
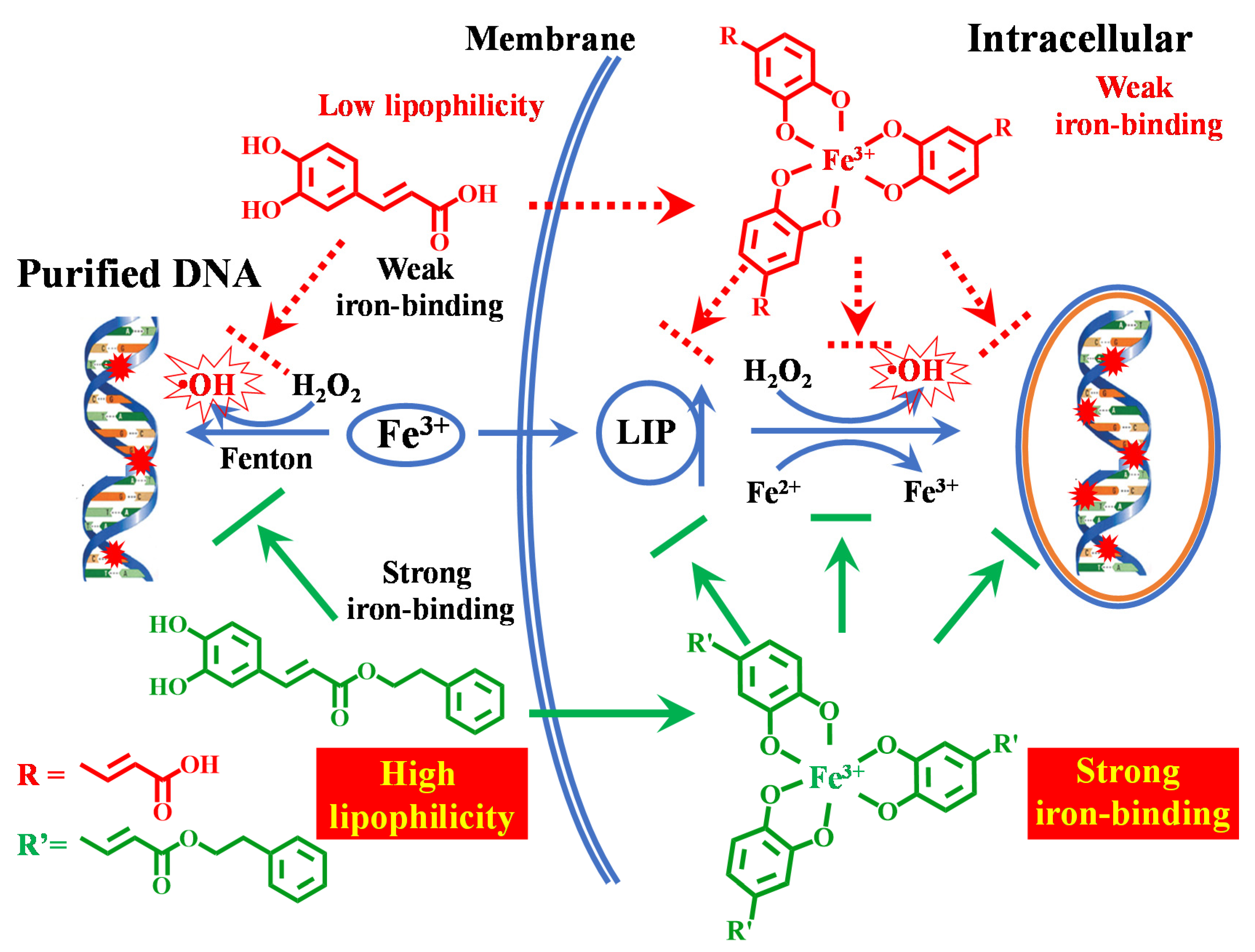
Caffeic Acid Phenyl Ester (CAPE) Protects against Iron-Mediated Cellular DNA Damage through Its Strong Iron-Binding Ability and High Lipophilicity

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Immunofluorescence Analysis of Intracellular γ-H2AX Generation in HeLa Cells
2.3. Labile Iron Pool (LIP) Determination
2.4. DNA Strand Breakage
2.5. Electron Spin Resonance (ESR) Spin-Trapping Studies
2.6. UV–Visible Spectra Measurement
2.7. Calcein Fluorescence Detection
2.8. Fourier Transform Ion Cyclotron Resonance Mass (FT-ICR-MS) Detection
2.9. Measurement of the Fe(III)-Binding Affinity with Ligands Including CAPE, CA, CAME, and CAEE
2.10. Cyclic Voltammetry (CV) Measurement
2.11. Partitioning Study
2.12. Statistical Analysis
3. Results and Discussion
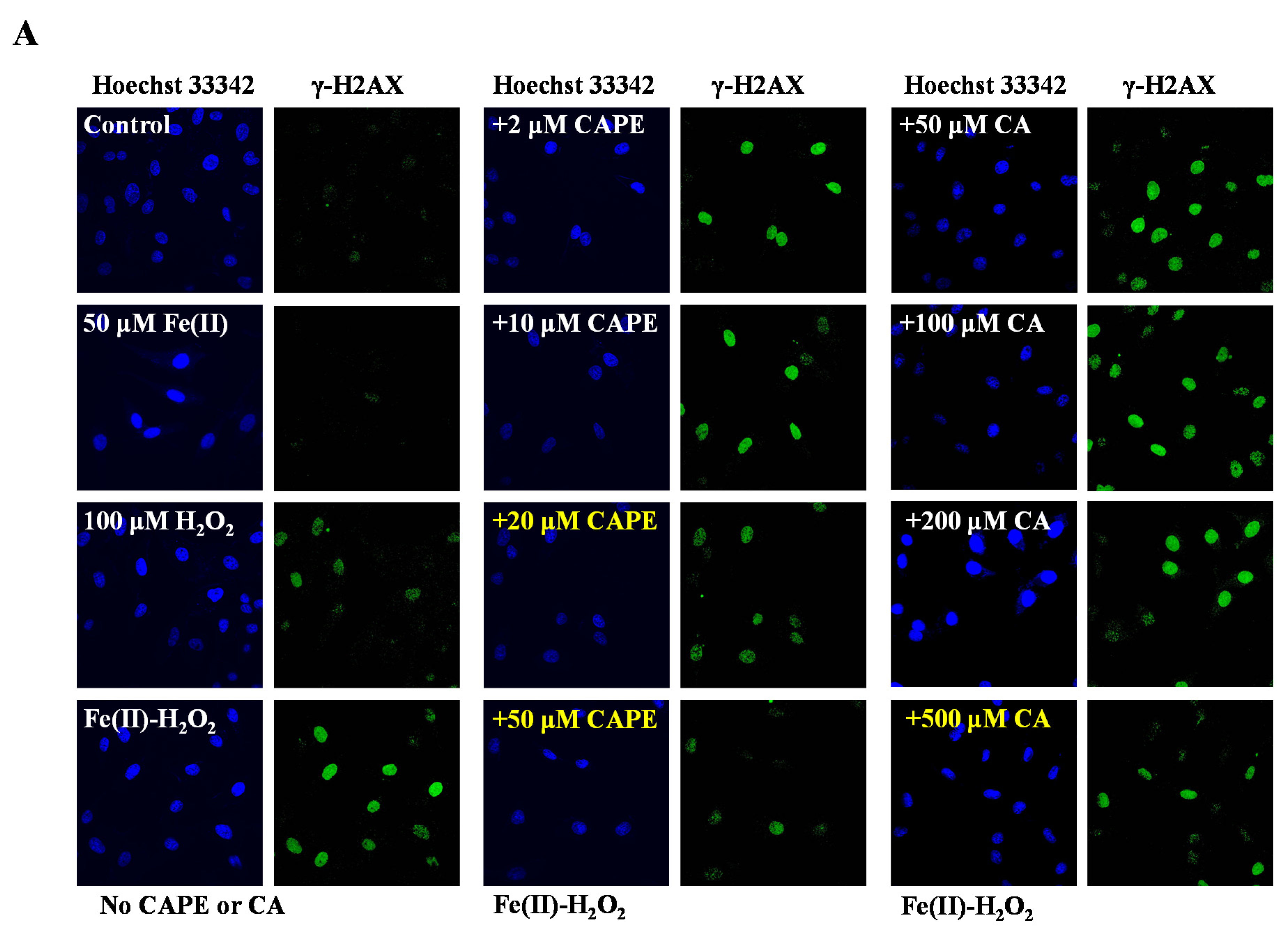
3.1. CAPE Was Found to Be the Most Potent in Protecting Against Iron-Mediated Cellular DNA Damage
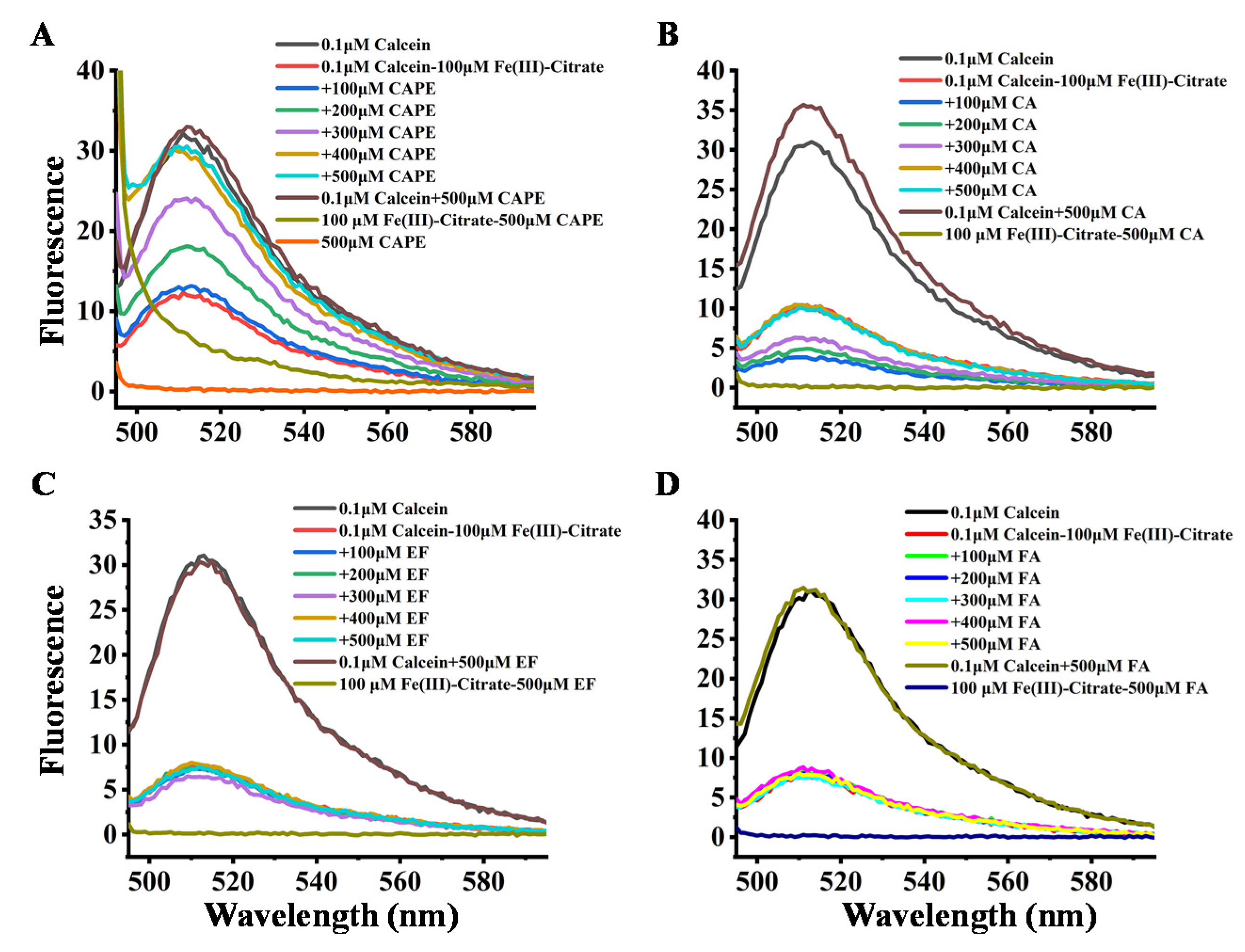
3.2. CAPE Was Found to Be the Most Potent in Decreasing LIP Levels Induced by Iron Overload
3.3. CAPE Was Found to Be More Effective Than CA in Protecting Against Iron-Mediated DNA Damage as Measured by DNA Strand Breaks
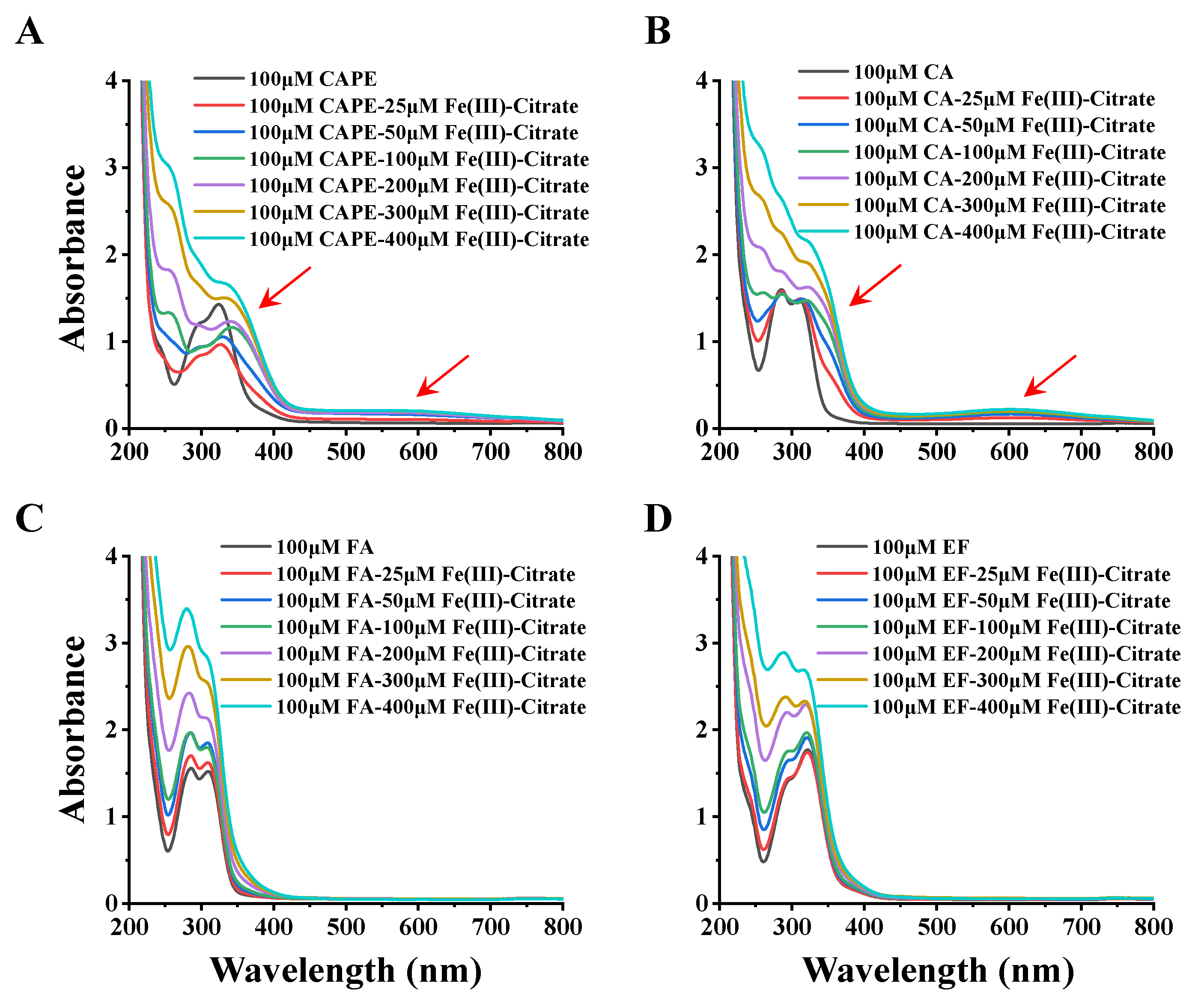
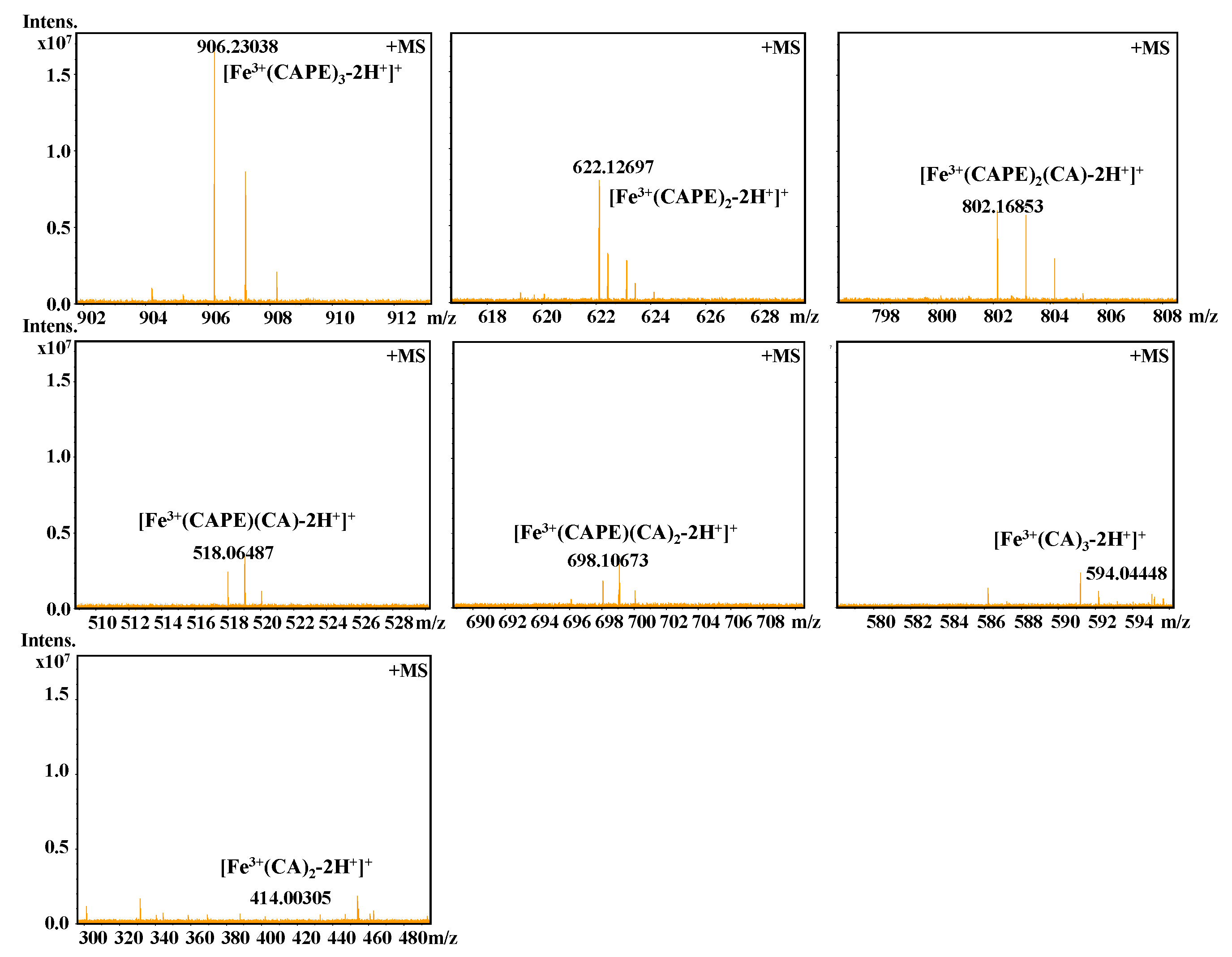
3.4. CAPE Was Found to Have Strong Iron-Binding Ability, Leading to the Formation of Redox-Inactive Iron–CAPE Complex
3.5. High Lipophilicity of CAPE Enhanced Its Protective Effect Against Iron-Mediated Cellular DNA Damage
3.6. Iron-Chelating Ability of CAPE, but Not Its Radical Scavenging Ability, Is Responsible for Its Protection Against Iron-Mediated DNA Damage
3.7. Potential Biological Implications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fonseca-Nunes, A.; Jakszyn, P.; Agudo, A. Iron and Cancer Risk-A Systematic Review and Meta-analysis of the Epidemiological Evidence. Cancer Epidemiol. Biomark. Prev. 2014, 23, 12–31. [Google Scholar] [CrossRef]
- Zhao, B.L.; Yang, Y.; Wang, X.L.; Chong, Z.C.; Yin, R.C.; Song, S.H.; Zhao, C.; Li, C.P.; Huang, H.; Sun, B.F.; et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and Tet-dependent mechanism. Nucleic Acids Res. 2014, 42, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.Z.; Frei, B. Biochemical and physiological interactions of vitamin C and iron: Pro- or antioxidant? In The Antioxidant Vitamins C and E; Packer, L., Traber, M.G., Kraemer, K., Frei, B., Eds.; Amer Oil Chemists Soc: Champaign, IL, USA, 2002; pp. 