
Specificity of Human Sulfiredoxin for Reductant and Peroxiredoxin Oligomeric State

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Crystallization and Structure Determination
2.3. Preparative Scale Hyperoxidation
2.4. SEC-MALS Analysis to Measure Prx Size Distribution
2.5. HPLC Srx Activity and Binding Assays
2.6. HRP Assay to Measure Peroxidase Activity
2.7. ESI-TOF MS Srx Activity Assay
3. Results
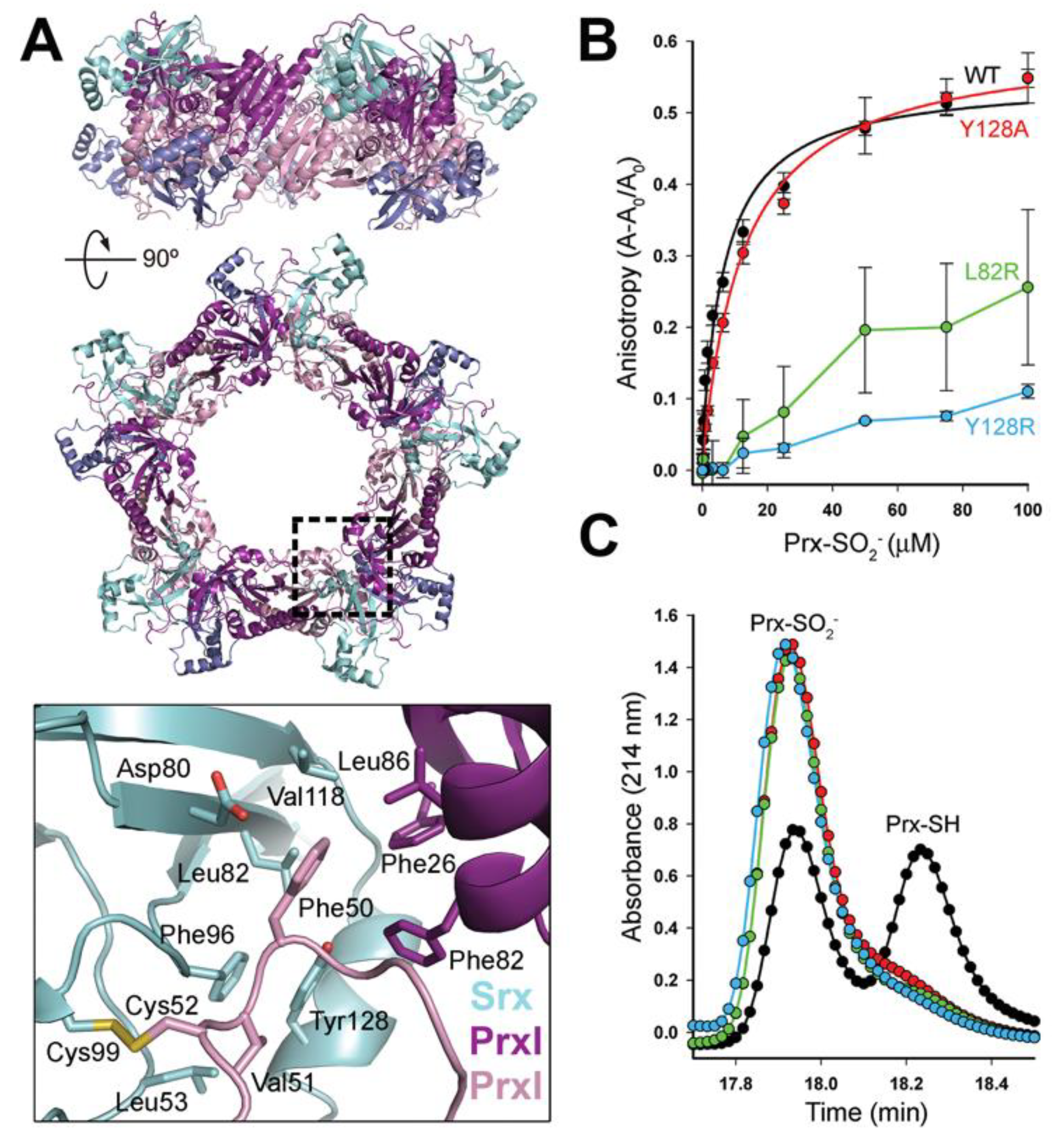
3.1. Decameric Srx-Prx1 Complex Reveals Extended Binding Interface
3.2. Dimer-Dimer Interface Disruption Decreases Srx Binding and Repair of Prx1
3.3. Higher Order Oligomeric State of Prx2 and Prx3 Is Also Required by Srx
3.4. C-Terminal Sequence Differences between Prx2 and Prx3 Impact the Rate of Repair by Srx
3.5. Human Srx Can Utilize GSH and H2S as Reducing Source
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hall, A.; Nelson, K.; Poole, L.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.; Schröder, E.; Harris, J.R.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Kil, I.S.; Lee, S.K.; Ryu, K.W.; Woo, H.A.; Hu, M.-C.; Bae, S.H.; Rhee, S.G. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Hopkins, B.L.; Nadler, M.; Skoko, J.J.; Bertomeu, T.; Pelosi, A.; Shafaei, P.M.; Levine, K.; Schempf, A.; Pennarun, B.; Yang, B.; et al. A peroxidase peroxiredoxin 1-specific redox regulation of the novel FOXO3 microRNA target let-7. Antioxid. Redox Signal. 2018, 28, 62–77. [Google Scholar] [CrossRef]
- Stöcker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 protects telomeres from oxidative damage and preserves telomeric DNA for extension by telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658–669. [Google Scholar] [CrossRef]
- Ahmed, W.; Lingner, J. Impact of oxidative stress on telomere biology. Differentiation 2018, 99, 21–27. [Google Scholar] [CrossRef]
- Majerska, J.; Feretzaki, M.; Glousker, G.; Lingner, J. Transformation-induced stress at telomeres is counteracted through changes in the telomeric proteome including SAMHD1. Life Sci. Alliance 2018, 1, e201800121. [Google Scholar] [CrossRef] [PubMed]
- Power, J.H.T.; Asad, S.; Chataway, T.; Chegini, F.; Manavis, J.; Temlett, J.A.; Jensen, P.H.; Blumbergs, P.C.; Gai, W.-P. Peroxiredoxin 6 in human brain: Molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol. 2008, 115, 611–622. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef]
- El Eter, E.; Al-Masri, A. Peroxiredoxin isoforms are associated with cardiovascular risk factors in type 2 diabetes mellitus. Braz. J. Med. Biol. Res. 2015, 48, 465–469. [Google Scholar] [CrossRef]
