
Vitamin K2 Modulates Organelle Damage and Tauopathy Induced by Streptozotocin and Menadione in SH-SY5Y Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture
2.2. Experimental Design for In Vitro Studies
2.3. Mitochondrial Activity (MTT) Assay
2.4. Reactive Oxygen Species
2.5. Lactate Dehydrogenase (LDH) Assay
2.6. Organelle Dysfunction Studies
2.6.1. Measurement of Mitochondrial Membrane Potential by the JC1 Assay
2.6.2. Detection of Endoplasmic Reticulum Stress Marker (CHOP and p-IRE1α) by Immunofluorescence
2.7. Detection of GSK3α/β by Immunofluorescence
2.8. Detection of Neurodegenerative Marker Proteins
Detection of Human (Total) Tau and Aβ42 Levels by ELISA
2.9. Statistical Analysis
3. Results
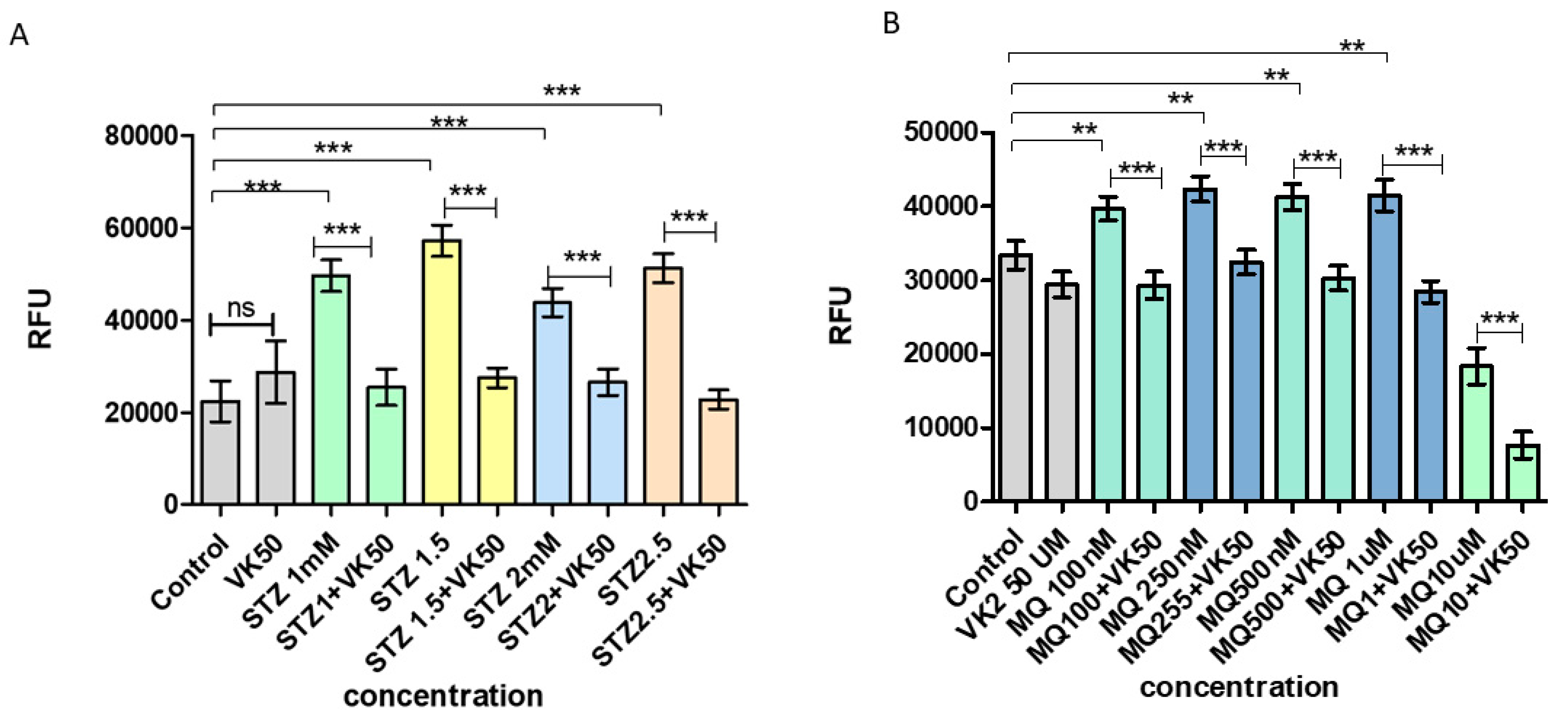
3.1. Vitamin K2 Protects the SH-SY5Y Cells from Streptozotocin- and Menadione-Induced Decrease in Cell Viability
3.2. Vitamin K2 Reduces Cell Cytotoxicity Induced by Streptozotocin and Menadione
3.3. Vitamin K2 Attenuated ROS Production Induced by Streptozotocin and Menadione
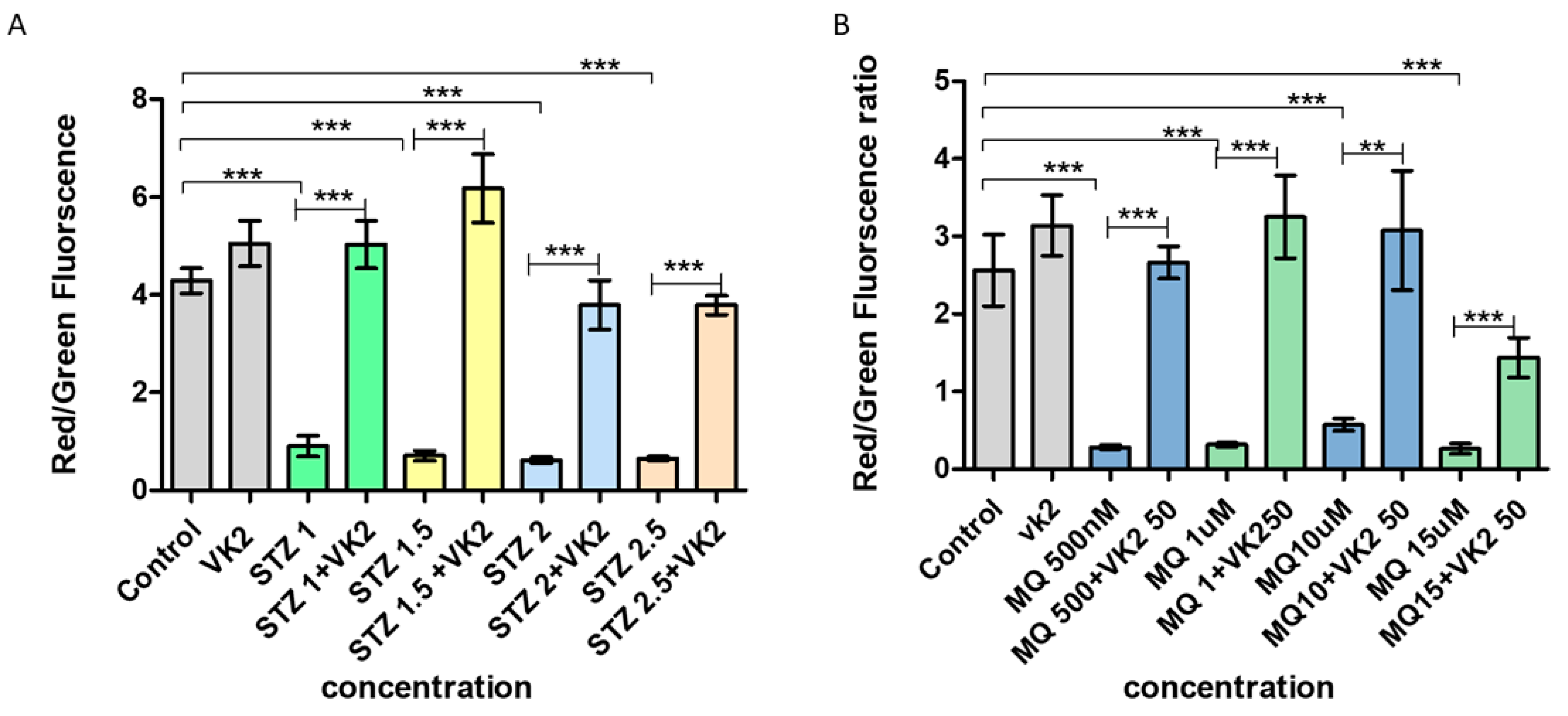
3.4. Vitamin K2 Inhibited Mitochondrial Depolarization Induced by Streptozotocin and Menadione
3.5. Vitamin K2 Reduced Endoplasmic Reticulum Stress-Induced by Streptozotocin and Menadione
3.6. Vitamin K2 Reduced Endoplasmic Reticulum Stress-Induced UPR Signaling Mediated by Streptozotocin and Menadione
3.7. Vitamin K2 Reduced the Expression of p GSK3α/β Induced by Streptozotocin and Menadione
3.8. Effect of Vitamin K2 on Intracellular Total Tau Protein Expression
3.9. Effect of Vitamin K2 on Secreted Aβ42
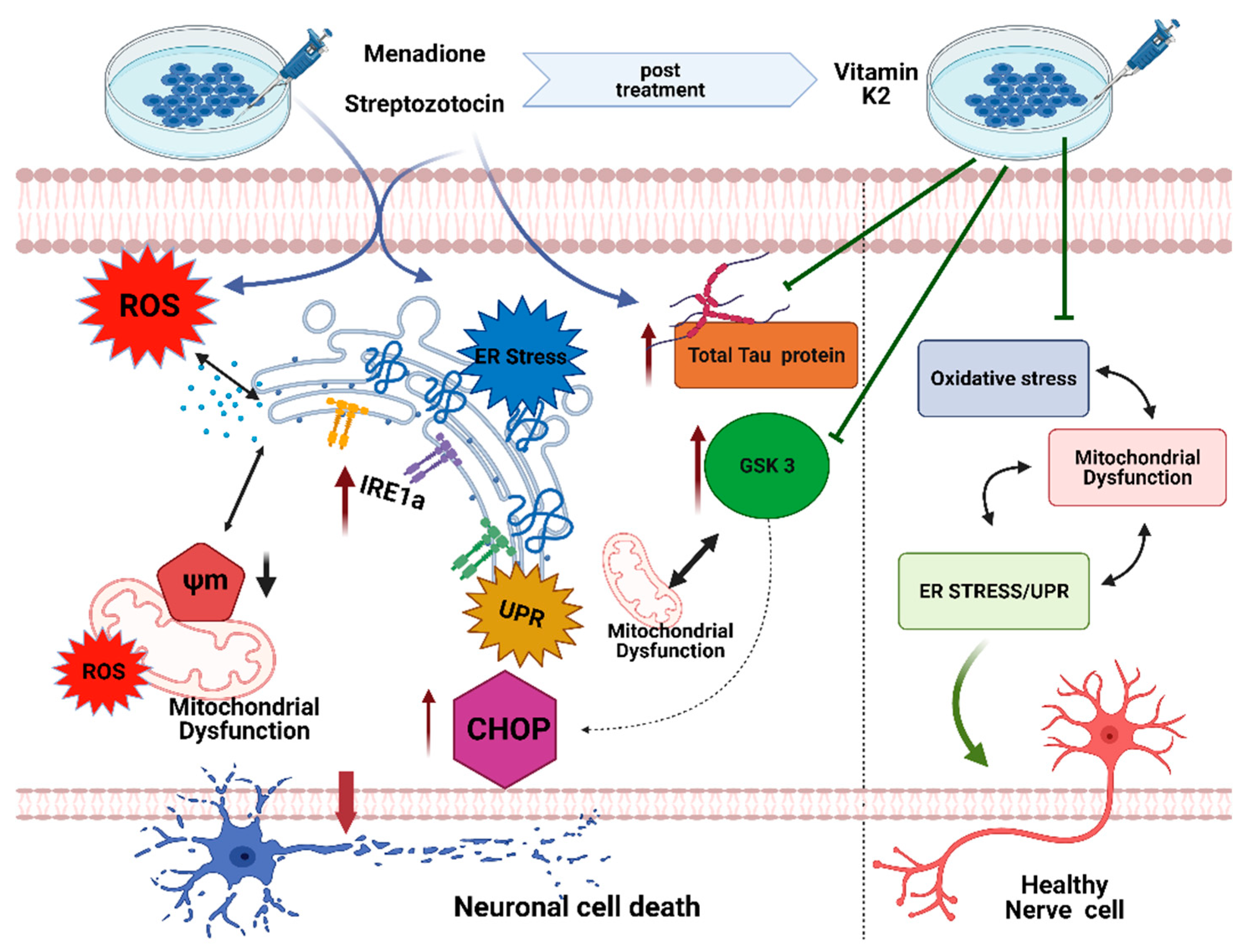
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corrada, M.; Hayden, K.M.; Paganini-Hill, A.; Bullain, S.S.; Demoss, J.; Aguirre, C.; Brookmeyer, R.; Kawas, C.H. Age of onset of hypertension and risk of dementia in the oldest-old: The 901 Study. Alzheimer’s Dement. 2017, 13, 103–110. [Google Scholar] [CrossRef]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling Introduction: Neurodegeneration in disease. J. Cell. Mol. Med 2008, 12, 2263–2280. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Wimo, A.; Winblad, B.; Aguero-Torres, H.; Von Strauss, E. The magnitude of dementia occurrence in the world. Alzheimer Dis. Assoc. Disord. 2003, 17, 63–67. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Y.; Shi, L.; Zhong, D.; Li, Y.; Li, J.; Jin, R. AMPK: Potential therapeutic target for Alzheimer’s disease. Curr. Protein Pept. Sci. 2020, 21, 66–77. [Google Scholar] [CrossRef]
- Abyadeh, M.; Gupta, V.; Chitranshi, N.; Gupta, V.; Wu, Y.; Saks, D.; Wander Wall, R.; Fitzhenry, M.J.; Basavarajappa, D.; You, Y.; et al. Mitochondrial dysfunction in Alzheimer’s disease-a proteomics perspective Mitochondrial dysfunction in Alzheimer’s disease-a proteomics perspective. Expert Rev. Proteom. 2021, 1–10. [Google Scholar] [CrossRef]
- Moltedo, O.; Remondelli, P.; Amodio, G. The mitochondria–endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef]
- Martucciello, S.; Masullo, M.; Cerulli, A.; Piacente, S. Molecular Sciences Natural Products Targeting ER Stress, and the Functional Link to Mitochondria. Int. J. Mol. Sci. 2020, 21, 1905. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicine 2021, 9, 156. [Google Scholar] [CrossRef]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.; Riaz, T.; Kim, H.; Molecular, H.C. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Park, J.J. Molecular Sciences Can Co-Activation of Nrf2 and Neurotrophic Signaling Pathway Slow Alzheimer’s Disease? Int. J. Mol. Sci. 2017, 18, 1168. [Google Scholar] [CrossRef]
- Butterfield, D.; Neuroscience, B.H. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Reddy, P. Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer’s disease: Implications for synaptic dysfunction and neuronal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1913–1921. [Google Scholar] [CrossRef]
- Raina, P.; Santaguida, P.; Ismaila, A.; Patterson, C.; Cowan, D.; Levine, M.; Booker, L.; Oremus, M. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: Evidence review for a clinical practice guideline. Ann. Intern. Med. 2008, 148, 379–397. [Google Scholar] [CrossRef]
