
The Use of Antioxidants as Potential Co-Adjuvants to Treat Chronic Chagas Disease


Abstract
1. Introduction
2. Mitochondrial Biogenesis under Cellular Stress Response
3. NRF2-KEAP1 Signaling under Oxidative Stress
4. Antioxidant Drugs
4.1. VitC/VitE
4.2. Melatonin
4.3. Curcumin
4.4. Resveratrol
4.5. Astaxanthin
4.6. Phenyl-alfa-tert-butyl Nitrone
4.7. Tempol
4.8. Desferrioxamine
4.9. Honokiol
4.10. Phytochemical Compounds
4.11. Apocynin
5. Therapeutic Uses of Antioxidants and Anti-Inflammatories for Chagas Disease Myocarditis
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Echeverría, L.E.; Marcus, R.; Novick, G.; Sosa-Estani, S.; Ralston, K.; Zaidel, E.J.; Forsyth, C.; RIbeiro, A.L.P.; Mendoza, I.; Falconi, M.L.; et al. WHF IASC Roadmap on Chagas Disease. Glob. Heart. 2020, 15, 26. [Google Scholar] [CrossRef]
- Chao, C.; Leone, J.L.; Vigliano, C.A. Chagas disease: Historic perspective. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165689. [Google Scholar] [CrossRef]
- Chadalawada, S.; Sillau, S.; Archuleta, S.; Mundo, W.; Bandali, M.; Parra-Henao, G.; Rodriguez-Morales, A.J.; Villamil-Gomez, W.E.; Suárez, J.A.; Shapiro, L.; et al. Risk of Chronic Cardiomyopathy Among Patients with the Acute Phase or Indeterminate Form of Chagas Disease: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2015072. [Google Scholar] [CrossRef]
- Rojas, L.Z.; Glisic, M.; Pletsch-Borba, L.; Echeverría, L.E.; Bramer, W.M.; Bano, A.; Stringa, N.; Zaciragic, A.; Kraja, B.; Asllanaj, E.; et al. Electrocardiographic abnormalities in Chagas disease in the general population: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2018, 12, e0006567. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, I.A. Epidemiology and dynamics of the vectorial transmission of Chagas disease. Mem. Inst. Oswaldo Cruz 1999, 94, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Chuit, R.; Meiss, R.; Salvatella, R. Epidemiology of Chagas Disease. In Chagas Disease: A Clinical Approach, 1st ed.; Altcheh, J.M., Freilij, H., Eds.; Springer: Cham, Switzerland, 2019; pp. 91–109. [Google Scholar]
- Cucunubá, Z.M.; Nouvellet, P.; Peterson, J.K.; Bartsch, S.M.; Lee, B.Y.; Dobson, A.P.; Basáñez, M.G. Complementary Paths to Chagas Disease Elimination: The Impact of Combining Vector Control with Etiological Treatment. Clin. Infect. Dis. 2018, 66, S293–S300. [Google Scholar] [CrossRef] [PubMed]
- Ortu, G.; Williams, O. Neglected tropical diseases: Exploring long term practical approaches to achieve sustainable disease elimination and beyond. Infect. Dis. Poverty 2017, 6, 147. [Google Scholar] [CrossRef]
- Zingales, B.; Andrade, S.G.; Briones, M.R.; Campbell, D.A.; Chiari, E.; Fernandes, O.; Guhl, F.; Lages-Silva, E.; Macedo, A.M.; Machado, C.R.; et al. Second Satellite Meeting. A new consensus for Trypanosoma cruzi intraspecific nomenclature: Second revision meeting recommends TcI to TcVI. Mem. Inst. Oswaldo Cruz 2009, 104, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Zingales, B.; Miles, M.A.; Campbell, D.A.; Tibayrenc, M.; Macedo, A.M.; Teixeira, M.M.; Schijman, A.G.; Llewellyn, M.S.; Lages-Silva, E.; Machado, C.R.; et al. The revised Trypanosoma cruzi subspecific nomenclature: Rationale, epidemiological relevance and research applications. Infect. Genet. Evol. 2012, 12, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Messenger, L.A.; Miles, M.A.; Bern, C. Between a bug and a hard place: Trypanosoma cruzi genetic diversity and the clinical outcomes of Chagas disease. Expert Rev. Anti Infect. Ther. 2015, 13, 995–1029. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Valdéz, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R.L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. Elife 2018, 7, e34039. [Google Scholar] [CrossRef]
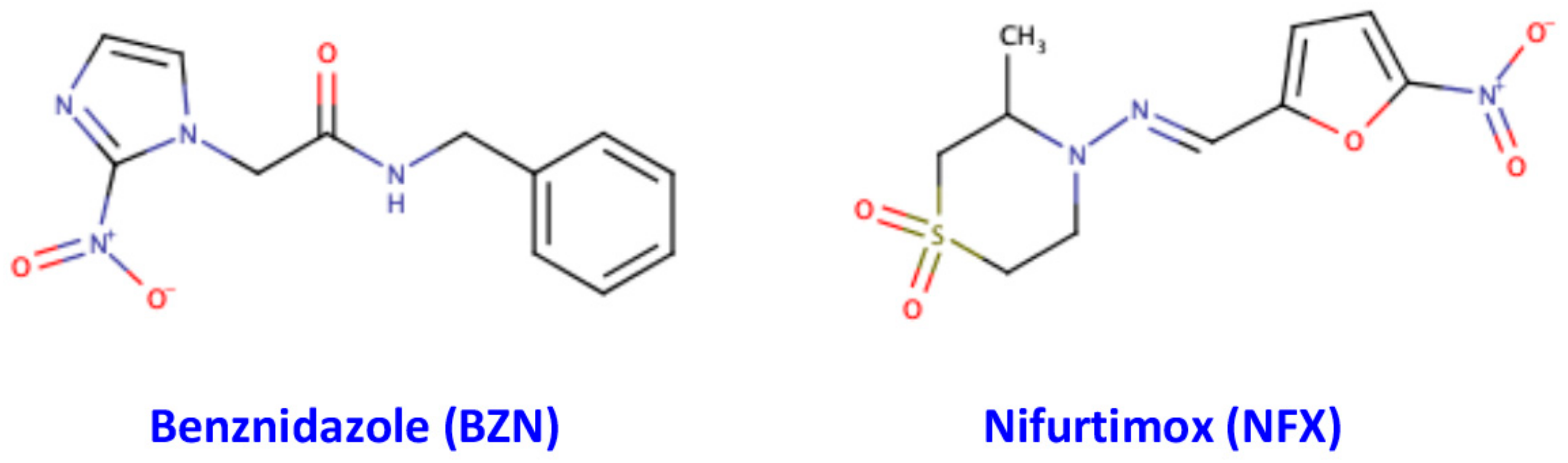
- Ribeiro, V.; Dias, N.; Paiva, T.; Hagström-Bex, L.; Nitz, N.; Pratesi, R.; Hecht, M. Current trends in the pharmacological management of Chagas disease. Int. J. Parasitol. Drugs Drug Resist. 2020, 12, 7–17. [Google Scholar] [CrossRef]
