
Hydroquinone Exposure Worsens Rheumatoid Arthritis through the Activation of the Aryl Hydrocarbon Receptor and Interleukin-17 Pathways

 , , , , and
, , , , and 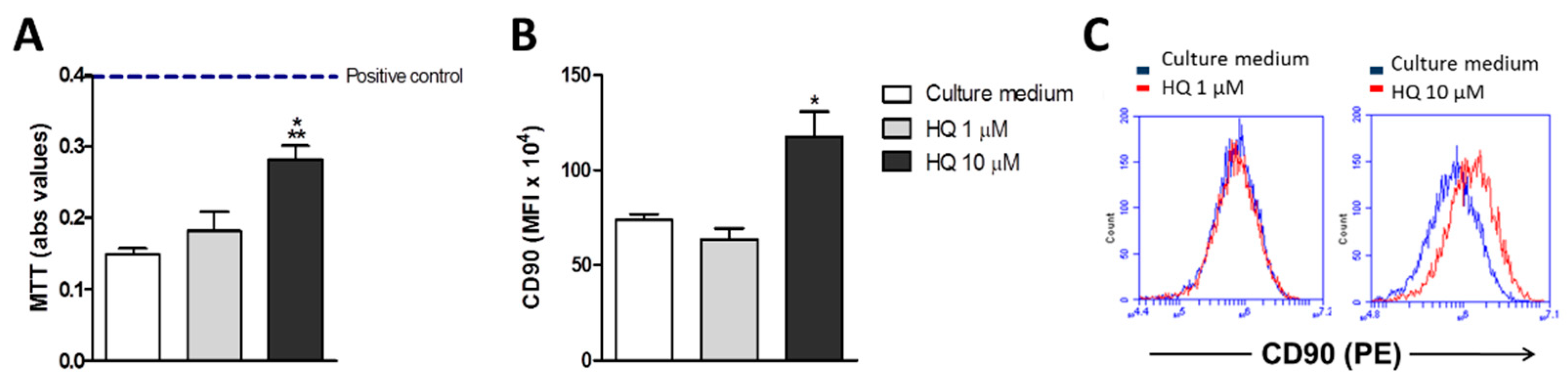{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Phenotypical Characterization
2.3. Animals
2.4. In Vivo Hydroquinone (HQ) Exposure
2.5. Antigen-Induced Arthritis (AIA)
2.6. Assessment of Articular Hyperalgesia and Edema
2.7. Synovial Fluid Collection and Measurement of Cell Influx
2.8. Histopathological and Immunofluorescence Analyses
2.9. Cell Viability
2.10. Cytokine Quantification
2.11. Protein Expression and Cell Frequency Measurements
2.12. In Vitro Detection of Reactive Oxygen Species
2.13. Influence of AhR Antagonist in RAHFLS Proliferation, TNFR Expression and ROS Generation
2.14. Quantification of mRNA Expression
2.15. Statistical Analysis
3. Results
3.1. In Vitro HQ Exposure Induces RAHFLS Activation
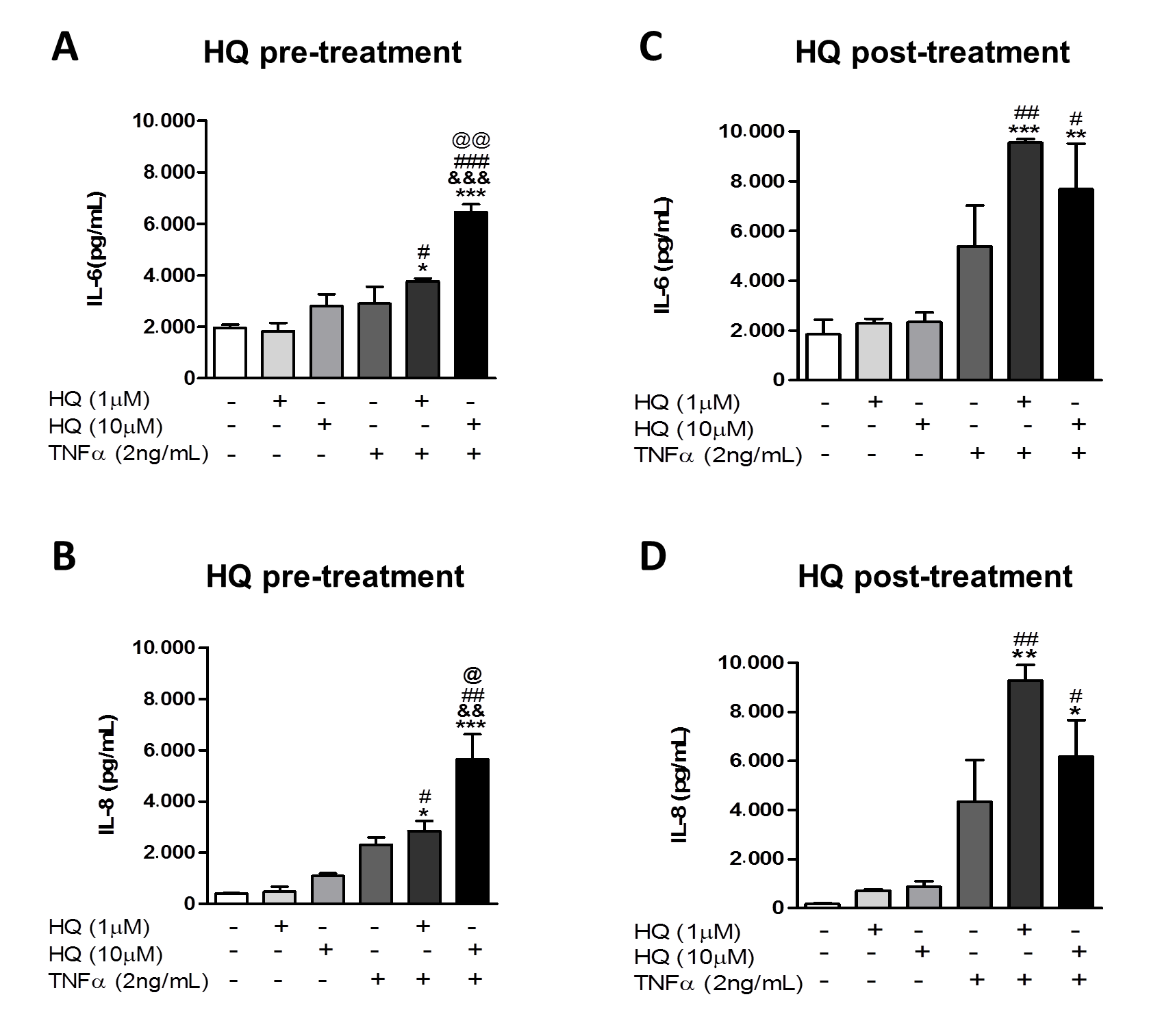
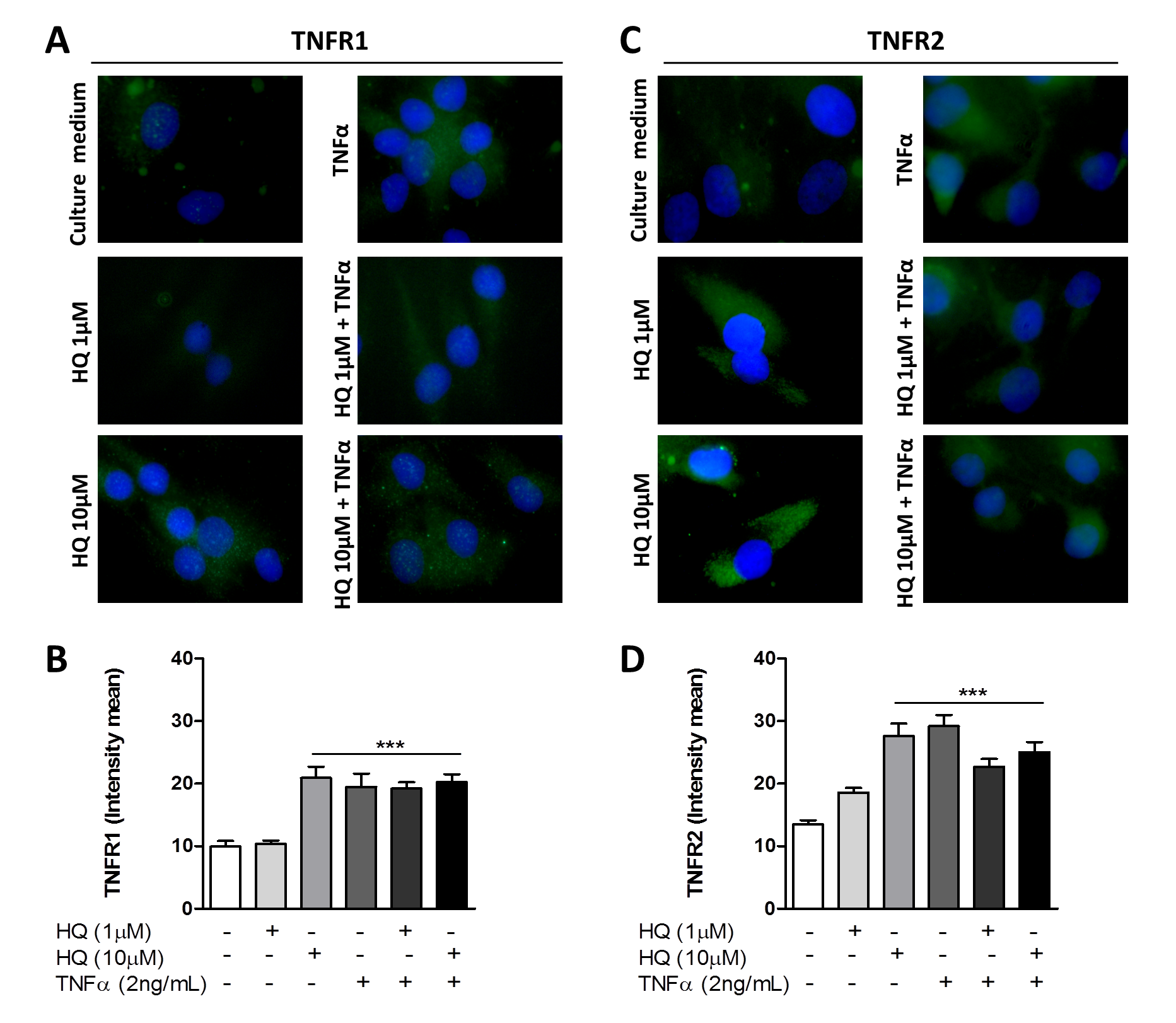
3.2. In Vitro HQ Exposure Potentiates the Cytokine Secretion and Enhances TNFR1 and TNFR2 Expression in TNF-α-Treated RAHFLS
3.3. In Vitro HQ Exposure Triggers the ROS Generation in RAHFLS
3.4. AhR Activity Is Involved in the HQ-Induced RAHFLS Proliferation and TNFRs Expression
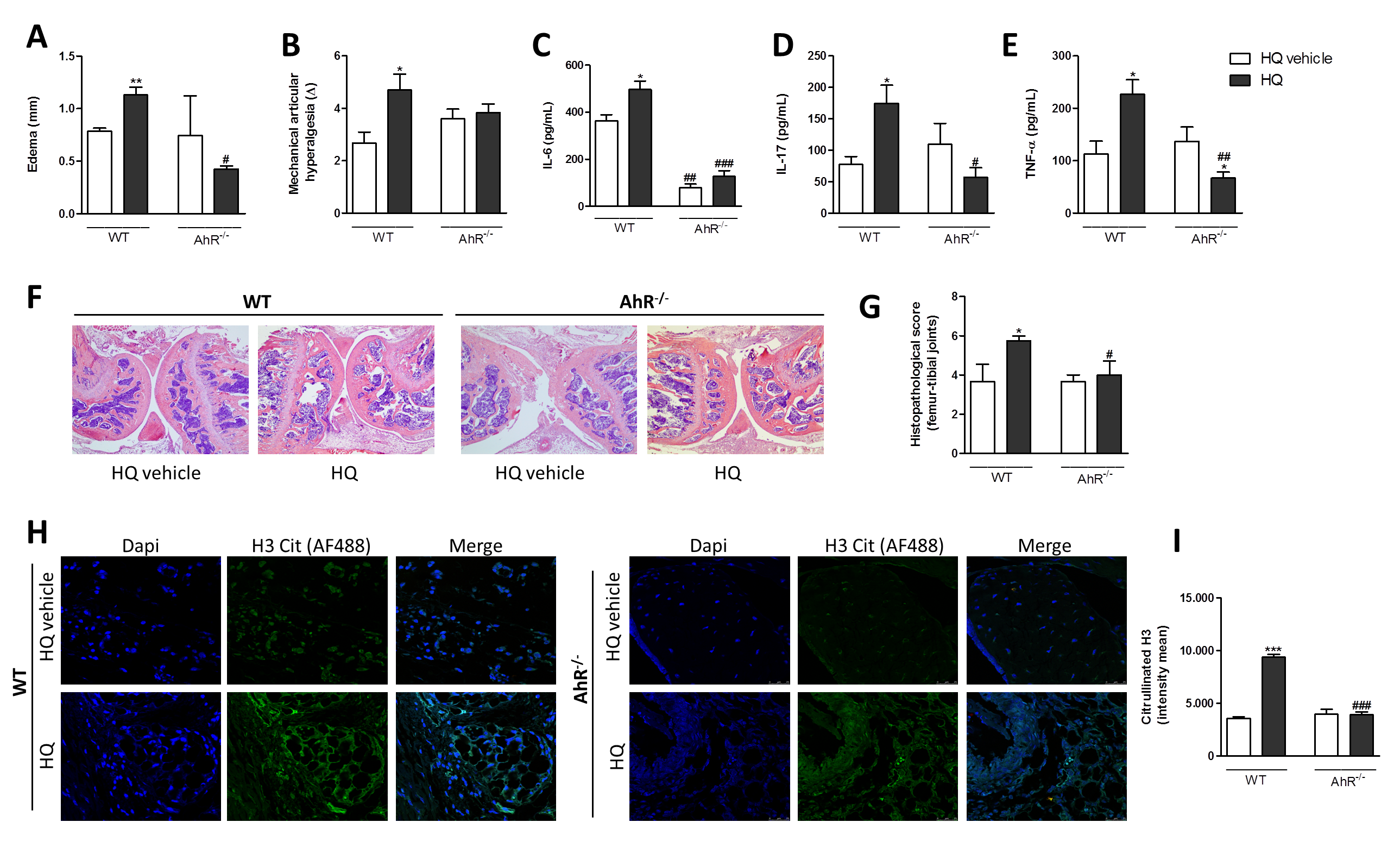
3.5. AhR and IL-17 Are Involved in the AIA Worsening upon an In Vivo HQ Exposure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nat. Cell Biol. 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Bessis, N.; Decker, P.; Assier, E.; Semerano, L.; Boissier, M.-C. Arthritis models: Usefulness and interpretation. Semin. Immunopathol. 2017, 39, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Youn, J. Regulation of TNF-α-mediated hyperplasia through TNF receptors, TRAFs, and NF-κB in synoviocytes obtained from patients with rheumatoid arthritis. Immunol. Lett. 2002, 83, 85–93. [Google Scholar] [CrossRef]
- NiuBei, Q.; Cai, B.; Huang, Z.-C.; Shi, Y.-Y.; Wang, L.-L. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol. Int. 2011, 32, 2731–2736. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E. The IL-23–IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef]