- Stancill, J.S.; Broniowska, K.A.; Oleson, B.J.; Naatz, A.; Corbett, J.A. Pancreatic beta-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J. Biol. Chem. 2019, 294, 4843–4853. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.; Furdui, C.M.; Loeser, R.F. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signaling in aging chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654. [Google Scholar] [CrossRef]
- Gertz, M.; Fischer, F.; Leipelt, M.; Wolters, D.; Steegborn, C. Identification of Peroxiredoxin 1 as a novel interaction partner for the lifespan regulator protein p66Shc. Aging 2009, 1, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Radyuk, S.; Michalak, K.; Klichko, V.I.; Benes, J.; Rebrin, I.; Sohal, R.S.; Orr, W.C. Peroxiredoxin 5 confers protection against oxidative stress and apoptosis and also promotes longevity in Drosophila. Biochem. J. 2009, 419, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-G.; Wang, L.; Kaifu, T.; Li, J.; Li, X.; Li, L. Featured Article: Accelerated decline of physical strength in peroxiredoxin-3 knockout mice. Exp. Biol. Med. 2016, 241, 1395–1400. [Google Scholar] [CrossRef]
- Argyropoulou, V.; Goemaere, J.; Clippe, A.; Lefort, C.; Tissir, F.; Schakman, O.; Gailly, P.; Ahn, M.-T.; Guiot, Y.; Galant, C.; et al. Peroxiredoxin-5 as a novel actor in inflammation and tumor suppression. Free Radic. Biol. Med. 2016, 100, S92. [Google Scholar] [CrossRef]
- Egler, R.; Fernandes, E.; Rothermund, K.; Sereika, S.; De Souza-Pinto, N.; Jaruga, P.; Dizdaroglu, M.; Prochownik, E.V. Regulation of reactive oxygen species, DNA damage and c-Myc function by peroxiredoxin 1. Oncogene 2005, 24, 8038–8050. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nat. Cell Biol. 2003, 424, 561–565. [Google Scholar] [CrossRef]
- Abbasi, A.; Corpeleijn, E.; Postmus, D.; Gansevoort, R.T.; de Jong, P.E.; Gans, R.; Struck, J.; Schulte, J.; Hillege, H.L.; van der Harst, P.; et al. Peroxiredoxin 4, a novel circulating biomarker for oxidative stress and the risk of incident cardiovascular disease and all-cause mortality. J. Am. Hear. Assoc. 2012, 1, e002956. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-C.; Chiu, C.-C.; Chang, H.-W.; Wang, Y.-S.; Syue, H.-H.; Song, Y.-C.; Weng, Z.-H.; Tai, M.-H.; Wu, C.-Y. Prdx1-encoded peroxiredoxin is important for vascular development in zebrafish. FEBS Lett. 2017, 591, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Messier, T.; Milczarek, S.; Saaman, A.; Beuschel, S.; Gandhi, U.; Heintz, N.; Smalley, T.; Lowther, W.T.; Cunniff, B. Unique cellular and biochemical features of human mitochondrial peroxiredoxin 3 establish the molecular basis for its specific reaction with thiostrepton. Antioxidants 2021, 10, 150. [Google Scholar] [CrossRef]
- Poynton, R.A.; Peskin, A.; Haynes, A.C.; Lowther, W.T.; Hampton, M.B.; Winterbourn, C.C. Kinetic analysis of structural influences on the susceptibility of peroxiredoxins 2 and 3 to hyperoxidation. Biochem. J. 2016, 473, 411–421. [Google Scholar] [CrossRef]