32–49. [Google Scholar]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Munoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Jankowska, E.A.; van Veldhuisen, D.J.; Ponikowski, P.; Anker, S.D. Iron deficiency and cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Doulias, P.T.; Nousis, L.; Zhu, B.Z.; Frei, B.; Galaris, D. Protection by tropolones against H2O2-induced DNA damage and apoptosis in cultured Jurkat cells. Free Radic. Res. 2005, 39, 125–135. [Google Scholar] [CrossRef]
- Barbouti, A.; Doulias, P.T.; Zhu, B.Z.; Frei, B.; Galaris, D. Intracellular iron, but not copper, plays a critical role in hydrogen peroxide-induced DNA damage. Free Radic. Biol. Med. 2001, 31, 490–498. [Google Scholar] [CrossRef]
- Galaris, D.; Zhu, B.Z.; Frei, B. On the role of iron and copper ions in hydrogen peroxide-induced cellular DNA damage. Free Radic. Biol. Med. 2002, 32, 198–199. [Google Scholar] [CrossRef]
- Suh, J.; Zhu, B.Z.; Frei, B. Ascorbate does not act as a pro-oxidant towards lipids and proteins in human plasma exposed to redox-active transition metal ions and hydrogen peroxide. Free Radic. Biol. Med. 2003, 34, 1306–1314. [Google Scholar] [CrossRef]
- Levy, S.; Shechtman, S.; Zhu, B.Z.; Stadtman, E.R.; Stadler, R.; Chevion, M. Synergism between the toxicity of chlorophenols and iron complexes. Environ. Toxicol. Chem. 2007, 26, 218–224. [Google Scholar] [CrossRef]
- Qu, N.; Guo, L.H.; Zhu, B.Z. An electrochemical biosensor for the detection of tyrosine oxidation induced by Fenton reaction. Biosens. Bioelectron. 2011, 26, 2292–2296. [Google Scholar] [CrossRef]
- Chevion, M.; Berenshtein, E.; Zhu, B.Z. The Role of Transition Metal Ions in Free Radical-Mediated Damage. In Reactive Oxygen Species in Biological Systems: An Interdisciplinary Approach; Gilbert, D.L., Colton, C.A., Eds.; Springer US: Boston, MA, USA, 2002; pp. 103–131. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Gimeno, C.J.; Ames, B.N. Urinary 8-hydroxy-2’-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc. Natl. Acad. Sci. USA 1989, 86, 9697–9701. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Perri, L.; Nocella, C.; Violi, F. Antioxidant and antiplatelet activity by polyphenol-rich nutrients: Focus on extra virgin olive oil and cocoa. Br. J. Clin. Pharmacol. 2017, 83, 96–102. [Google Scholar] [CrossRef]
- Murador, D.; Braga, A.R.; Da Cunha, D.; De Rosso, V. Alterations in phenolic compound levels and antioxidant activity in response to cooking technique effects: A meta-analytic investigation. Crit. Rev. Food Sci. Nutr. 2018, 58, 169–177. [Google Scholar] [CrossRef]