- Radner, H.; Aletaha, D. Anti-TNF in rheumatoid arthritis: An overview. Wien. Med. Wochenschr. 2015, 165, 3–9. [Google Scholar] [CrossRef]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent Pro- and Antiinflammatory Roles for IL-23 and IL-12 in Joint Autoimmune Inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Sigaux, J.; Biton, J.; André, E.; Semerano, L.; Boissier, M.-C. Air pollution as a determinant of rheumatoid arthritis. Jt. Bone Spine 2019, 86, 37–42. [Google Scholar] [CrossRef]
- Sokolove, J.; Wagner, C.A.; Lahey, L.J.; Sayles, H.; Duryee, M.; Reimold, A.M.; Kerr, G.; Robinson, W.H.; Cannon, G.W.; Thiele, G.M.; et al. Increased inflammation and disease activity among current cigarette smokers with rheumatoid arthritis: A cross-sectional analysis of US veterans. Rheumatology 2016, 55, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Tobon, G.; Youinou, P.; Saraux, A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J. Autoimmun. 2010, 35, 10–14. [Google Scholar] [CrossRef]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Harris, H.E.; Ulfgren, A.-K.; Rantapää-Dahlqvist, S.; Eklund, A.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2005, 54, 38–46. [Google Scholar] [CrossRef]
- Stabbert, R.; Dempsey, R.; Diekmann, J.; Euchenhofer, C.; Hagemeister, T.; Haussmann, H.-J.; Knorr, A.; Mueller, B.P.; Pospisil, P.; Reininghaus, W.; et al. Studies on the contributions of smoke constituents, individually and in mixtures, in a range of in vitro bioactivity assays. Toxicol. Vitr. 2017, 42, 222–246. [Google Scholar] [CrossRef]
- Arimilli, S.; Schmidt, E.; Damratoski, B.E.; Prasad, G.L. Role of Oxidative Stress in the Suppression of Immune Responses in Peripheral Blood Mononuclear Cells Exposed to Combustible Tobacco Product Preparation. Inflammation 2017, 40, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Pulliero, A. Molecular damage and lung tumors in cigarette smoke-exposed mice. Ann. N. Y. Acad. Sci. 2015, 1340, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Scharf, P.; da Rocha, G.H.; Sandri, S.; Heluany, C.S.; Filho, W.R.P.; Farsky, S.H. Immunotoxic mechanisms of cigarette smoke and heat-not-burn tobacco vapor on Jurkat T cell functions. Environ. Pollut. 2021, 268, 115863. [Google Scholar] [CrossRef]
- Adachi, M.; Okamoto, S.; Chujyo, S.; Arakawa, T.; Yokoyama, M.; Yamada, K.; Hayashi, A.; Akita, K.; Takeno, M.; Itoh, S.; et al. Cigarette Smoke Condensate Extracts Induce IL-1-Beta Production from Rheumatoid Arthritis Patient-Derived Synoviocytes, but Not Osteoarthritis Patient-Derived Synoviocytes, Through Aryl Hydrocarbon Receptor-Dependent NF-Kappa-B Activation and Novel NF-Kappa-B Sites. J. Interf. Cytokine Res. 2013, 33, 297–307. [Google Scholar] [CrossRef]