- Zetterberg, H.; Psychiatry, B.B. Biomarkers for Alzheimer’s disease—Preparing for a new era of disease-modifying therapies. Mol. Psychiatry 2020, 26, 296–308. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Jan, A.T.; Azam, M.; Rahman, S.; Almigeiti, A.M.S.; Choi, D.H.; Lee, E.J.; Haq, Q.M.R.; Choi, I. Perspective insights into disease progression, diagnostics, and therapeutic approaches in Alzheimer’s disease: A judicious update. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Simes, D.C.; Viegas, C.S.B.; Araújo, N.; Marreiros, C. Vitamin K as a Diet Supplement with Impact in Human Health: Current Evidence in Age-Related Diseases. Nutrients 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.P.; Duan, L.; Practice, S.L. Effect of vitamin K2 on type 2 diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2018, 136, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Booth, S.L.; Van Den Heuvel, E.G.H.M.; Stoecklin, E.; Baka, A.; Vermeer, C. The role of menaquinones (vitamin K 2 ) in human health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Alisi, L.; Cao, R.; De Angelis, C.; Cafolla, A.; Caramia, F.; Cartocci, G.; Librando, A.; Fiorelli, M. The relationships between vitamin k and cognition: A review of current evidence. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Shirakawa, H.; Hiwatashi, K.; Furukawa, Y.; Mizutani, T.; Komai, M. Bioscience, Biotechnology, and Biochemistry Vitamin K Suppresses Lipopolysaccharide-Induced Inflammation in the Rat. Taylor Fr. 2006, 70, 926–932. [Google Scholar] [CrossRef]
- Carrié, I.; Bélanger, E.; Portoukalian, J.; Rochford, J.; Ferland, G. Lifelong low-phylloquinone intake is associated with cognitive impairments in old rats 1-3. J. Nutr. 2011, 141, 1495–1501. [Google Scholar] [CrossRef]
- Presse, N.; Shatenstein, B.; Kergoat, M.J.; Ferland, G. Low vitamin K intakes in community-dwelling elders at an early stage of Alzheimer’s disease. J. Am. Diet. Assoc. 2008, 108, 2095–2099. [Google Scholar] [CrossRef]
- Hadipour, E.; Tayarani-Najaran, Z.; Fereidoni, M. Vitamin K2 protects PC12 cells against Aβ (1-42) and H2O2-induced apoptosis via p38 MAP kinase pathway*. Nutr. Neurosci. 2020, 23, 343–352. [Google Scholar] [CrossRef]