- Sales, P.A., Jr.; Molina, I.; Fonseca Murta, S.M.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Carneiro, C.M. Experimental and Clinical Treatment of Chagas Disease: A Review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Bot, C.; Wilkinson, S.R. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 2011, 286, 13088–13095. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.M.; Garcia Bournissen, F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef]
- Rodriques Coura, J.; de Castro, S.L. A critical review on Chagas disease chemotherapy. Mem. Inst. Oswaldo Cruz 2002, 97, 3–24. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef]
- Zingales, B.; Araujo, R.G.; Moreno, M.; Franco, J.; Aguiar, P.H.; Nunes, S.L.; Silva, M.N.; Ienne, S.; Machado, C.R.; Brandão, A. A novel ABCG-like transporter of Trypanosoma cruzi is involved in natural resistance to benznidazole. Mem. Inst. Oswaldo Cruz 2015, 110, 433–444. [Google Scholar] [CrossRef]
- Franco, J.; Ferreira, R.C.; Ienne, S.; Zingales, B. ABCG-like transporter of Trypanosoma cruzi involved in benznidazole resistance: Gene polymorphisms disclose inter-strain intragenic recombination in hybrid isolates. Infect. Genet. Evol. 2015, 31, 198–208. [Google Scholar] [CrossRef]
- Garcia, S.; Ramos, C.O.; Senra, J.F.; Vilas-Boas, F.; Rodrigues, M.M.; Campos-de-Carvalho, A.C.; Ribeiro-Dos-Santos, R.; Soares, M.B. Treatment with benznidazole during the chronic phase of experimental Chagas’ disease decreases cardiac alterations. Antimicrob Agents Chemother. 2005, 49, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, E.; Brum-Soares, L.; Reis, R.; Cubides, J.C. Chagas disease: Review of needs, neglect, and obstacles to treatment access in Latin America. Rev. Soc. Bras. Med. Trop. 2017, 50, 296–300. [Google Scholar] [CrossRef]
- Vermelho, A.B.; Rodrigues, G.C.; Supuran, C.T. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin. Drug Discov. 2020, 15, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Marin-Neto, J.A.; Rassi, A., Jr.; Morillo, C.A.; Avezum, A.; Connolly, S.J.; Sosa-Estani, S.; Rosas, F.; Yusuf, S.; BENEFIT Investigators. Rationale and design of a randomized placebo-controlled trial assessing the effects of etiologic treatment in Chagas’ cardiomyopathy: The BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT). Am. Heart J. 2008, 156, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Marin, J.A.N.; Rassi, A. Chronic Chagas cardiomyopathy: A review of the main pathogenic mechanisms and the efficacy of aetiological treatment following the BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) trial. Mem. Inst. Oswaldo Cruz. 2017, 112, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Suzuki, J. Oxidative stress and myocarditis. Curr Pharm Des. 2016, 22, 450–471. [Google Scholar] [CrossRef]
- Wan, X.; Gupta, S.; Zago, M.P.; Davidson, M.M.; Dousset, P.; Amoroso, A.; Garg, N.J. Defects of mtDNA replication impaired mitochondrial biogenesis during Trypanosoma cruzi infection in human cardiomyocytes and chagasic patients: The role of Nrf1/2 and antioxidant response. J. Am. Heart Assoc. 2012, 1, e003855. [Google Scholar] [CrossRef]
- Gupta, S.; Bhatia, V.; Wen, J.J.; Wu, Y.; Huang, M.H.; Garg, N.J. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic. Biol. Med. 2009, 47, 1414–1421. [Google Scholar] [CrossRef]
- Clayton, D.A. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000, 15, 11–17. [Google Scholar] [CrossRef]
- Falkenberg, M. Mitochondrial DNA replication in mammalian cells: Overview of the pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar] [CrossRef]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef]
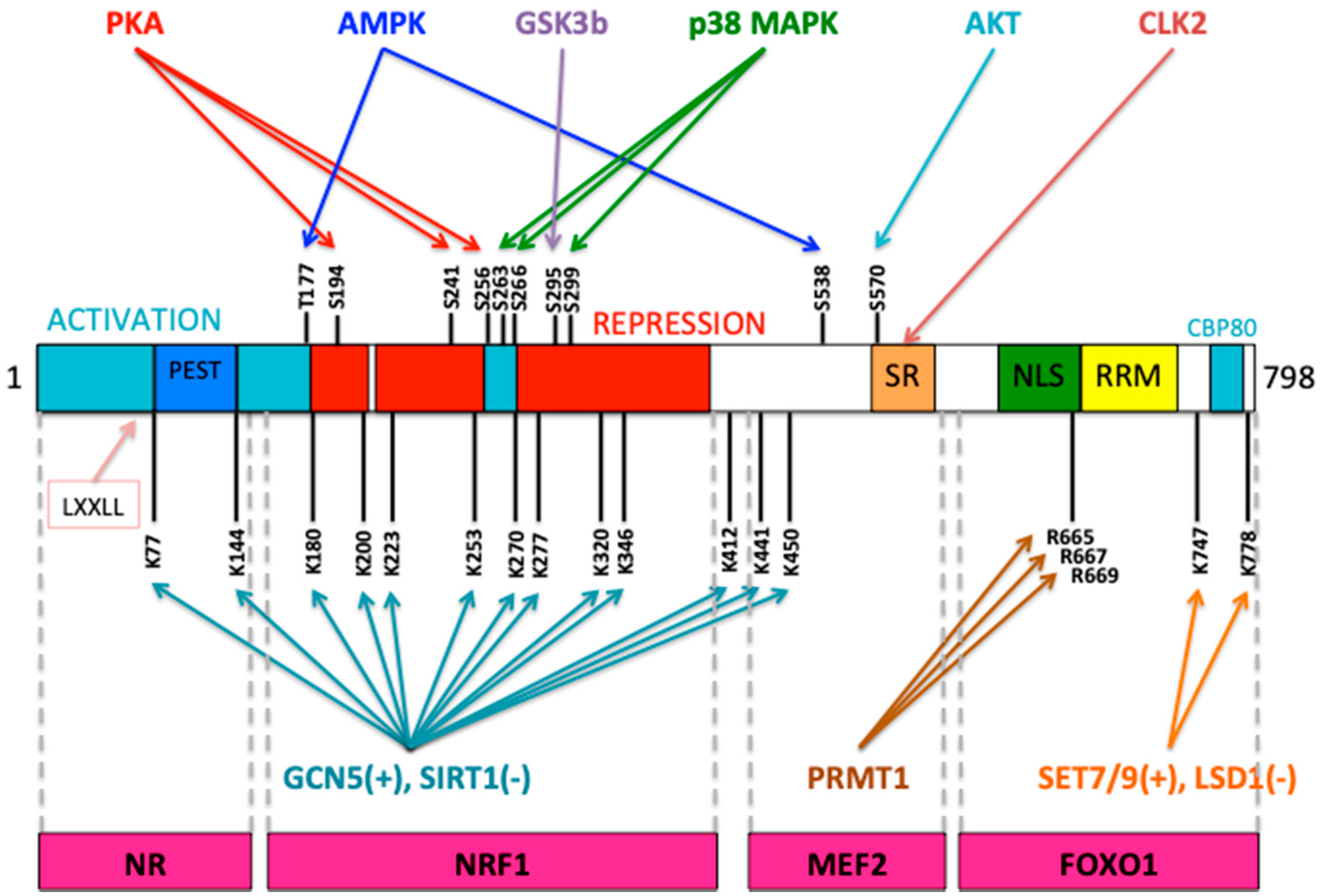
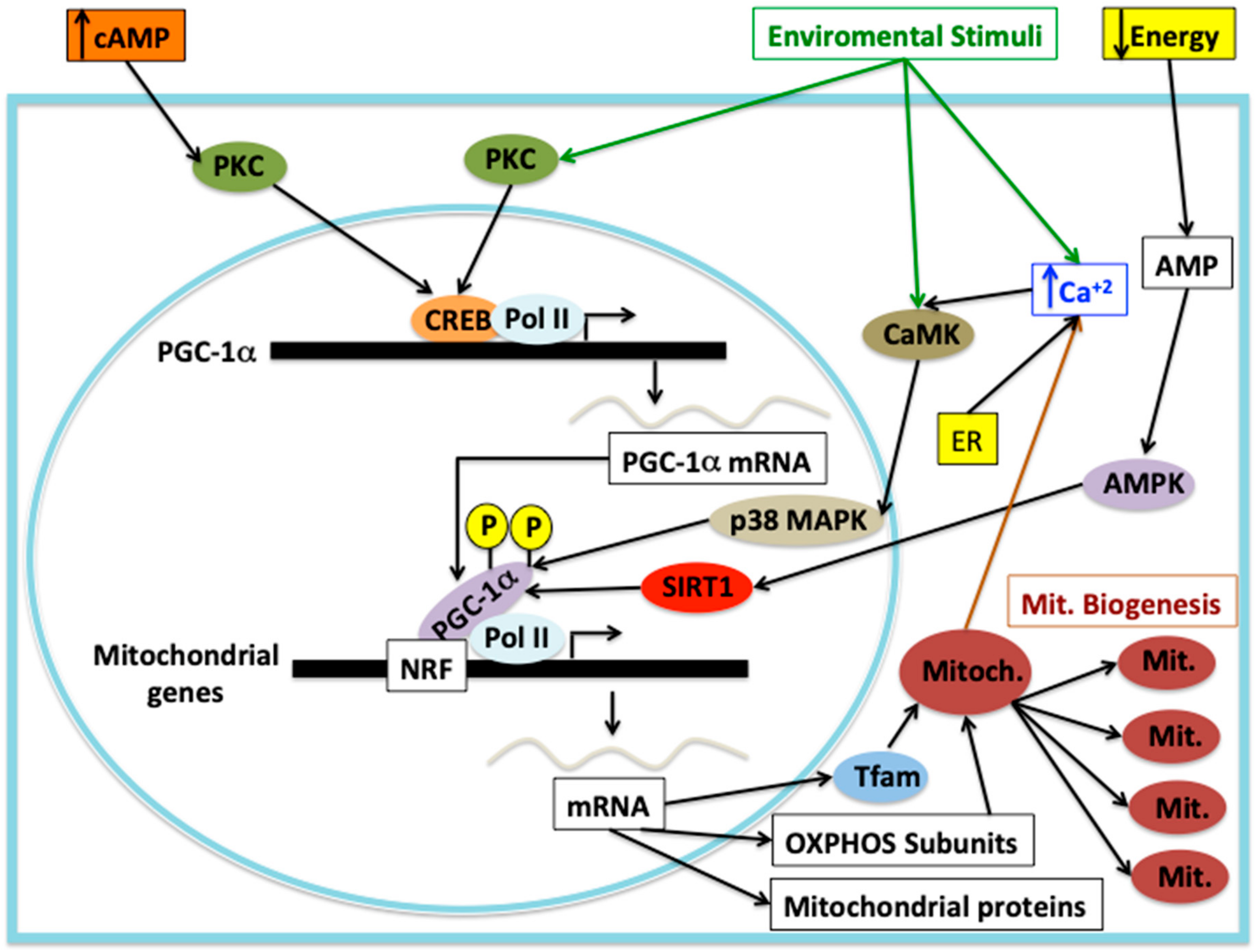
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Wenz, T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion 2013, 13, 134–142. [Google Scholar] [CrossRef]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.N.; Clark, J.P.; Anderson, R.M. Mitochondrial regulator PGC-1a-Modulating the modulator. Curr. Opin. Endocr. Metab. Res. 2019, 5, 37–44. [Google Scholar] [CrossRef]
- Dhar, S.S.; Ongwijitwat, S.; Wong-Riley, M.T.T. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J. Biol. Chem. 2008, 283, 3120–3129. [Google Scholar] [CrossRef]