- Kishimoto, T.; Nguyen, N.T.; Nakahama, T.; Nguyen, H.C.; Tran, T.T.; Le, V.S.; Chu, H.H. Aryl hydrocarbon receptor antagonism and its role in rheumatoid arthritis. J. Exp. Pharmacol. 2015, 7, 29–35. [Google Scholar] [CrossRef]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. 2018, 20, 119. [Google Scholar] [CrossRef] [PubMed]
- Kazantseva, M.G.; Highton, J.; Stamp, L.K.; A Hessian, P. Dendritic cells provide a potential link between smoking and inflammation in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R208. [Google Scholar] [CrossRef]
- Mescher, M.; Haarmann-Stemmann, T. Modulation of CYP1A1 metabolism: From adverse health effects to chemoprevention and therapeutic options. Pharmacol. Ther. 2018, 187, 71–87. [Google Scholar] [CrossRef]
- Nakahama, T.; Kimura, A.; Nguyen, N.T.; Chinen, I.; Hanieh, H.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 14222–14227. [Google Scholar] [CrossRef]
- McGregor, D. Hydroquinone: An Evaluation of the Human Risks from its Carcinogenic and Mutagenic Properties. Crit. Rev. Toxicol. 2007, 37, 887–914. [Google Scholar] [CrossRef] [PubMed]
- Fabris, A.L.; Nunes, A.V.; Schuch, V.; De Paula-Silva, M.; Rocha, G.; Nakaya, H.I.; Ho, P.L.; Silveira, E.L.; Farsky, S.H.P.; Rocha, G. Hydroquinone exposure alters the morphology of lymphoid organs in vaccinated C57Bl/6 mice. Environ. Pollut. 2020, 257, 113554. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.T.; Shimada, A.L.B.; Hebeda, C.B.; de Oliveira, T.F.; Loureiro, A.P.D.M.; Filho, W.D.R.P.; Santos, A.M.D.A.; de Lima, W.T.; Farsky, S.H.P. In vivo hydroquinone exposure alters circulating neutrophil activities and impairs LPS-induced lung inflammation in mice. Toxicology 2011, 288, 1–7. [Google Scholar] [CrossRef]
- Shimada, A.L.B.; Lino-Dos-Santos-Franco, A.; Bolonheis, S.M.; Nakasato, A.; Damazo, A.S.; Tavares-De-Lima, W.; Farsky, S.H.P. In vivo hydroquinone exposure causes tracheal hyperresponsiveness due to TNF secretion by epithelial cells. Toxicol. Lett. 2012, 211, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Cousins, S.W.; Csaky, K.G.; Striker, G.; Marin-Castaño, M.E. Cigarette Smoke-Related Hydroquinone Induces Filamentous Actin Reorganization and Heat Shock Protein 27 Phosphorylation through p38 and Extracellular Signal-Regulated Kinase 1/2 in Retinal Pigment Epithelium: Implications for Age-Related Macular Degeneration. Am. J. Pathol. 2010, 177, 1198–1213. [Google Scholar] [CrossRef]