- Psomiadou, E.; Tsimidou, M. Stability of virgin olive oil. 1. Autoxidation studies. J. Agric. Food Chem. 2002, 50, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Hanasaki, Y.; Ogawa, S.; Fukui, S. The correlation between active oxygens scavenging and antioxidative effects of flavonoids. Free Radic. Biol. Med. 1994, 16, 845–850. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Wei, X.L.; Fang, Y.P.; Gan, R.Y.; Wang, M.; Ge, Y.Y.; Zhang, D.; Cheng, L.Z.; Corke, H. Nanochemoprevention with therapeutic benefits: An updated review focused on epigallocatechin gallate delivery. Crit. Rev. Food Sci. Nutr. 2020, 60, 1243–1264. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.Y.; Wang, W.W.; Ye, J.F.; Li, S.S.; Wu, Q.; Yin, D.D.; Li, B.; Xu, Y.J.; Wang, L.S. Polyphenol profile and antioxidant activity of the fruit and leaf of Vaccinium glaucoalbum from the Tibetan Himalayas. Food Chem. 2017, 219, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.Z.; Mao, L.; Fan, R.M.; Zhu, J.G.; Zhang, Y.N.; Wang, J.; Kalyanaraman, B.; Frei, B. Ergothioneine Prevents Copper-Induced Oxidative Damage to DNA and Protein by Forming a Redox-Inactive Ergothioneine-Copper Complex. Chem. Res. Toxicol. 2011, 24, 30–34. [Google Scholar] [CrossRef]
- Sheng, Z.G.; Li, Y.; Fan, R.M.; Chao, X.J.; Zhu, B.Z. Lethal synergism between organic and inorganic wood preservatives via formation of an unusual lipophilic ternary complex. Toxicol. Appl. Pharmacol. 2013, 266, 335–344. [Google Scholar] [CrossRef]
- Mao, L.; Huang, C.H.; Shao, J.; Qin, L.; Xu, D.; Shao, B.; Zhu, B.Z. An unexpected antioxidant and redox activity for the classic copper-chelating drug penicillamine. Free Radic. Biol. Med. 2020, 147, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Lopes, G.K.; Schulman, H.M.; Hermes-Lima, M. Polyphenol tannic acid inhibits hydroxyl radical formation from Fenton reaction by complexing ferrous ions. Biochim. Biophys. Acta BBA Gen. Subj. 1999, 1472, 142–152. [Google Scholar] [CrossRef]
- Sestili, P.; Diamantini, G.; Bedini, A.; Cerioni, L.; Tommasini, I.; Tarzia, G.; Cantoni, O. Plant-derived phenolic compounds prevent the DNA single-strand breakage and cytotoxicity induced by tert-butylhydroperoxide via an iron-chelating mechanism. Biochem. J. 2002, 364, 121–128. [Google Scholar] [CrossRef]