- Huang, S.-H.; Fang, S.-T.; Chen, Y.-C.; Grimm, W. Molecular Mechanism of Vitamin K2 Protection against Amyloid-β-Induced Cytotoxicity. Biomolecules 2021, 11, 423. [Google Scholar] [CrossRef]
- Yu, Y.X.; Li, Y.P.; Gao, F.; Hu, Q.S.; Zhang, Y.; Chen, D.; Wang, G.H. Vitamin K 2 suppresses rotenone-induced microglial activation in vitro. Acta Pharmacol. Sin. 2016, 37, 1178–1189. [Google Scholar] [CrossRef]
- Chiang, M.F.; Liu, W.-K.; Yen, S.-H. Reversible Heat Stress-related Loss of Phosphorylated Alzheimer-type Epitopes in Tau Proteins of Human Neuroblastoma Cells. J. Neurosci. 1993, 13, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Ko, L.; Liu, W.; Georgieff, I.; Research, S.Y. Modulated induction of tau proteins in cultured human neuroblastoma cells. Brain Res. 1996, 707, 256–265. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Osmanovic-Barilar, J.; Brückner, M.K.; Hoyer, S.; Arendt, T.; Riederer, P. Cerebral amyloid angiopathy in streptozotocin rat model of sporadic Alzheimer’s disease: A long-term follow up study. J. Neural Transm. 2011, 118, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Biswas, J.; Gupta, S.; Verma, D.K.; Gupta, P.; Singh, A.; Tiwari, S.; Goswami, P.; Sharma, S.; Singh, S. Involvement of glucose related energy crisis and endoplasmic reticulum stress: Insinuation of streptozotocin induced Alzheimer’s like pathology. Cell. Signal. 2018, 42, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; Sciences, A.J. Streptozotocin-induced cytotoxicity, oxidative stress and mitochondrial dysfunction in human hepatoma HepG2 cells. Int. J. Mol. Sci. 2012, 13, 5751–5767. [Google Scholar] [CrossRef]
- Kesari, K.K.; Dhasmana, A.; Shandilya, S.; Prabhakar, N.; Shaukat, A.; Dou, J.; Rosenholm, J.M.; Vuorinen, T.; Ruokolainen, J. Plant-derived natural biomolecule picein attenuates menadione induced oxidative stress on neuroblastoma cell mitochondria. Antioxidants 2020, 9, 552. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef]
- Niwa, M.; Sidrauski, C.; Kaufman, R.J.; Walter, P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell 1999, 99, 691–702. [Google Scholar] [CrossRef]