- Ongwijitwat, S.; Wong-Riley, M.T. Is nuclear respiratory factor 2 a master transcriptional coordinator for all ten nuclear-encoded cytochrome c oxidase subunits in neurons? Gene 2005, 360, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Ongwijitwat, S.; Liang, H.L.; Graboyes, E.M.; Wong-Riley, M.T. Nuclear respiratory factor 2 senses changing cellular energy demands and its silencing down-regulates cytochrome oxidase and other target gene mRNAs. Gene 2006, 374, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell. 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Charos, A.E.; Reed, B.D.; Raha, D.; Szekely, A.M.; Weissman, S.M.; Snyder, M. A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells. Genome Res. 2012, 22, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Agarwal, S.; Ganesh, S. Perinuclear mitochondrial clustering, increased ROS levels, and HIF1 are required for the activation of HSF1 by heat stress. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef]
- Herzig, R.P.; Scacco, S.; Scarpulla, R.C. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J. Biol. Chem. 2000, 275, 13134–13141. [Google Scholar] [CrossRef]
- Vercauteren, K.; Pasko, R.A.; Gleyzer, N.; Marino, V.M.; Scarpulla, R.C. PGC-1-related coactivator: Immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol. Cell Biol. 2006, 26, 7409–7419. [Google Scholar] [CrossRef]
- Lee, J.; Kim, C.H.; Simon, D.K.; Aminova, L.R.; Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Lonze, B.E.; Kim, K.S.; Ginty, D.D.; et al. Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J. Biol. Chem. 2005, 280, 40398–40401. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Impey, S.; Ratan, R.R.; Ferrante, R.J. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 13915–13920. [Google Scholar] [CrossRef]
- Popov, D.V.; Lysenko, E.A.; Kuzmin, I.V.; Vinogradova, V.; Grigoriev, A.I. Regulation of PGC-1α Isoform Expression in Skeletal Muscles. Acta Nat. 2015, 7, 48–59. [Google Scholar] [CrossRef]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-Gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Leone, T.C.; Zechner, C.; Schaeffer, P.J.; Kelly, S.M.; Flanagan, D.P.; Medeiros, D.M.; Kovacs, A.; Kelly, D.P. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008, 22, 1948–1961. [Google Scholar] [CrossRef]
- Dorn, G.W., II; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.H.; Iyer, V.R. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-Box usage in vivo. PLoS ONE 2008, 3, e1798. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
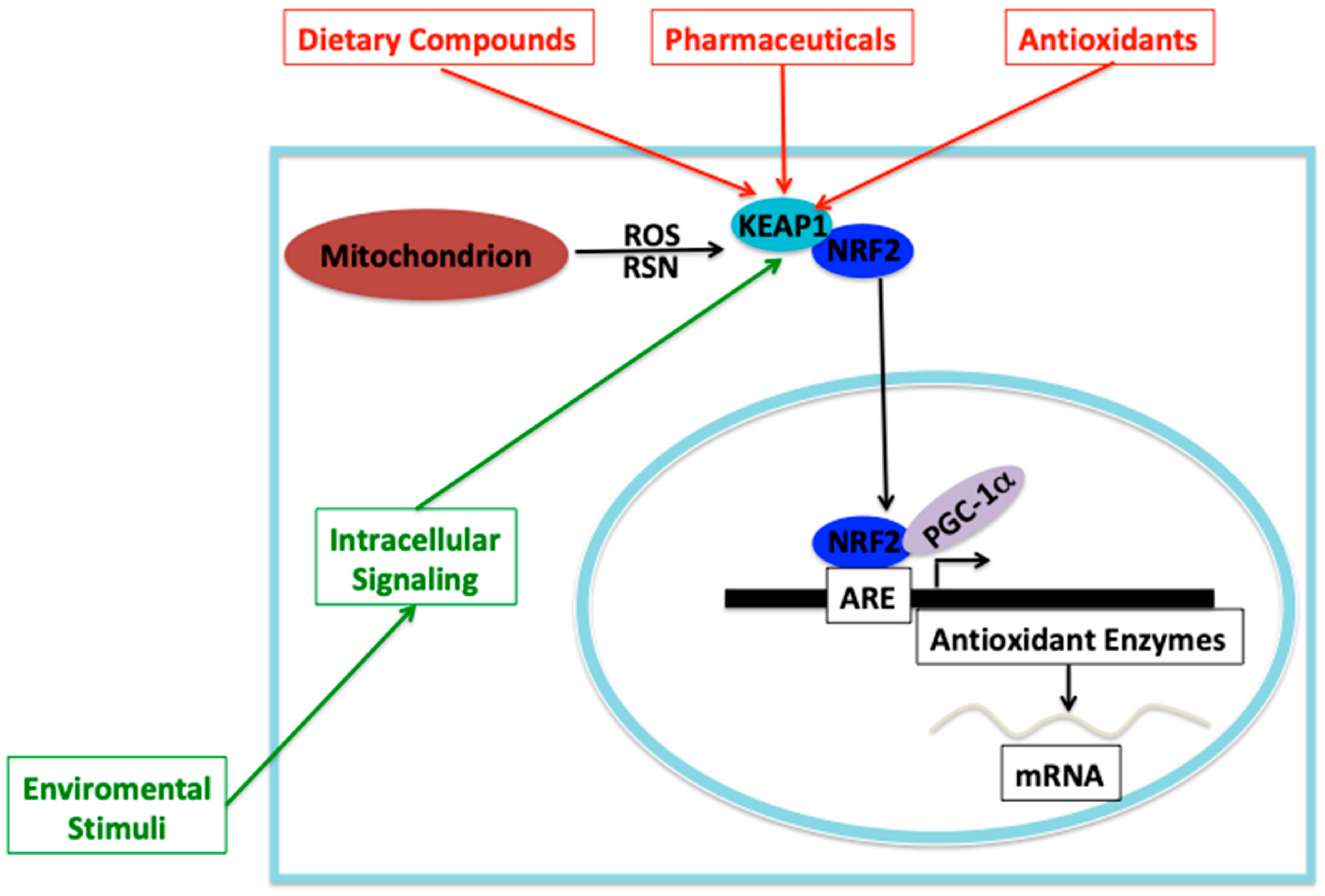
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidant. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef]
- Wilson, A.J.; Kerns, J.K.; Callahan, J.F.; Moody, C.J. Keap calm, and carry on covalently. J. Med. Chem. 2013, 56, 7463–7476. [Google Scholar] [CrossRef]
- Yang, L.; Palliyaguru, D.L.; Kensler, T.W. Frugal chemoprevention: Targeting Nrf2 with foods rich in sulforaphane. Semin. Oncol. 2016, 43, 146–153. [Google Scholar] [CrossRef]
- Hahn, M.E.; Timme-Laragy, A.R.; Karchner, S.I.; Stegeman, J.J. Nrf2 and Nrf2-related proteins in development and developmental toxicity: Insights from studies in zebrafish (Danio rerio). Free Radic. Biol. Med. 2015, 88, 275–289. [Google Scholar] [CrossRef]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Paiva, C.N.; Medei, E.; Bozza, M.T. ROS and Trypanosoma cruzi: Fuel to infection, poison to the heart. PLoS Pathog. 2018, 14, e1006928. [Google Scholar] [CrossRef]
- Goes, G.R.; Rocha, P.S.; Diniz, A.R.; Aguiar, P.H.; Machado, C.R.; Vieira, L.Q. Trypanosoma cruzi Needs a Signal Provided by Reactive Oxygen Species to Infect Macrophages. PLoS Negl. Trop. Dis. 2016, 10, e0004555. [Google Scholar] [CrossRef]
- Da Silva Augusto, L.; Moretti, N.S.; Ramos, T.C.; de Jesus, T.C.; Zhang, M.; Castilho, B.A.; Schenkman, S. A membrane-bound eIF2 alpha kinase located in endosomes is regulated by heme and controls differentiation and ROS levels in Trypanosoma cruzi. PLoS Pathog. 2015, 11, e1004618. [Google Scholar] [CrossRef]