- Alam, M.A.; Subhan, N.; Hossain, H.; Hossain, M.; Reza, H.M.; Rahman, M.M.; Ullah, M.O. Hydroxycinnamic acid derivatives: A potential class of natural compounds for the management of lipid metabolism and obesity. Nutr. Metab. 2016, 13, 13. [Google Scholar] [CrossRef]
- Bijalwan, V.; Ali, U.; Kesarwani, A.K.; Yadav, K.; Mazumder, K. Hydroxycinnamic acid bound arabinoxylans from millet brans-structural features and antioxidant activity. Int. J. Biol. Macromol. 2016, 88, 296–305. [Google Scholar] [CrossRef]
- Firat, F.; Ozgul, M.; Uluer, E.T.; Inan, S. Effects of caffeic acid phenethyl ester (CAPE) on angiogenesis, apoptosis and oxidative stress in various cancer cell lines. Biotech. Histochem. 2019, 94, 491–497. [Google Scholar] [CrossRef]
- Kuo, Y.Y.; Jim, W.T.; Su, L.C.; Chung, C.J.; Lin, C.Y.; Huo, C.; Tseng, J.C.; Huang, S.H.; Lai, C.J.; Chen, B.C.; et al. Caffeic Acid Phenethyl Ester Is a Potential Therapeutic Agent for Oral Cancer. Int. J. Mol. Sci. 2015, 16, 10748–10766. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, A.; Koltuksuz, U.; Ozen, S.; Uz, E.; Ciralik, H.; Akyol, O. The effects of caffeic acid phenethyl ester (CAPE) on spinal cord ischemia/reperfusion injury in rabbits. Eur. J. Cardiothorac. Surg. 1999, 16, 458–463. [Google Scholar] [CrossRef]
- Irmak, M.K.; Fadillioglu, E.; Sogut, S.; Erdogan, H.; Gulec, M.; Ozer, M.; Yagmurca, M.; Gozukara, M.E. Effects of caffeic acid phenethyl ester and alpha-tocopherol on reperfusion injury in rat brain. Cell Biochem. Funct. Cell. Biochem. Its Modul. Act. Agents Dis. 2003, 21, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Longo, R.; Vanella, A. Antioxidant activity of propolis: Role of caffeic acid phenethyl ester and galangin. Fitoterapia 2002, 73, S21–S29. [Google Scholar] [CrossRef]
- Xu, D.; Huang, C.H.; Xie, L.N.; Shao, B.; Mao, L.; Shao, J.; Kalyanaraman, B.; Zhu, B.Z. Mechanism of unprecedented hydroxyl radical production and site-specific oxidative DNA damage by photoactivation of the classic arylhydroxamic acid carcinogens. Carcinogenesis 2019, 40, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Mao, L.; Shao, J.; Huang, C.H.; Qin, L.; Huang, R.; Sheng, Z.G.; Cao, D.; Zhang, Z.Q.; Lin, L.; et al. Mechanism of synergistic DNA damage induced by caffeic acid phenethyl ester (CAPE) and Cu(II): Competitive binding between CAPE and DNA with Cu(II)/Cu(I). Free Radic. Biol. Med. 2020, 159, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Chao, X.J.; Tang, M.; Huang, R.; Huang, C.H.; Shao, J.; Yan, Z.Y.; Zhu, B.Z. Targeted live-cell nuclear delivery of the DNA ‘light-switching’ Ru(II) complex via ion-pairing with chlorophenolate counter-anions: The critical role of binding stability and lipophilicity of the ion-pairing complexes. Nucleic Acids Res. 2019, 47, 10520–10528. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Tang, M.; Huang, C.H.; Chao, X.J.; Yan, Z.Y.; Shao, J.; Zhu, B.Z. What Are the Major Physicochemical Factors in Determining the Preferential Nuclear Uptake of the DNA "Light-Switching" Ru(II)-Polypyridyl Complex in Live Cells via Ion-Pairing with Chlorophenolate Counter-Anions? J. Phys. Chem. Lett. 2019, 10, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Jacobs, A. An intracellular transit iron pool. Ciba Found. Symp. 1976, 91–106. [Google Scholar] [CrossRef]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Jacobs, A. Low molecular weight intracellular iron transport compounds. Blood 1977, 50, 433–439. [Google Scholar] [CrossRef]
- Grootveld, M.; Bell, J.D.; Halliwell, B.; Aruoma, O.; Bomford, A.; Sadler, P.J. Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy. J. Biol. Chem. 1989, 264, 4417–4422. [Google Scholar] [CrossRef]
- Adam, F.I.; Bounds, P.L.; Kissner, R.; Koppenol, W.H. Redox properties and activity of iron-citrate complexes: Evidence for redox cycling. Chem. Res. Toxicol. 2015, 28, 604–614. [Google Scholar] [CrossRef]
- Petrat, F.; Rauen, U.; de Groot, H. Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK. Hepatology 1999, 29, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Breuer, W.; Ermers, M.J.J.; Pootrakul, P.; Abramov, A.; Hershko, C.; Cabantchik, Z.I. Desferrioxamine-chelatable iron, a component of serum non-transferrin-bound iron, used for assessing chelation therapy. Blood 2001, 97, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Breuer, W.; Epsztejn, S.; Millgram, P.; Cabantchik, I.Z. Transport of iron and other transition metals into cells as revealed by a fluorescent probe. Am. J. Physiol. Cell Physiol. 1995, 268, C1354–C1361. [Google Scholar] [CrossRef]
- Gao, G.; Zhang, N.; Wang, Y.-Q.; Wu, Q.; Yu, P.; Shi, Z.-H.; Duan, X.-L.; Zhao, B.-L.; Wu, W.-S.; Chang, Y.-Z. Mitochondrial ferritin protects hydrogen peroxide-induced neuronal cell damage. Aging Dis. 2017, 8, 458. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.P.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.F.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Wang, M.Y.; Fu, X.R.; Peng, Y.; Gao, G.F.; Fan, Y.M.; Duan, X.L.; Zhao, B.L.; Chang, Y.Z.; Shi, Z.H. Neuroprotective effects of ginkgetin against neuroinjury in Parkinson’s disease model induced by MPTP via chelating iron. Free Radic. Res. 2015, 49, 1069–1080. [Google Scholar] [CrossRef]