- Delacourte, A. Tauopathies: Recent insights into old diseases. Folia Neuropathol. 2005, 43, 244–257. [Google Scholar] [PubMed]
- Wei, L.; Lu, Y.; He, S.; Jin, X.; Zeng, L.; Zhang, S.; Chen, Y.; Tian, B.; Mai, G.; Yang, G.; et al. Induction of diabetes with signs of autoimmunity in primates by the injection of multiple-low-dose streptozotocin. Biochem. Biophys. Res. Commun. 2011, 412, 373–378. [Google Scholar] [CrossRef]
- Falone, S.; Sannino, A.; Romeo, S.; Zeni, O.; Santini, S.; Rispoli, R.; Amicarelli, F.; Scarfì, M.R. Protective effect of 1950 MHz electromagnetic field in human neuroblastoma cells challenged with menadione. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lobner, D. Comparison of the LDH and MTT assays for quantifying cell death: Validity for neuronal apoptosis? J. Neurosci. Methods 2000, 96, 147–152. [Google Scholar] [CrossRef]
- Kidd, V.J. Proteolytic activities that mediate apoptosis. Annu. Rev. Physiol. 1998, 60, 533–573. [Google Scholar] [CrossRef]
- Chen, L.; Shi, X.; Weng, S.J.; Xie, J.; Tang, J.H.; Yan, D.Y.; Wang, B.Z.; Xie, Z.J.; Wu, Z.Y.; Yang, L. Vitamin K2 Can Rescue the Dexamethasone-Induced Downregulation of Osteoblast Autophagy and Mitophagy Thereby Restoring Osteoblast Function In Vitro and In Vivo. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Qiu, C.; Zheng, H.; Tao, H.; Yu, W.; Jiang, X.; Li, A.; Jin, H.; Lv, A.; Li, H. Vitamin K2 inhibits rat vascular smooth muscle cell calcification by restoring the Gas6/Axl/Akt anti-apoptotic pathway. Mol. Cell. Biochem. 2017, 433, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Quadros Gomes, B.A.; Bastos Silva, J.P.; Rodrigues Romeiro, C.F.; dos Santos, S.M.; Rodrigues, C.A.; Gonçalves, P.R.; Sakai, J.T.; Santos Mendes, P.F.; Pompeu Varela, E.L.; Monteiro, M.C. Neuroprotective mechanisms of resveratrol in Alzheimer’s disease: Role of SIRT1. Oxidative Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Shimura, T.; Kunugita, N. Mitochondrial reactive oxygen species-mediated genomic instability in low-dose irradiated human cells through nuclear retention of cyclin D1. Cell Cycle 2016, 15, 1410–1414. [Google Scholar] [CrossRef]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Nahdi, A.M.T.A.; John, A.; Raza, H. Elucidation of Molecular Mechanisms of Streptozotocin-Induced Oxidative Stress, Apoptosis, and Mitochondrial Dysfunction in Rin-5F Pancreatic β-Cells. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Ramesh, T. Oxidative stress and hepatocellular mitochondrial dysfunction attenuated by asiatic acid in streptozotocin-induced diabetic rats. J. King Saud Univ. Sci. 2021, 33, 101369. [Google Scholar] [CrossRef]
- Lin, X.; Wen, X.; Wei, Z.; Guo, K.; Shi, F.; Huang, T.; Wang, W.; Zheng, J. Vitamin K2protects against Aβ42-induced neurotoxicity by activating autophagy and improving mitochondrial function in Drosophila. Neuroreport 2021, 431–437. [Google Scholar] [CrossRef]
- Prasuhn, J.; Kasten, M.; Vos, M.; König, I.R.; Schmid, S.M.; Wilms, B.; Klein, C.; Brüggemann, N. The Use of Vitamin K2 in Patients With Parkinson’s Disease and Mitochondrial Dysfunction (PD-K2): A Theranostic Pilot Study in a Placebo-Controlled Parallel Group Design. Front. Neurol. 2021, 11. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Akha, A.A.S.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, 2024–2041. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef]
- Murray, H.C.; Dieriks, B.V.; Swanson, M.E.V.; Anekal, P.V.; Turner, C.; Faull, R.L.M.; Belluscio, L.; Koretsky, A.; Curtis, M.A. The unfolded protein response is activated in the olfactory system in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef]
- Hiroi, T.; Wei, H.; Hough, C.; Leeds, P.; Chuang, D.M. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: Roles of intracellular calcium, GRP78 and Bcl-2. Pharm. J. 2005, 5, 102–111. [Google Scholar] [CrossRef]
- Wang, Q.M.; Fiol, C.J.; Depaoli-Roach, A.A.; Roach, P.J. Glycogen Synthase Kinase-3P Is a Dual Specificity Kinase Differentially Regulated by Tyrosine and Serinemhreonine Phosphorylation. J. Biol. Chem. 1994, 269, 14566–14574. [Google Scholar] [CrossRef]
- Song, H.C.; Chen, Y.; Chen, Y.; Park, J.; Zheng, M.; Surh, Y.J.; Kim, U.H.; Park, J.W.; Yu, R.; Chung, H.T.; et al. GSK-3β inhibition by curcumin mitigates amyloidogenesis via TFEB activation and anti-oxidative activity in human neuroblastoma cells. Free Radic. Res. 2020. [Google Scholar] [CrossRef]