- Rim, T.H.; Cheng, C.-Y.; Kim, D.W.; Kim, S.S.; Wong, T.Y. A nationwide cohort study of cigarette smoking and risk of neovascular age-related macular degeneration in East Asian men. Br. J. Ophthalmol. 2017, 101, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Heluany, C.S.; Kupa, L.D.V.K.; Viana, M.N.; Fernandes, C.M.; Farsky, S.H.P. Hydroquinone exposure worsens the symptomatology of rheumatoid arthritis. Chem. Interact. 2018, 291, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Heluany, C.S.; Kupa, L.D.V.K.; Viana, M.N.; Fernandes, C.M.; Silveira, E.L.V.; Farsky, S.H.P. In vivo exposure to hydroquinone during the early phase of collagen-induced arthritis aggravates the disease. Toxicology 2018, 408, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Hülso, C.; Liu, Y.; Gasparini, S.J.; Fong-Yee, C.; Tu, J.; Stoner, S.; Stewart, P.M.; Raza, K.; Cooper, M.S.; et al. Characterisation of fibroblast-like synoviocytes from a murine model of joint inflammation. Arthritis Res. Ther. 2013, 15, R24. [Google Scholar] [CrossRef]
- Fueldner, C.; Mittag, A.; Knauer, J.; Biskop, M.; Hepp, P.; Scholz, R.; Wagner, U.; Sack, U.; Emmrich, F.; Tárnok, A.; et al. Identification and evaluation of novel synovial tissue biomarkers in rheumatoid arthritis by laser scanning cytometry. Arthritis Res. Ther. 2012, 14, R8. [Google Scholar] [CrossRef]
- Pinto, L.G.; Cunha, T.; Vieira, S.M.; Lemos, H.D.P.; Cunha, F.Q.; Ferreira, S.H. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain 2010, 148, 247–256. [Google Scholar] [CrossRef]
- Horobin, R. How Romanowsky stains work and why they remain valuable—Including a proposed universal Romanowsky staining mechanism and a rational troubleshooting scheme. Biotech. Histochem. 2011, 86, 36–51. [Google Scholar] [CrossRef]
- Williams, A.S.; Richards, P.J.; Thomas, E.; Carty, S.; Mari, A.N.; Goodfellow, R.M.; Dent, C.M.; Williams, B.D.; Jones, S.A.; Topley, N. Interferon-g protects against the development of structural damage in experi-mental arthritis by regulating polymorphonuclear neutrophil influx into diseased joints. Arthritis Rheum. 2007, 56, 2244–2254. [Google Scholar] [CrossRef]