- Bullock, J.E.; Vagnini, M.T.; Ramanan, C.; Co, D.T.; Wilson, T.M.; Dicke, J.W.; Marks, T.J.; Wasielewski, M.R. Photophysics and redox properties of rylene imide and diimide dyes alkylated ortho to the imide groups. J. Phys. Chem. B 2010, 114, 1794–1802. [Google Scholar] [CrossRef]
- Yang, S.; Bai, G.; Chen, L.; Shen, Q.; Diao, X.; Zhao, G. The interaction of phenolic acids with Fe(III) in the presence of citrate as studied by isothermal titration calorimetry. Food Chem. 2014, 157, 302–309. [Google Scholar] [CrossRef]
- Park, J.; Morimoto, Y.; Lee, Y.M.; You, Y.; Nam, W.; Fukuzumi, S. Scandium ion-enhanced oxidative dimerization and N-demethylation of N,N-dimethylanilines by a non-heme iron(IV)-oxo complex. Inorg. Chem. 2011, 50, 11612–11622. [Google Scholar] [CrossRef]
- Lee, C.Y.; Nanah, C.N.; Held, R.A.; Clark, A.R.; Huynh, U.G.; Maraskine, M.C.; Uzarski, R.L.; McCracken, J.; Sharma, A. Effect of electron donating groups on polyphenol-based antioxidant dendrimers. Biochimie 2015, 111, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Nakanishi, I.; Ohkubo, K.; Ohno, A.; Mizuno, M.; Fukuzumi, S.; Matsumoto, K.I.; Fukuhara, K. Synthesis and radical-scavenging activity of C-methylated fisetin analogues. Bioorg. Med. Chem. 2019, 27, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Yavin, E.; Kikkiri, R.; Gil, S.; Arad-Yellin, R.; Yavin, E.; Shanzer, A. Synthesis and biological evaluation of lipophilic iron chelators as protective agents from oxidative stress. Org. Biomol. Chem. 2005, 3, 2685–2687. [Google Scholar] [CrossRef] [PubMed]
- Gerogianni, P.S.; Chatziathanasiadou, M.V.; Diamantis, D.A.; Tzakos, A.G.; Galaris, D. Lipophilic ester and amide derivatives of rosmarinic acid protect cells against H2O2-induced DNA damage and apoptosis: The potential role of intracellular accumulation and labile iron chelation. Redox Biol. 2018, 15, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Imbert, D.; Baret, P.; Gaude, D.; Gautier-Luneau, I.; Gellon, G.; Thomas, F.; Serratrice, G.; Pierre, J.L. Hydrophilic and lipophilic iron chelators with the same complexing abilities. Chem. Eur. J. 2002, 8, 1091–1100. [Google Scholar] [CrossRef]