- Garcia, E.S.; Genta, F.A.; de Azambuja, P.; Schaub, G.A. Interactions between intestinal compounds of triatomines and Trypanosoma cruzi. Trends Parasitol. 2010, 26, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hou, Y.; Liu, C.; Li, Y.; Guo, W.; Wu, J.L.; Xu, D.; You, X.; Pan, Y.; Chen, Y. Identification of an adaptor protein that facilitates Nrf2-Keap1 complex formation and modulates antioxidant response. Free Radic Biol. Med. 2016, 97, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Porter, C.; Garg, N.J. Inhibition of NFE2L2-Antioxidant Response Element Pathway by Mitochondrial Reactive Oxygen Species Contributes to Development of Cardiomyopathy and Left Ventricular Dysfunction in Chagas Disease. Antioxid. Redox. Signal 2017, 27, 550–566. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Gupta, S.; Davidson, M.; Garg, N.J. Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes. J. Biol. Chem. 2010, 285, 11596–11606. [Google Scholar] [CrossRef]
- Vyatkina, G.; Bhatia, V.; Gerstner, A.; Papaconstantinou, J.; Garg, N. Impaired mitochondrial respiratory chain and bioenergetics during chagasic cardiomyopathy development. Biochim. Biophys. Acta 2004, 1689, 162–173. [Google Scholar] [CrossRef]
- Dhiman, M.; Coronado, Y.A.; Vallejo, C.K.; Petersen, J.R.; Ejilemele, A.; Nuñez, S.; Zago, M.P.; Spratt, H.; Garg, N.J. nnate immune responses and antioxidant/oxidant imbalance are major determinants of human Chagas disease. PLoS Negl. Trop. Dis. 2013, 7, e2364. [Google Scholar] [CrossRef]
- De Oliveira, T.B.; Pedrosa, R.C.; Filho, D.W. Oxidative stress in chronic cardiopathy associated with Chagas disease. Int. J. Cardiol. 2007, 116, 357–363. [Google Scholar] [CrossRef]
- Wen, J.J.; Garg, N.J. Manganese superoxide dismutase deficiency exacerbates the mitochondrial ROS production and oxidative damage in Chagas disease. PLoS Negl. Trop. Dis. 2018, 12, e0006687. [Google Scholar] [CrossRef]
- Andrade, L.O.; Galvão, L.M.; Meirelles, M.D.E.N.; Chiari, E.; Pena, S.D.; Macedo, A.M. Differential tissue tropism of Trypanosoma cruzi strains: An in vitro study. Mem. Inst. Oswaldo. Cruz. 2010, 105, 834–837. [Google Scholar] [CrossRef]
- Rios, L.E.; Vázquez-Chagoyán, J.C.; Pacheco, A.O.; Zago, M.P.; Garg, N.J. Immunity and vaccine development efforts against Trypanosoma cruzi. Acta Trop. 2019, 200, 105168. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M.; Luthringer, D.J.; Kim, S.A.; Garg, N.J.; Engman, D.M. Pathology and Pathogenesis of Chagas Heart Disease. Annu. Rev. Pathol. 2019, 14, 421–447. [Google Scholar] [CrossRef]
- Guiñazú, N.; Carrera-Silva, E.A.; Becerra, M.C.; Pellegrini, A.; Albesa, I.; Gea, S. Induction of NADPH oxidase activity and reactive oxygen species production by a single Trypanosoma cruzi antigen. Int. J. Parasitol. 2010, 40, 1531–1538. [Google Scholar] [CrossRef]
- Koo, S.J.; Szczesny, B.; Wan, X.; Putluri, N.; Garg, N.J. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front. Immunol. 2018, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.N.; Peluffo, G.; Piacenza, L.; Radi, R. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: Consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J. Biol. Chem. 2011, 286, 6627–6640. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Garg, N.J. Mitochondrial generation of reactive oxygen species is enhanced at the Q(o) site of the complex III in the myocardium of Trypanosoma cruzi-infected mice: Beneficial effects of an antioxidant. J Bioenerg Biomembr. 2008, 40, 587–598. [Google Scholar] [CrossRef]
- Wen, J.J.; Dhiman, M.; Whorton, E.B.; Garg, N.J. Tissue-Specific oxidative imbalance and mitochondrial dysfunction during Trypanosoma cruzi infection in mice. Microbes Infect. 2008, 10, 1201–1209. [Google Scholar] [CrossRef]
- Wan, X.; Wen, J.J.; Koo, S.J.; Liang, L.Y.; Garg, N.J. SIRT1-PGC1α-NFκB Pathway of Oxidative and Inflammatory Stress during Trypanosoma cruzi Infection: Benefits of SIRT1-Targeted Therapy in Improving Heart Function in Chagas Disease. PLoS Pathog. 2016, 12, e1005954. [Google Scholar] [CrossRef]
- Lopez, M.; Tanowitz, H.B.; Garg, N.J. Pathogenesis of Chronic Chagas Disease: Macrophages, Mitochondria, and Oxidative Stress. Curr. Clin. Microbiol. Rep. 2018, 5, 45–54. [Google Scholar] [CrossRef]