- Xiao, X.; Feng, Y.-P.; Du, B.; Sun, H.-R.; Ding, Y.-Q.; Qi, J.-G. Antibody incubation at 37 °C improves fluorescent immunolabeling in free-floating thick tissue sections. Biotechniques 2017, 62, 115–122. [Google Scholar] [CrossRef]
- Katsikis, P.D.; Chu, C.Q.; Brennan, F.M.; Maini, R.N.; Feldmann, M. Immunoregulartory role of interleukin 10 in rheumatoid arthritis. J. Exp. Med. 1995, 179, 1517–1527. [Google Scholar] [CrossRef]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in toll-like receptor 4-dependent activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef]
- Pryor, W.A.; Stone, K. Oxidants in Cigarette Smoke Radicals, Hydrogen Peroxide, Peroxynitrate, and Peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27. [Google Scholar] [CrossRef]
- Mao, J.; Dai, W.; Zhang, S.; Sun, L.; Wang, H.; Gao, Y.; Wang, J.; Zhang, F. Quinone–thioether metabolites of hydroquinone play a dual role in promoting a vicious cycle of ROS generation: In vitro and in silico insights. Arch. Toxicol. 2019, 93, 1297–1309. [Google Scholar] [CrossRef]
- Peng, C.; Arthur, D.; Liu, F.; Lee, J.; Xia, Q.; Lavin, M.F.; Ng, J.C. Genotoxicity of hydroquinone in A549 cells. Cell Biol. Toxicol. 2013, 29, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Go, R.-E.; Hwang, K.-A.; Choi, K.-C. Cytochrome P450 1 family and cancers. J. Steroid Biochem. Mol. Biol. 2015, 147, 24–30. [Google Scholar] [CrossRef]
- Zhao, C.N.; Xu, Z.; Wu, G.C.; Mao, Y.M.; Liu, L.N.; Dan, Y.L.; Tao, S.S.; Zhang, Q.; Sam, N.B.; Fan, Y.G.; et al. Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 2019, 18, 607–614. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Triggianese, P.; Conigliaro, P.; Candi, E.; Melino, G.; Perricone, R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis. 2015, 6, e1887. [Google Scholar] [CrossRef] [PubMed]
- Whysner, J.; Verna, L.; English, J.; Williams, G. Analysis of Studies Related to Tumorigenicity Induced by Hydroquinone. Regul. Toxicol. Pharmacol. 1995, 21, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Simmonds, R.; Foxwell, B.M. Signalling, inflammation and arthritis: NF- B and its relevance to arthritis and inflammation. Rheumatology 2008, 47, 584–590. [Google Scholar] [CrossRef]