- Meares, G.P.; Mines, M.A.; Beurel, E.; Eom, T.Y.; Song, L.; Zmijewska, A.A.; Jope, R.S. Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER) stress-induced CHOP expression in neuronal cells. Exp. Cell Res. 2011, 317, 1621–1628. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Jensen, M.; Basun, H.; Lannfelt, L. Increased cerebrospinal fluid tau in patients with Alzheimer’s disease. Neurosci. Lett. 1995, 186, 189–191. [Google Scholar] [CrossRef]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Londos, E.; Blennow, K.; Minthon, L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol. 2006, 5, 228–234. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, Y.; Tan, H.; Boukhali, M.; Khatri, A.; Yu, Y.; Hua, F.; Liu, L.; Li, M.; Yang, G.; et al. Tau Contributes to Sevoflurane-induced Neurocognitive Impairment in Neonatal Mice. Anesthesiology 2020, 133, 595–610. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Mett, J.; Hartmann, T. The impact of Vitamin E and other fat-soluble vitamins on Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1785. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shandilya, S.; Kesari, K.K.; Ruokolainen, J. Vitamin K2 Modulates Organelle Damage and Tauopathy Induced by Streptozotocin and Menadione in SH-SY5Y Cells. Antioxidants 2021, 10, 983. https://doi.org/10.3390/antiox10060983
Shandilya S, Kesari KK, Ruokolainen J. Vitamin K2 Modulates Organelle Damage and Tauopathy Induced by Streptozotocin and Menadione in SH-SY5Y Cells. Antioxidants. 2021; 10(6):983. https://doi.org/10.3390/antiox10060983
Chicago/Turabian StyleShandilya, Shruti, Kavindra Kumar Kesari, and Janne Ruokolainen. 2021. "Vitamin K2 Modulates Organelle Damage and Tauopathy Induced by Streptozotocin and Menadione in SH-SY5Y Cells" Antioxidants 10, no. 6: 983. https://doi.org/10.3390/antiox10060983
APA StyleShandilya, S., Kesari, K. K., & Ruokolainen, J. (2021). Vitamin K2 Modulates Organelle Damage and Tauopathy Induced by Streptozotocin and Menadione in SH-SY5Y Cells. Antioxidants, 10(6), 983. https://doi.org/10.3390/antiox10060983