
Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model

 , ,
, ,
Abstract
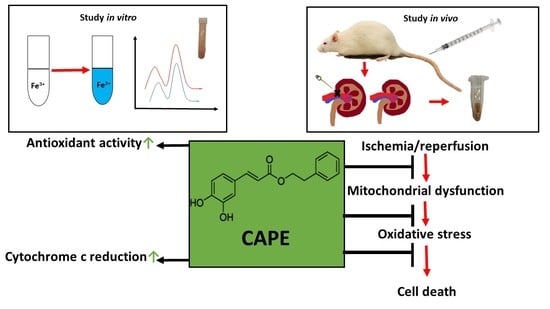
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Model
2.2. Preparation of Isolated Kidney Mitochondria
2.3. Measurement of Mitochondrial Respiration
2.4. Measurement of Antioxidant Activity of CAPE
2.5. Measurement of Cytochrome c Reduction Level of CAPE
2.6. Measurement of Mitochondrial Respiratory Chain Complex I, II+III, II (SDH)
2.7. Measurement of H2O2 Generation in Kidney Mitochondria
2.8. Measurement of Lactate Dehydrogenase Activity
2.9. Statistical Analysis
3. Results
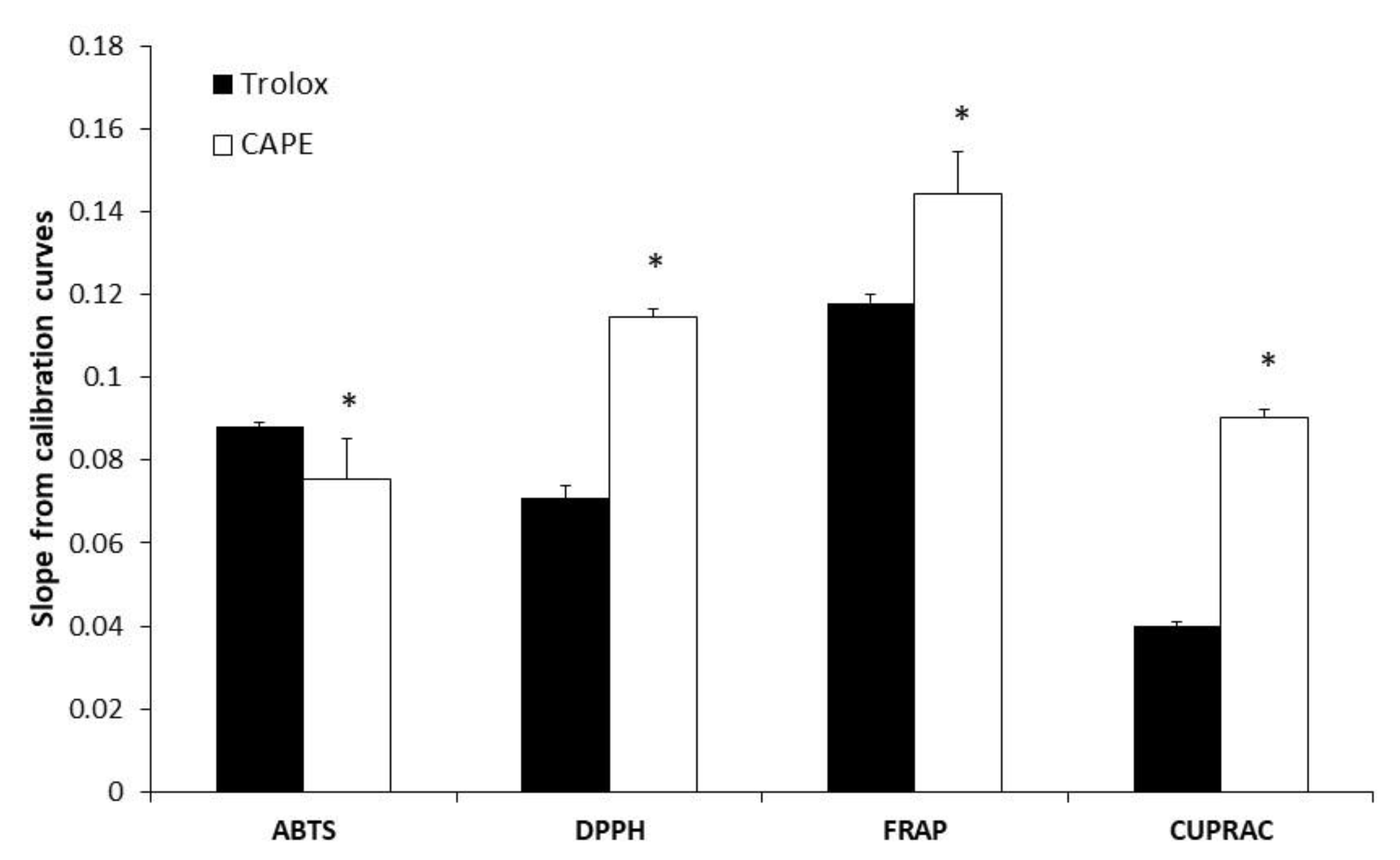
3.1. Evaluation of Antioxidant (Antiradical and Reducing) Activity of CAPE