- Pérez-Fuentes, R.; Guégan, J.F.; Barnabé, C.; López-Colombo, A.; Salgado-Rosas, H.; Torres-Rasgado, E.; Briones, B.; Romero-Díaz, M.; Ramos-Jiménez, J.; Sánchez-Guillén, M.C. Severity of chronic Chagas disease is associated with cytokine/antioxidant imbalance in chronically infected individuals. Int. J. Parasitol. 2003, 33, 293–299. [Google Scholar] [CrossRef]
- Machado, F.S.; Tanowitz, H.B.; Ribeiro, A.L. Pathogenesis of chagas cardiomyopathy: Role of inflammation and oxidative stress. J. Am. Heart Assoc. 2013, 2, e000539. [Google Scholar] [CrossRef]
- Gupta, S.; Dhiman, M.; Wen, J.J.; Garg, N.J. ROS signalling of inflammatory cytokines during Trypanosoma cruzi infection. Adv. Parasitol. 2011, 76, 153–170. [Google Scholar] [CrossRef]
- Puente, V.; Demaria, A.; Frank, F.M.; Batlle, A.; Lombardo, M.E. Anti-Parasitic effect of vitamin C alone and in combination with benznidazole against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2018, 12, e0006764. [Google Scholar] [CrossRef] [PubMed]
- Providello, M.V.; Carneiro, Z.A.; Portapilla, G.B.; do Vale, G.T.; Camargo, R.S.; Tirapelli, C.R.; de Albuquerque, S. Benefits of Ascorbic Acid in Association with Low-Dose Benznidazole in Treatment of Chagas Disease. Antimicrob. Agents Chemother. 2018, 62, e00514-18. [Google Scholar] [CrossRef] [PubMed]
- Gusmão, A.S.; Castanho, R.E.; Andrade, R.F.; Farsetti, C.M.; Mathias, A.B.; Therezo, A.L.; Martins, L.P. Vitamin C effects in mice experimentally infected with Trypanosoma cruzi QM2 strain. Rev. Soc. Bras. Med. Trop. 2012, 45, 51–54. [Google Scholar] [CrossRef]
- Marim, R.G.; Gusmão, A.S.; Castanho, R.E.; Deminice, R.; Therezo, A.L.; Jordão Júnior, A.A.; Martins, L.P. Effects of vitamin C supplementation on acute phase Chagas disease in experimentally infected mice with Trypanosoma cruzi QM1 strain. Rev. Inst. Med. Trop. Sao Paulo 2012, 54, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, J.R.P.T.; Castanho, R.E.P.; Rocha, H., Jr.; Pagliari, C.; Duarte, M.I.S.; Therezo, A.L.S.; Chagas, E.F.B.; Martins, L.P.A. Paradoxical effects of vitamin C in Chagas disease. Parasitol. Int. 2018, 67, 547–555. [Google Scholar] [CrossRef]
- Tieghi, T.M.; Manca, C.C.; Garcia, L.C.T.; Castanho, R.E.P.; Therezo, A.L.S.; Frei, F.; Taipeiro, E.F.; Martins, L.P.A. Evaluation of antioxidant therapy in experimental Chagas disease. Rev. Soc. Bras. Med. Trop. 2017, 50, 184–193. [Google Scholar] [CrossRef]
- Novaes, R.D.; Santos, E.C.; Fialho, M.D.C.Q.; Gonçalves, W.G.; Sequetto, P.L.; Talvani, A.; Gonçalves, R.V. Nonsteroidal anti-inflammatory is more effective than anti-oxidant therapy in counteracting oxidative/nitrosative stress and heart disease in T. cruzi-infected mice. Parasitology 2017, 144, 904–916. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Collin, F. Melatonin: Action as antioxidant and potential applications in human disease and aging. Toxicology 2010, 278, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Santello, F.H.; Frare, E.O.; dos Santos, C.D.; Toldo, M.P.; Kawasse, L.M.; Zucoloto, S.; do Prado, J.C., Jr. Melatonin treatment reduces the severity of experimental Trypanosoma cruzi infection. J. Pineal Res. 2007, 42, 359–363. [Google Scholar] [CrossRef]
- Brazão, V.; Santello, F.H.; Colato, R.P.; Mazotti, T.T.; Tazinafo, L.F.; Toldo, M.P.A.; do Vale, G.T.; Tirapelli, C.R.; do Prado, J.C., Jr. Melatonin: Antioxidant and modulatory properties in age-related changes during Trypanosoma cruzi infection. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Brazão, V.; Santello, F.H.; Colato, R.P.; do Prado, J.C., Jr. T. cruzi infection among aged rats: Melatonin as a promising therapeutic molecule. Exp. Gerontol. 2020, 135, 110922. [Google Scholar] [CrossRef]
- Zheng, D.; Huang, C.; Huang, H.; Zhao, Y.; Khan, M.R.U.; Zhao, H.; Huang, L. Antibacterial Mechanism of Curcumin: A Review. Chem. Biodivers. 2020, 17, e2000171. [Google Scholar] [CrossRef]