- Kitsati, N.; Fokas, D.; Ouzouni, M.-D.; Mantzaris, M.D.; Barbouti, A.; Galaris, D. Lipophilic caffeic acid derivatives protect cells against H2O2-Induced DNA damage by chelating intracellular labile iron. J. Agric. Food Chem. 2012, 60, 7873–7879. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Giménez, R. Beneficial effects of green tea—A review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef]
- Vinson, J.A.; Su, X.; Zubik, L.; Bose, P. Phenol antioxidant quantity and quality in foods: Fruits. J. Agric. Food Chem. 2001, 49, 5315–5321. [Google Scholar] [CrossRef]
- Vinson, J.A.; Hao, Y.; Su, X.; Zubik, L. Phenol antioxidant quantity and quality in foods: Vegetables. J. Agric. Food Chem. 1998, 46, 3630–3634. [Google Scholar] [CrossRef]
- Vinson, J.A.; Proch, J.; Zubik, L. Phenol antioxidant quantity and quality in foods: Cocoa, dark chocolate, and milk chocolate. J. Agric. Food Chem. 1999, 47, 4821–4824. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.Z.; Chao, X.J.; Huang, C.H.; Li, Y. Delivering the cell-impermeable DNA ’light-switching’ Ru(II) complexes preferentially into live-cell nucleus via an unprecedented ion-pairing method. Chem. Sci. 2016, 7, 4016–4023. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.M.; Lu, L.; Long, Y.; Wang, T.; Liu, L.; Chen, Q.; Wang, R. Free radical scavenging and antioxidative activities of caffeic acid phenethyl ester (CAPE) and its related compounds in solution and membranes: A structure-activity insight. Food Chem. 2007, 105, 107–115. [Google Scholar] [CrossRef]
- Rice-evans, C.A.; Miller, N.J.; Bolwell, P.G.; Bramley, P.M.; Pridham, J.B. The relative antioxidant activities of plant-derived polyphenolic flavonoids. Free Radic. Res. 1995, 22, 375–383. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Role of free radicals and catalytic metal ions in human disease: An overview. Methods Enzymol. 1990, 186, 1–85. [Google Scholar]
- Khokhar, S.; Apenten, R.K.O. Iron binding characteristics of phenolic compounds: Some tentative structure–activity relations. Food Chem. 2003, 81, 133–140. [Google Scholar] [CrossRef]
- Sestili, P.; Guidarelli, A.; Dachà, M.; Cantoni, O. Quercetin prevents DNA single strand breakage and cytotoxicity caused by tert-butylhydroperoxide: Free radical scavenging versus iron chelating mechanism. Free Radic. Biol. Med. 1998, 25, 196–200. [Google Scholar] [CrossRef]
- Aherne, S.A.; O’Brien, N.M. Mechanism of protection by the flavonoids, quercetin and rutin, against tert-butylhydroperoxide-and menadione-induced DNA single strand breaks in Caco-2 cells. Free Radic. Biol. Med. 2000, 29, 507–514. [Google Scholar] [CrossRef]