- Hebeda, C.B.; Pinedo, F.J.; Bolonheis, S.M.; Ferreira, Z.F.; Muscará, M.N.; Teixeira, S.; Farsky, S.H.P. Intracellular mechanisms of hydroquinone toxicity on endotoxin-activated neutrophils. Arch. Toxicol. 2012, 86, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Pyatt, D.W.; Stillman, W.S.; Irons, R.D. Hydroquinone, a Reactive Metabolite of Benzene, Inhibits NF-κB in Primary Human CD4+T Lymphocytes. Toxicol. Appl. Pharmacol. 1998, 149, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Proske, M.; Duffey, M.; Stangl, H.; Martinez, G.F.; Peters, N.; Kraske, A.; Rainer, H.S.; Bethea, J.R.; Kontermann, R.E.; et al. Selective activation of tumor necrosis factor receptor II induces antiinflamatory re-sponses and alleviates experimental arthritis. Arthritis Rheum. 2018, 70, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Santinon, F.; Batignes, M.; Mebrek, M.L.; Biton, J.; Clavel, G.; Hervé, R.; Lemeiter, D.; Breckler, M.; Busato, F.; Tost, J.; et al. Involvement of Tumor Necrosis Factor Receptor Type II in FoxP3 Stability and as a Marker of Treg Cells Specifically Expanded by Anti–Tumor Necrosis Factor Treatments in Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Baka, Z.; Buzás, E.; Nagy, G. Rheumatoid arthritis and smoking: Putting the pieces together. Arthritis Res. Ther. 2009, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Okamoto, H.; Iwamoto, T.; Toyama, Y.; Tomatsu, T.; Yamanaka, H.; Momohara, S. A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology 2008, 47, 1317–1322. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nakahama, T.; Kishimoto, T. Aryl hydrocarbon receptor and experimental autoimmune arthritis. Semin. Immunopathol. 2013, 35, 637–644. [Google Scholar] [CrossRef]
- Lahoti, T.S.; John, K.; Hughes, J.M.; Kusnadi, A.; Murray, I.A.; Krishnegowda, G.; Amin, S.; Perdew, G.H. Aryl hydrocarbon receptor antagonism mitigates cytokine-mediated inflammatory signalling in primary human fibroblast-like synoviocytes. Ann. Rheum. Dis. 2013, 72, 1708–1716. [Google Scholar] [CrossRef]
- Abiko, Y.; Puga, A.; Kumagai, Y. Covalent binding of quinones activates the Ah receptor in Hepa1c1c7 cells. J. Toxicol. Sci. 2015, 40, 873–886. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Yoon, B.-I.; Li, G.-X.; Fujii-Kuriyama, Y.; Kaneko, T.; Kanno, J.; Inoue, T. Benzene-induced hematopoietic toxicity transmitted by AhR in wild-type mouse and nullified by repopulation with AhR-deficient bone marrow cells: Time after benzene treatment and recovery. Chemosphere 2008, 73, S290–S294. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Inoue, T. Benzene-induced bone-marrow toxicity: A hematopoietic stem-cell-specific, aryl hydrocarbon receptor-mediated adverse effect. Chem. Interact. 2010, 184, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.-I.; Hirabayashi, Y.; Kawasaki, Y.; Kodama, Y.; Kaneko, T.; Kanno, J.; Kim, D.-Y.; Fujii-Kuriyama, Y.; Inoue, T. Aryl hydrocarbon receptor mediates benzene-induced hematotoxicity. Toxicol. Sci. 2002, 70, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Fortunati, N.; Guaraldi, F.; Zunino, V.; Penner, F.; D’Angelo, V.; Zenga, F.; Giraldi, F.P.; Catalano, M.G.; Arvat, E. Effects of environmental pollutants on signaling pathways in rat pituitary GH3 adenoma cells. Environ. Res. 2017, 158, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Tapella, L.; Sesta, A.; Cassarino, M.F.; Zunino, V.; Catalano, M.G.; Giraldi, F.P. Benzene and 2-ethyl-phthalate induce proliferation in normal rat pituitary cells. Pituitary 2016, 20, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Badham, H.J.; Winn, L.M. Investigating the role of the aryl hydrocarbon receptor in benzene-initiated toxicity in vitro. Toxicology 2007, 229, 177–185. [Google Scholar] [CrossRef]
- Desai, P.B.; Manjunath, S.; Kadi, S.; Chetana, K.; Vanishree, J. Oxidative stress and enzymatic antioxidant status in rheumatoid arthritis: A case control study. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 959–967. [Google Scholar]
- Brand, D.D. Rodent models of rheumatoid arthritis. Comp. Med. 2005, 55, 114–122. [Google Scholar]
- Nickdel, M.; Conigliaro, P.; Valesini, G.; Hutchison, S.; Benson, R.; Bundick, R.; Leishman, A.; McInnes, I.; Brewer, J.; Garside, P. Dissecting the contribution of innate and antigen-specific pathways to the breach of self-tolerance observed in a murine model of arthritis. Ann. Rheum. Dis. 2008, 68, 1059–1066. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nat. Cell Biol. 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Miyashita, T.; Yamamoto, Y.; Munesue, S.; Harashima, A.; Takayama, H.; Fushida, S.; Ohta, T. Citrullinated Histone H3: Early Biomarker of Neutrophil Extracellular Traps in Septic Liver Damage. J. Surg. Res. 2019, 234, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Tsourouktsoglou, T.-D.; Warnatsch, A.; Ioannou, M.; Hoving, D.; Wang, Q.; Papayannopoulos, V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020, 31, 107602. [Google Scholar] [CrossRef] [PubMed]