- Selvam, C.; Prabu, S.L.; Jordan, B.C.; Purushothaman, Y.; Umamaheswari, A.; Hosseini Zare, M.S.; Thilagavathi, R. Molecular mechanisms of curcumin and its analogs in colon cancer prevention and treatment. Life Sci. 2019, 239, 117032. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sureda, A.; Devkota, H.P.; Pittalà, V.; Barreca, D.; Silva, A.S.; Tewari, D.; Xu, S.; Nabavi, S.M. Curcumin, the golden spice in treating cardiovascular diseases. Biotechnol. Adv. 2020, 38, 107343. [Google Scholar] [CrossRef]
- Nagajyothi, F.; Zhao, D.; Weiss, L.M.; Tanowitz, H.B. Curcumin treatment provides protection against Trypanosoma cruzi infection. Parasitol. Res. 2012, 110, 2491–2499. [Google Scholar] [CrossRef]
- Novaes, R.D.; Sartini, M.V.; Rodrigues, J.P.; Gonçalves, R.V.; Santos, E.C.; Souza, R.L.; Caldas, I.S. Curcumin Enhances the Anti-Trypanosoma cruzi Activity of Benznidazole-Based Chemotherapy in Acute Experimental Chagas Disease. Antimicrob Agents Chemother. 2016, 60, 3355–3364. [Google Scholar] [CrossRef]
- Hernández, M.; Wicz, S.; Santamaría, M.H.; Corral, R.S. Curcumin exerts anti-inflammatory and vasoprotective effects through amelioration of NFAT-dependent endothelin-1 production in mice with acute Chagas cardiomyopathy. Mem. Inst. Oswaldo Cruz 2018, 113, e180171. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.; Wicz, S.; Pérez Caballero, E.; Santamaría, M.H.; Corral, R.S. Dual chemotherapy with benznidazole at suboptimal dose plus curcumin nanoparticles mitigates Trypanosoma cruzi-elicited chronic cardiomyopathy. Parasitol. Int. 2021, 81, 102248. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont-Rousselot, D. Resveratrol and Cardiovascular Diseases. Nutrients 2016, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef]
- Vilar-Pereira, G.; Carneiro, V.C.; Mata-Santos, H.; Vicentino, A.R.; Ramos, I.P.; Giarola, N.L.; Feijó, D.F.; Meyer-Fernandes, J.R.; Paula-Neto, H.A.; Medei, E.; et al. Resveratrol Reverses Functional Chagas Heart Disease in Mice. PLoS Pathog. 2016, 12, e1005947. [Google Scholar] [CrossRef]
- Fracasso, M.; Dutra da Silva, A.; Bottari, N.B.; Monteiro, S.G.; Garzon, L.R.; Farias de Souza, L.A.; Schetinger, M.R.C.; da Silva, A.S. Resveratrol impacts in oxidative stress in liver during Trypanosoma cruzi infection. Microb. Pathog. 2021, 153, 104800. [Google Scholar] [CrossRef]
- Kohandel, Z.; Farkhondeh, T.; Aschner, M.; Samarghandian, S. Nrf2 a molecular therapeutic target for Astaxanthin. Biomed. Pharmacother. 2021, 137, 111374. [Google Scholar] [CrossRef]
- Horta, A.L.; Figueiredo, V.P.; Leite, A.L.J.; Costa, G.P.; Menezes, A.P.J.; Ramos, C.O.; Pedrosa, T.C.F.; Bezerra, F.S.; Vieira, P.M.A.; Talvani, A. The β-blocker carvedilol and the benznidazole modulate the cardiac immune response in the acute infection induced by Colombian strain of the Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 2018, 113, e180271. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Gupta, S.; Guan, Z.; Dhiman, M.; Condon, D.; Lui, C.; Garg, N.J. Phenyl-Alpha-Tert-Butyl-Nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic Rats. J. Am. Coll. Cardiol. 2010, 55, 2499–2508. [Google Scholar] [CrossRef]
- Wen, J.J.; Bhatia, V.; Popov, V.L.; Garg, N.J. Phenyl-Alpha-Tert-Butyl nitrone reverses mitochondrial decay in acute Chagas’ disease. Am. J. Pathol. 2006, 169, 1953–1964. [Google Scholar] [CrossRef][Green Version]
- Kotake, Y.; Sang, H.; Miyajima, T.; Wallis, G.L. Inhibition of NF-kappaB, iNOS mRNA, COX2 mRNA, and COX catalytic activity by phenyl-N-tert-butylnitrone (PBN). Biochim. Biophys. Acta 1998, 1448, 77–84. [Google Scholar] [CrossRef]
- Ho, E.; Chen, G.; Bray, T.M. Alpha-Phenyl-Tert-Butylnitrone (PBN) inhibits NFkappaB activation offering protection against chemically induced diabetes. Free Radic. Biol. Med. 2000, 28, 604–614. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-Targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.; Richmond, R.; Halliwell, B. Inhibition of the iron-catalysed formation of hydroxyl radicals from superoxide and of lipid peroxidation by desferrioxamine. Biochem. J. 1979, 184, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Basaran, U.N.; Ayvaz, S.; Aksu, B.; Karaca, T.; Cemek, M.; Karaboga, I.; Inan, M.; Aksu, F.; Pul, M. Desferrioxamine reduces oxidative stress in the lung contusion. Sci. World J. 2013, 2013, 376959. [Google Scholar] [CrossRef]