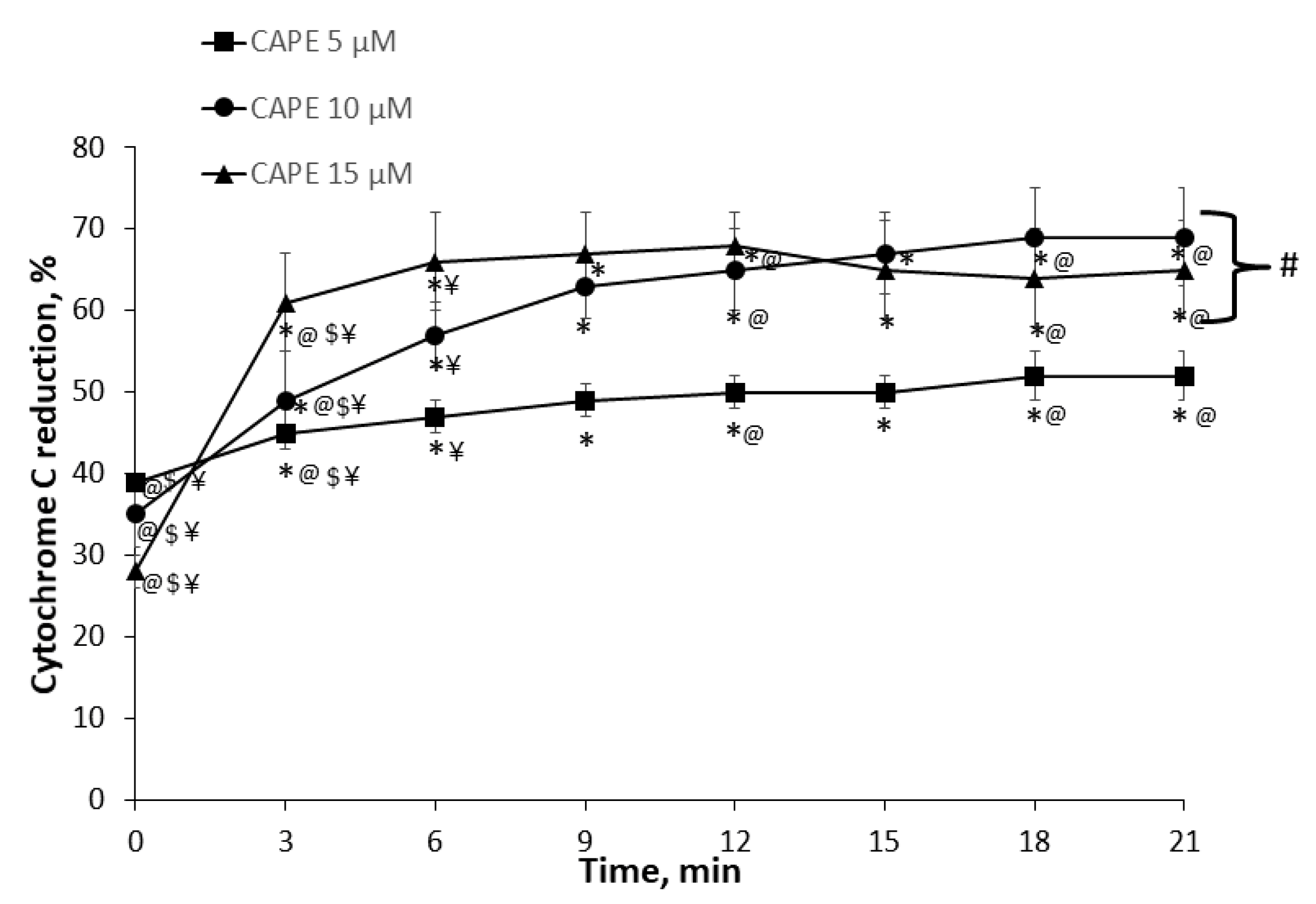
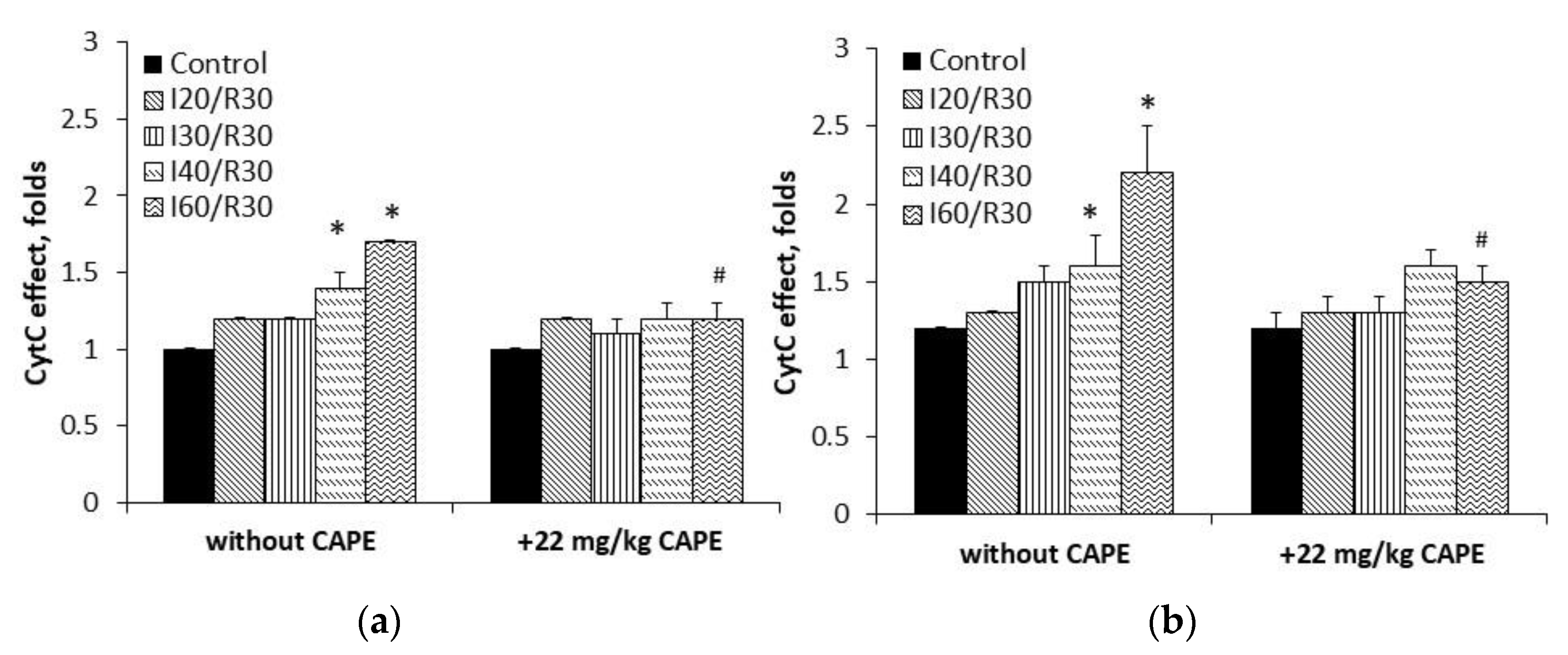
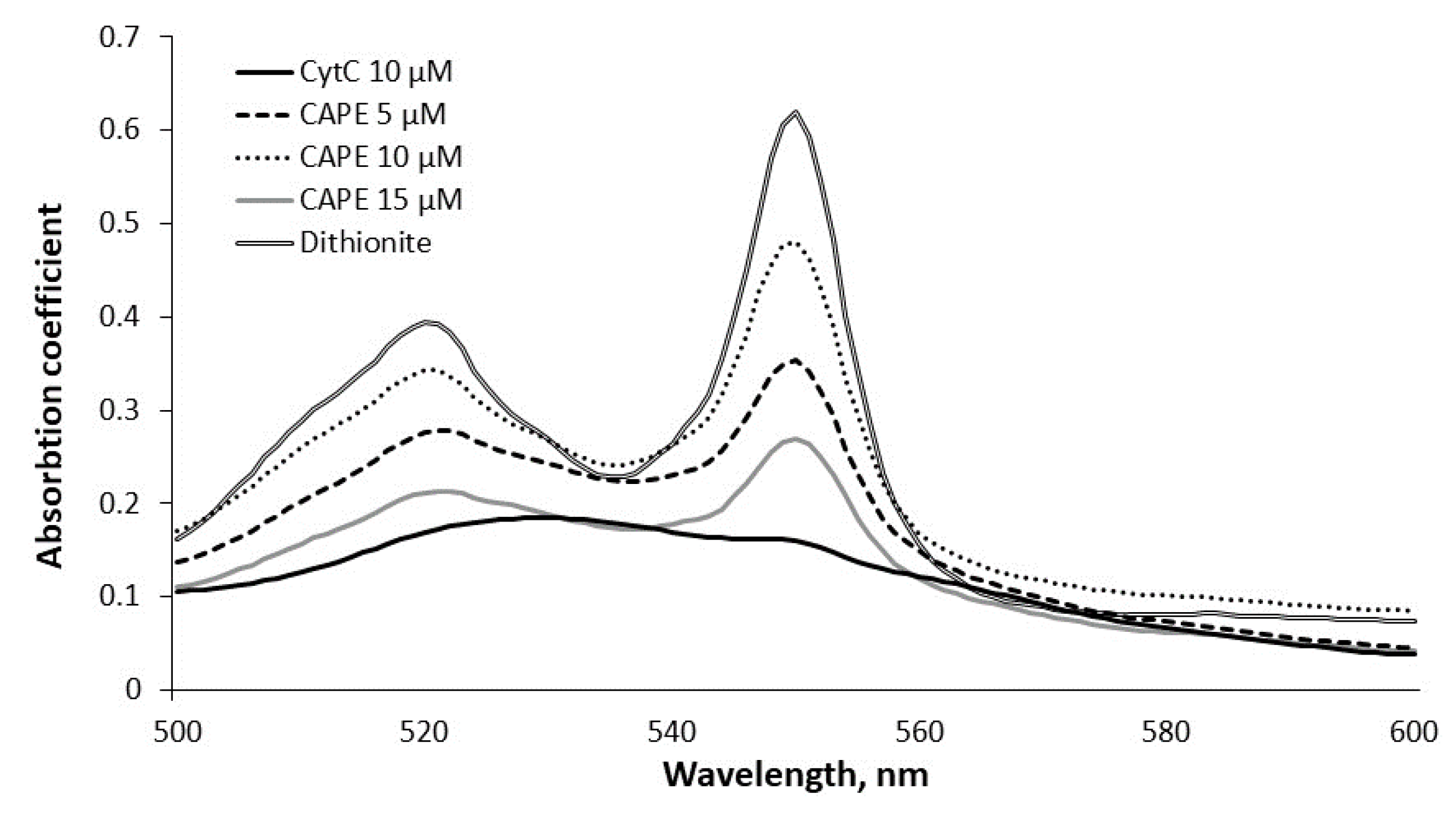
3.2. Measurement of Cytochrome c Reduction Level of CAPE