- Coleman, J.B.; Gilfor, D.; Farber, J.L. Dissociation of the accumulation of single-strand breaks in DNA from the killing of cultured hepatocytes by an oxidative stress. Mol. Pharmacol. 1989, 36, 193–200. [Google Scholar]
- Byrnes, R.W. Inhibition of hydroperoxide-induced DNA single-strand breakage by 1, 10-phenanthroline in HL-60 cells: Implications for iron speciation. Arch. Biochem. Biophys. 1996, 332, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D. Iron chelators as therapeutic agents for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 42, 267–281. [Google Scholar] [CrossRef]
- Richardson, D. Potential of iron chelators as effective antiproliferative agents. Can. J. Physiol. Pharmacol. 1997, 75, 1164–1180. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.L.; Greene, B.T.; Turner, J.; Torti, F.M.; Torti, S.V. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem. 2004, 4, 1623–1635. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | LogKow |
|---|---|
| CAPE | 2.01 ± 0.02 |
| CA | −0.98 ± 0.02 |
| CAME | 1.27 ± 0.06 |
| CAEE | 1.51 ± 0.03 |
| EF | 1.38 ± 0.02 |
| FA | −0.77 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, B.; Mao, L.; Tang, M.; Yan, Z.-Y.; Shao, J.; Huang, C.-H.; Sheng, Z.-G.; Zhu, B.-Z. Caffeic Acid Phenyl Ester (CAPE) Protects against Iron-Mediated Cellular DNA Damage through Its Strong Iron-Binding Ability and High Lipophilicity. Antioxidants 2021, 10, 798. https://doi.org/10.3390/antiox10050798
Shao B, Mao L, Tang M, Yan Z-Y, Shao J, Huang C-H, Sheng Z-G, Zhu B-Z. Caffeic Acid Phenyl Ester (CAPE) Protects against Iron-Mediated Cellular DNA Damage through Its Strong Iron-Binding Ability and High Lipophilicity. Antioxidants. 2021; 10(5):798. https://doi.org/10.3390/antiox10050798
Chicago/Turabian StyleShao, Bo, Li Mao, Miao Tang, Zhu-Ying Yan, Jie Shao, Chun-Hua Huang, Zhi-Guo Sheng, and Ben-Zhan Zhu. 2021. "Caffeic Acid Phenyl Ester (CAPE) Protects against Iron-Mediated Cellular DNA Damage through Its Strong Iron-Binding Ability and High Lipophilicity" Antioxidants 10, no. 5: 798. https://doi.org/10.3390/antiox10050798
APA StyleShao, B., Mao, L., Tang, M., Yan, Z.-Y., Shao, J., Huang, C.-H., Sheng, Z.-G., & Zhu, B.-Z. (2021). Caffeic Acid Phenyl Ester (CAPE) Protects against Iron-Mediated Cellular DNA Damage through Its Strong Iron-Binding Ability and High Lipophilicity. Antioxidants, 10(5), 798. https://doi.org/10.3390/antiox10050798