10081223