- Pastor-Flores, D.; Talwar, D.; Pedre, B.; Dick, T.P. Real-time monitoring of peroxiredoxin oligomerization dynamics in living cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16313–16323. [Google Scholar] [CrossRef]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar] [CrossRef]
- Haynes, A.C.; Qian, J.; Reisz, J.A.; Furdui, C.M.; Lowther, W.T. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J. Biol. Chem. 2013, 288, 29714–29723. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Peskin, A. Kinetic approaches to measuring peroxiredoxin reactivity. Mol. Cells 2016, 39, 26–30. [Google Scholar] [CrossRef]
- Forshaw, T.E.; Holmila, R.; Nelson, K.J.; Lewis, J.E.; Kemp, M.L.; Tsang, A.W.; Poole, L.B.; Lowther, W.T.; Furdui, C.M. Peroxiredoxins in cancer and response to radiation therapies. Antioxidants 2019, 8, 11. [Google Scholar] [CrossRef]
- Bolduc, J.; Nelson, K.J.; Haynes, A.C.; Lee, J.; Reisz, J.A.; Graff, A.H.; Clodfelter, J.E.; Parsonage, D.; Poole, L.; Furdui, C.M.; et al. Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Ledesma, S.; Randall, L.M.; Parsonage, D.; Rizza, J.D.; Karplus, P.A.; Poole, L.; DeNicola, A.; Ferrer-Sueta, G. Differential kinetics of two-cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 2018, 57, 3416–3424. [Google Scholar] [CrossRef]
- Cao, Z.; Roszak, A.W.; Gourlay, L.J.; Lindsay, J.G.; Isaacs, N.W. Bovine mitochondrial peroxiredoxin III forms a two-ring catenane. Structure 2005, 13, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine–sulphinic acid by S. cerevisiae sulphiredoxin. Nat. Cell Biol. 2003, 425, 980–984. [Google Scholar] [CrossRef]
- Jönsson, T.J.; Johnson, L.C.; Lowther, W.T. Structure of the sulphiredoxin–peroxiredoxin complex reveals an essential repair embrace. Nat. Cell Biol. 2008, 451, 98–101. [Google Scholar] [CrossRef]
- Jönsson, T.J.; Johnson, L.C.; Lowther, W. Protein Engineering of the Quaternary Sulfiredoxin·peroxiredoxin enzyme-substrate complex reveals the molecular basis for cysteine sulfinic acid phosphorylation. J. Biol. Chem. 2009, 284, 33305–33310. [Google Scholar] [CrossRef]
- Boukhenouna, S.; Mazon, H.; Branlant, G.; Jacob, C.; Toledano, M.B.; Rahuel-Clermont, S. Evidence that glutathione and the glutathione system efficiently recycle 1-Cys sulfiredoxin in vivo. Antioxid. Redox Signal. 2015, 22, 731–743. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, Z. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 63, 32–41. [Google Scholar] [CrossRef]