- Arantes, J.M.; Pedrosa, M.L.; Martins, H.R.; Veloso, V.M.; de Lana, M.; Bahia, M.T.; Tafuri, W.L.; Carneiro, C.M. Trypanosoma cruzi: Treatment with the iron chelator desferrioxamine reduces parasitemia and mortality in experimentally infected mice. Exp. Parasitol. 2007, 117, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Arantes, J.M.; Francisco, A.F.; de Abreu Vieira, P.M.; Silva, M.; Araújo, M.S.; de Carvalho, A.T.; Pedrosa, M.L.; Carneiro, C.M.; Tafuri, W.L.; Martins-Filho, O.A.; et al. Trypanosoma cruzi: Desferrioxamine decreases mortality and parasitemia in infected mice through a trypanostatic effect. Exp. Parasitol. 2011, 128, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.F.; de Abreu Vieira, P.M.; Arantes, J.M.; Pedrosa, M.L.; Martins, H.R.; Silva, M.; Veloso, V.M.; de Lana, M.; Bahia, M.T.; Tafuri, W.L.; et al. Trypanosoma cruzi: Effect of benznidazole therapy combined with the iron chelator desferrioxamine in infected mice. Exp. Parasitol. 2008, 120, 314–319. [Google Scholar] [CrossRef]
- Wen, J.J.; Vyatkina, G.; Garg, N. Oxidative damage during chagasic cardiomyopathy development: Role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radic. Biol. Med. 2004, 37, 1821–1833. [Google Scholar] [CrossRef]
- Caballero, E.P.; Mariz-Ponte, N.; Rigazio, C.S.; Santamaría, M.H.; Corral, R.S. Honokiol attenuates oxidative stress-dependent heart dysfunction in chronic Chagas disease by targeting AMPK/NFE2L2/SIRT3 signaling pathway. Free Radic. Biol. Med. 2020, 156, 113–124. [Google Scholar] [CrossRef]
- Montenote, M.C.; Wajsman, V.Z.; Konno, Y.T.; Ferreira, P.C.; Silva, R.M.G.; Therezo, A.L.S.; Silva, L.P.; Martins, L.P.A. Antioxidant effect of Morus nigra on Chagas disease progression. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e73. [Google Scholar] [CrossRef]
- Castañeda, J.S.; Suta-Velásquez, M.; Mateus, J.; Pardo-Rodriguez, D.; Puerta, C.J.; Cuéllar, A.; Robles, J.; Cuervo, C. Preliminary chemical characterization of ethanolic extracts from Colombian plants with promising anti-Trypanosoma cruzi activity. Exp. Parasitol. 2021, 223, 108079. [Google Scholar] [CrossRef]
- Stefanska, J.; Pawliczak, R. Apocynin: Molecular aptitudes. Mediat. Inflamm. 2008, 2008, 106507. [Google Scholar] [CrossRef] [PubMed]
- Heumüller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schröder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, M.; Garg, N.J. NADPH oxidase inhibition ameliorates Trypanosoma cruzi-induced myocarditis during Chagas disease. J. Pathol. 2011, 225, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Villamil, J.P.; Bautista-Niño, P.K.; Serrano, N.C.; Rincon, M.Y.; Garg, N.J. Potential Role of Antioxidants as Adjunctive Therapy in Chagas Disease. Oxid Med. Cell Longev. 2020, 2020, 9081813. [Google Scholar] [CrossRef]
- Freitas, D.S.; Silva Godinho, A.S.; Mondêgo-Oliveira, R.; Cardoso, F.O.; Abreu-Silva, A.L.; Silva, L.A. Anti-inflammatory and antioxidant therapies for chagasic myocarditis: A systematic review. Parasitology 2020, 147, 603–610. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant | Oxidative Stress Marker | ||||||
|---|---|---|---|---|---|---|---|
| Mitochondrial Function | NO Production | Lipid Peroxidation | PCN | GTN | SOD | CAT | |
| ASTX Apocynin | X | ||||||
| * | * | ||||||
| Carvedilol | NS | X | Xa, *(−h) | X | |||
| Curcumin DFX Flavonoids | * | *(+) | *(−) | *(−) | |||
| X | X | *(+) | |||||
| X | |||||||
| HKL | * | *(+) | *(+) | ||||
| Melatonin | X | * | |||||
| PBN | * | ||||||
| Resveratrol | * | *(−) | |||||
| Tampol | * | * | |||||
| Vitamin C or E | X | X | X | ||||
| Vitamin C/E | NS | * | |||||
| Study | Design | Intervention | Results |
|---|---|---|---|
| Barbosa et al. 2016 | Prospective, open cohort | BZN (2 months) and vitamins C and E supplementation (6 months) |
|
| Budni et al. 2013 | Prospective, open cohort | Carvedilol (6 months) and 6 months wash out vitamins C and E (6 months) |
|
| Ribeiro et al. 2010 | Prospective, cohort | BZN (2 months) and vitamins C and E supplementation (6 months) |
|
| Maçao et al. 2007 | Prospective, cohort | Vitamins C and E supplementation (6 months) |
|