- Sohn, D.H.; Rhodes, C.; Onuma, K.; Zhao, X.; Sharpe, O.; Gazitt, T.; Shiao, R.; Fert-Bober, J.; Cheng, D.; Lahey, L.J.; et al. Local Joint Inflammation and Histone Citrullination in a Murine Model of the Transition from Preclinical Autoimmunity to Inflammatory Arthritis. Arthritis Rheumatol. 2015, 67, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lin, C.; Leso, A.; Nefedova, Y. Quantification of Citrullinated Histone H3 Bound DNA for Detection of Neutrophil Extracellular Traps. Cancers 2020, 12, 3424. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heluany, C.S.; Donate, P.B.; Schneider, A.H.; Fabris, A.L.; Gomes, R.A.; Villas-Boas, I.M.; Tambourgi, D.V.; Silva, T.A.d.; Trossini, G.H.G.; Nalesso, G.; et al. Hydroquinone Exposure Worsens Rheumatoid Arthritis through the Activation of the Aryl Hydrocarbon Receptor and Interleukin-17 Pathways. Antioxidants 2021, 10, 929. https://doi.org/10.3390/antiox10060929
Heluany CS, Donate PB, Schneider AH, Fabris AL, Gomes RA, Villas-Boas IM, Tambourgi DV, Silva TAd, Trossini GHG, Nalesso G, et al. Hydroquinone Exposure Worsens Rheumatoid Arthritis through the Activation of the Aryl Hydrocarbon Receptor and Interleukin-17 Pathways. Antioxidants. 2021; 10(6):929. https://doi.org/10.3390/antiox10060929
Chicago/Turabian StyleHeluany, Cintia Scucuglia, Paula Barbim Donate, Ayda Henriques Schneider, André Luis Fabris, Renan Augusto Gomes, Isadora Maria Villas-Boas, Denise Vilarinho Tambourgi, Tarcilia Aparecida da Silva, Gustavo Henrique Goulart Trossini, Giovanna Nalesso, and et al. 2021. "Hydroquinone Exposure Worsens Rheumatoid Arthritis through the Activation of the Aryl Hydrocarbon Receptor and Interleukin-17 Pathways" Antioxidants 10, no. 6: 929. https://doi.org/10.3390/antiox10060929
APA StyleHeluany, C. S., Donate, P. B., Schneider, A. H., Fabris, A. L., Gomes, R. A., Villas-Boas, I. M., Tambourgi, D. V., Silva, T. A. d., Trossini, G. H. G., Nalesso, G., Silveira, E. L. V., Cunha, F. Q., & Farsky, S. H. P. (2021). Hydroquinone Exposure Worsens Rheumatoid Arthritis through the Activation of the Aryl Hydrocarbon Receptor and Interleukin-17 Pathways. Antioxidants, 10(6), 929. https://doi.org/10.3390/antiox10060929