- Schröder, E.; Littlechil, J.; Lebedev, A.; Errington, N.; Vagin, A.; Isupov, M. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7Å resolution. Structure 2000, 8, 605–615. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K.D. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Laskowski, R.; MacArthur, M.W.; Moss, D.S.; Thornton, J. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Collaborative Computational Project. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.J.; Parsonage, D. Measurement of peroxiredoxin activity. Curr. Protoc. Toxicol. 2011, 49, 7.10.1–7.10.28. [Google Scholar] [CrossRef] [PubMed]
- Parsonage, D.; Youngblood, D.S.; Sarma, G.N.; Wood, Z.A.; Karplus, P.A.; Poole, L.B. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 2005, 44, 10583–10592. [Google Scholar] [CrossRef]
- Yewdall, N.A.; Venugopal, H.; Desfosses, A.; Abrishami, V.; Yosaatmadja, Y.; Hampton, M.B.; Gerrard, J.A.; Goldstone, D.C.; Mitra, A.K.; Radjainia, M. Structures of human peroxiredoxin 3 suggest self-chaperoning assembly that maintains catalytic state. Structure 2016, 24, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-S.; Jeong, W.; Woo, H.A.; Lee, S.M.; Park, S.; Rhee, S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004, 279, 50994–51001. [Google Scholar] [CrossRef]
- Jeong, W.; Park, S.J.; Chang, T.-S.; Lee, D.-Y.; Rhee, S.G. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J. Biol. Chem. 2006, 281, 14400–14407. [Google Scholar] [CrossRef]
- Bayer, S.; Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Interactions between peroxiredoxin 2, hemichrome and the erythrocyte membrane. Free Radic. Res. 2016, 50, 1329–1339. [Google Scholar] [CrossRef]
- Lim, J.C.; Choi, H.-I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.-S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.C.; Hah, Y.-S.; Kim, W.Y.; Jung, B.G.; Jang, H.H.; Lee, J.R.; Kim, S.Y.; Lee, Y.M.; Jeon, M.G.; Kim, C.W.; et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced Cell Death. J. Biol. Chem. 2005, 280, 28775–28784. [Google Scholar] [CrossRef] [PubMed]
- Noichri, Y.; Palais, G.; Ruby, V.; D’Autreaux, B.; Delaunay-Moisan, A.; Nyström, T.; Molin, M.; Toledano, M. In vivo parameters influencing 2-Cys Prx oligomerization: The role of enzyme sulfinylation. Redox Biol. 2015, 6, 326–333. [Google Scholar] [CrossRef]
- Ogasawara, Y.; Ohminato, T.; Nakamura, Y.; Ishii, K. Structural and functional analysis of native peroxiredoxin 2 in human red blood cells. Int. J. Biochem. Cell Biol. 2012, 44, 1072–1077. [Google Scholar] [CrossRef]