| Model | Age | T. cruzi Strain and Dose | Treatment | Antioxidant/ Oxidant Stress Marker | Tissue | Reference |
|---|---|---|---|---|---|---|
| C57BL/6 mice | 6–8 weeks | Sylvio × 10 (1 × 104) | 50 mg/kg PBN (i.p.) on alternate days for 3 weeks | Respiratory complex activities, MDA, GSH, ATP, H2O2 | Heart, heart mitochondria | [121] |
| SWRJ/W male mice | 4 weeks | Y (1 × 102) | 5 mg/50 μL/day desferrioxamine (i.p.) 14 days prior to infection and for 21 days i.p. | GSH, TBARS, PCN, nitrate/nitrite | Serum, liver | [129] |
| Sprague Dawley rats | 4–5 weeks | Sylvio × 10 (1 × 104) | 1.3 mM PBN and/or 0.7 mM benznidazole for three weeks in drinking water | ROS, TBARS | Heart, heart mitochondria | [120] |
| Sprague Dawley rats | 4–5 weeks | Sylvio × 10 (1 × 104) | 1.3 mM PBN and/or 0.7 mM benznidazole for three weeks in drinking water | PCN | Heart, heart mitochondria | [138] |
| CD1 mice | 6–8 weeks | Brazil (5 × 104) | 100 mg/kg/day curcumin for 35 days orally | mRNA levels of proteins/enzymes | Heart | [110] |
| SWR/J male mice | 3 weeks | QM1 (5 × 104) | 10 μL vitamin C (D50 mg or D500 mg) per day for 60 days or 180 days orally | TBARS, total peroxide, GSH | Plasma, heart, colon, skeletal muscle | [99] |
| Wistar male rats | NR | Y (1 × 105) | 5 mg/kg melatonin/day for 60 days orally | Nitrite production in macrophages, TBARS in plasma | Plasma, spleen | [138] |
| SWR/J female mice | 8–12 weeks | Y (2 × 103) | Curcumin (C) +/− Benznidazole (B) for 20 days by gavage. C100 (+/−B50–B100), B50–B100 only (mg/kg/day) | MDA, PCN | [111] | |
| BALB/c male and female mice | 5–7 weeks | Colombian (2 × 102) | 15 mg/kg trans-resveratrol (i.p.) or 40 mg/kg resveratrol, 500 mg/kg metformin, 100 mg/kg/Tempol or 25 mg/kg benznidazole for 30 days orally | TBARS | [116] | |
| Swiss SWR/J male mice | 52 weeks | Y (2 × 103) | 500 mg/day vitamin C/800 UI/day vitamin E for 15 days orally | TBARS, catalase, PCN, GST and SOD activities, nitrite/nitrate, 8-OHdG | [102] | |
| Swiss SWR/J male mice | 3 weeks | QM2 (5 × 104) | 500 mg/day vitamin C/800 UI/day vitamin E (individually and in combination) for 60 or 120 days | FRAPS, GSH, TBARS | [101] | |
| Swiss SWR/J male mice | 3 weeks | QM2 (5 × 104) | 20% blackberry plant extract (25–75 μL/day) for 180 days orally | TBARS, FRAPS, GSH, sulfhydryl groups | [132] | |
| BALB/c female mice | 4–6 weeks | Ninoa (10) | 10 mg/kg/day astaxanthin +/− 10 mg/kg/day nifurtimox for 60 days orally | MDA | [138] | |
| Swiss SWR/J male mice | 6 weeks | Y (1 × 104) | 7.14 mg/kg/day vitamin C +/− 100 mg/kg benznidazole for 15 days by gavage | TBARS, ROS | [97] | |
| Swiss SWR/J male mice | 3.5 weeks | QM2 (5 × 104) | 500 mg/day vitamin C for 60 days in drinking water | FRAPS, GSH, GST, plasma sulfhydryl group, nitrate/nitrite | [100] | |
| C57BL/6 male mice | 8–10 weeks | Colombian (50) | 25 mg/kg/day carvedilol +/− 100 mg/kg/day benznidazole for 23 days by gavage | SOD and CAT activities, TBARS, protein carbonyls | [119] | |
| C57BL/6 male and female mice | 6–8 weeks | Brazil (1 × 104) | 0.2 mg/kg/day honokiol or 100 mg/kg/day benzdidazole for 57 and 85 days | CAT, SOD activities, ROS, MDA | [131] | |
| C3H/HeN male mice | 6–8 weeks | Sylvio ×10 (1 × 104) | 1.5 mM apocynin for 150 days in drinking water | ROS | [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, E.; Rojas, D.A.; Urbina, F.; Solari, A. The Use of Antioxidants as Potential Co-Adjuvants to Treat Chronic Chagas Disease. Antioxidants 2021, 10, 1022. https://doi.org/10.3390/antiox10071022
Maldonado E, Rojas DA, Urbina F, Solari A. The Use of Antioxidants as Potential Co-Adjuvants to Treat Chronic Chagas Disease. Antioxidants. 2021; 10(7):1022. https://doi.org/10.3390/antiox10071022
Chicago/Turabian StyleMaldonado, Edio, Diego A. Rojas, Fabiola Urbina, and Aldo Solari. 2021. "The Use of Antioxidants as Potential Co-Adjuvants to Treat Chronic Chagas Disease" Antioxidants 10, no. 7: 1022. https://doi.org/10.3390/antiox10071022
APA StyleMaldonado, E., Rojas, D. A., Urbina, F., & Solari, A. (2021). The Use of Antioxidants as Potential Co-Adjuvants to Treat Chronic Chagas Disease. Antioxidants, 10(7), 1022. https://doi.org/10.3390/antiox10071022