- Angelucci, F.; Saccoccia, F.; Ardini, M.; Boumis, G.; Brunori, M.; Di Leandro, L.; Ippoliti, R.; Miele, A.E.; Natoli, G.; Scotti, S.; et al. Switching between the alternative structures and functions of a 2-Cys peroxiredoxin, by site-directed mutagenesis. J. Mol. Biol. 2013, 425, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.H.; Baek, J.Y.; Jeong, W.; Rhee, S.G.; Chang, T.-S. Sulfiredoxin translocation into mitochondria plays a crucial role in reducing hyperoxidized peroxiredoxin III. J. Biol. Chem. 2009, 284, 8470–8477. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kil, I.S. Mitochondrial H2O2 signaling is controlled by the concerted action of peroxiredoxin III and sulfiredoxin: Linking mitochondrial function to circadian rhythm. Free Radic. Biol. Med. 2016, 100, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef]
- Roussel, X.; Béchade, G.; Kriznik, A.; Van Dorsselaer, A.; Cianferani, S.; Branlant, G.; Rahuel-Clermont, S. Evidence for the formation of a covalent thiosulfinate intermediate with peroxiredoxin in the catalytic mechanism of sulfiredoxin. J. Biol. Chem. 2008, 283, 22371–22382. [Google Scholar] [CrossRef] [PubMed]
- Roussel, X.; Kriznik, A.; Richard, C.; Rahuel-Clermont, S.; Branlant, G. Catalytic mechanism of sulfiredoxin from Saccharomyces cerevisiae passes through an oxidized disulfide sulfiredoxin intermediate that is reduced by thioredoxin. J. Biol. Chem. 2009, 284, 33048–33055. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Coletta, C.; Erdélyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef] [PubMed]
- Benchoam, D.; Semelak, J.A.; Cuevasanta, E.; Mastrogiovanni, M.; Grassano, J.S.; Ferrer-Sueta, G.; Zeida, A.; Trujillo, M.; Möller, M.N.; Estrin, D.A.; et al. Acidity and nucleophilic reactivity of glutathione persulfide. J. Biol. Chem. 2020, 295, 15466–15481. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forshaw, T.E.; Reisz, J.A.; Nelson, K.J.; Gumpena, R.; Lawson, J.R.; Jönsson, T.J.; Wu, H.; Clodfelter, J.E.; Johnson, L.C.; Furdui, C.M.; et al. Specificity of Human Sulfiredoxin for Reductant and Peroxiredoxin Oligomeric State. Antioxidants 2021, 10, 946. https://doi.org/10.3390/antiox10060946
Forshaw TE, Reisz JA, Nelson KJ, Gumpena R, Lawson JR, Jönsson TJ, Wu H, Clodfelter JE, Johnson LC, Furdui CM, et al. Specificity of Human Sulfiredoxin for Reductant and Peroxiredoxin Oligomeric State. Antioxidants. 2021; 10(6):946. https://doi.org/10.3390/antiox10060946
Chicago/Turabian StyleForshaw, Tom E., Julie A. Reisz, Kimberly J. Nelson, Rajesh Gumpena, J. Reed Lawson, Thomas J. Jönsson, Hanzhi Wu, Jill E. Clodfelter, Lynnette C. Johnson, Cristina M. Furdui, and et al. 2021. "Specificity of Human Sulfiredoxin for Reductant and Peroxiredoxin Oligomeric State" Antioxidants 10, no. 6: 946. https://doi.org/10.3390/antiox10060946
APA StyleForshaw, T. E., Reisz, J. A., Nelson, K. J., Gumpena, R., Lawson, J. R., Jönsson, T. J., Wu, H., Clodfelter, J. E., Johnson, L. C., Furdui, C. M., & Lowther, W. T. (2021). Specificity of Human Sulfiredoxin for Reductant and Peroxiredoxin Oligomeric State. Antioxidants, 10(6), 946. https://doi.org/10.3390/antiox10060946