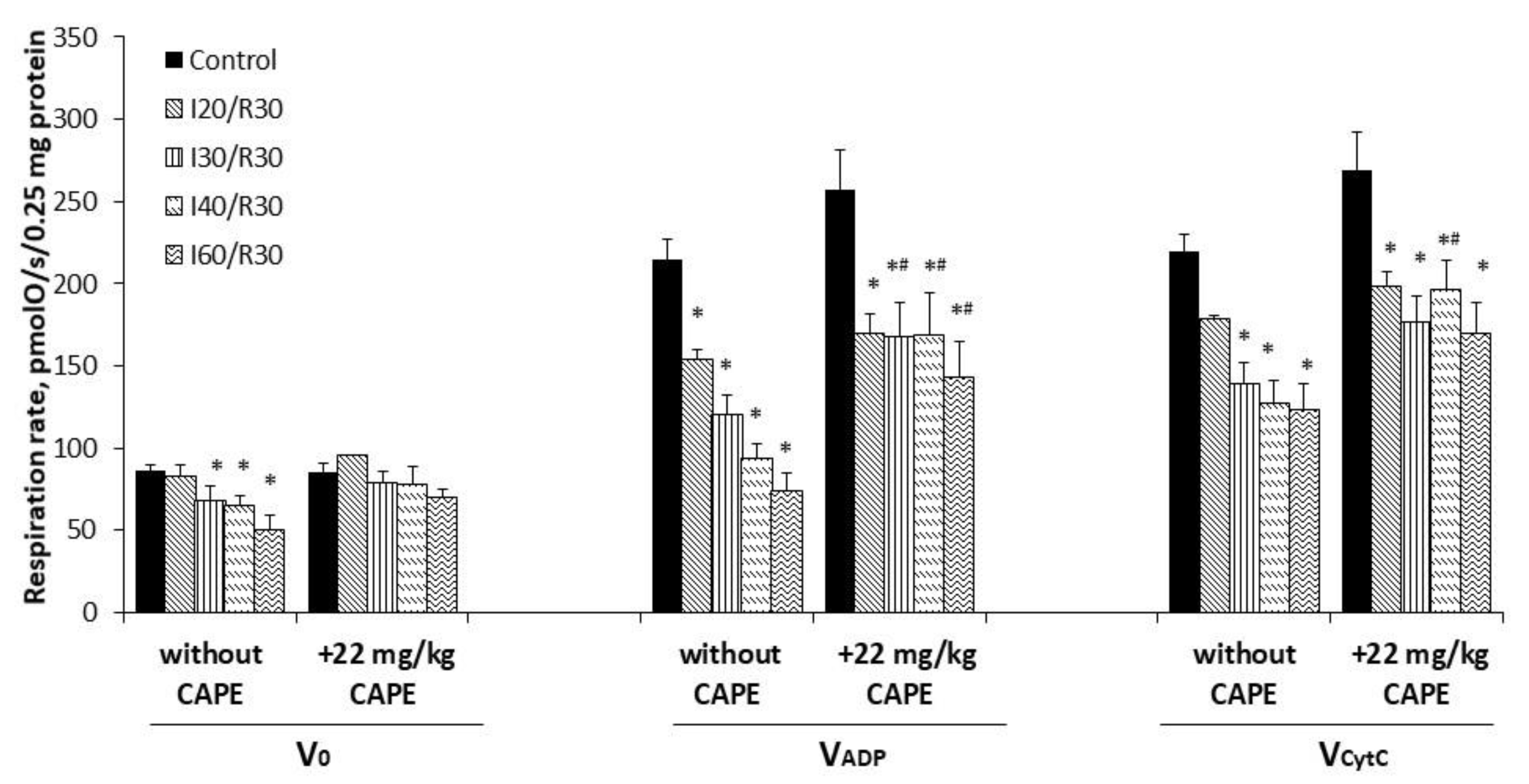
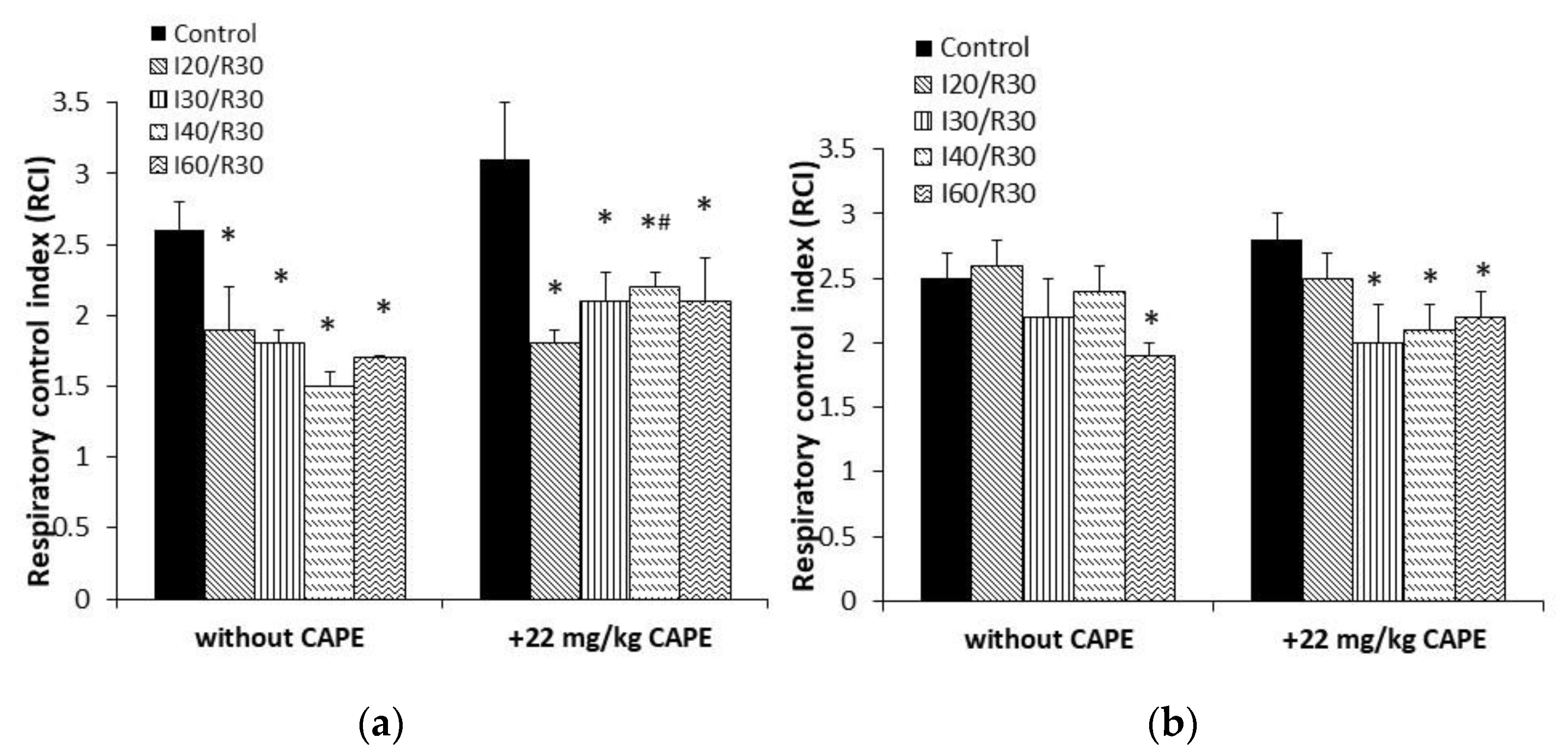
3.3. Effects of Ischemia/Reperfusion and Pretreatment with CAPE on Kidney Mitochondrial Respiration Rates
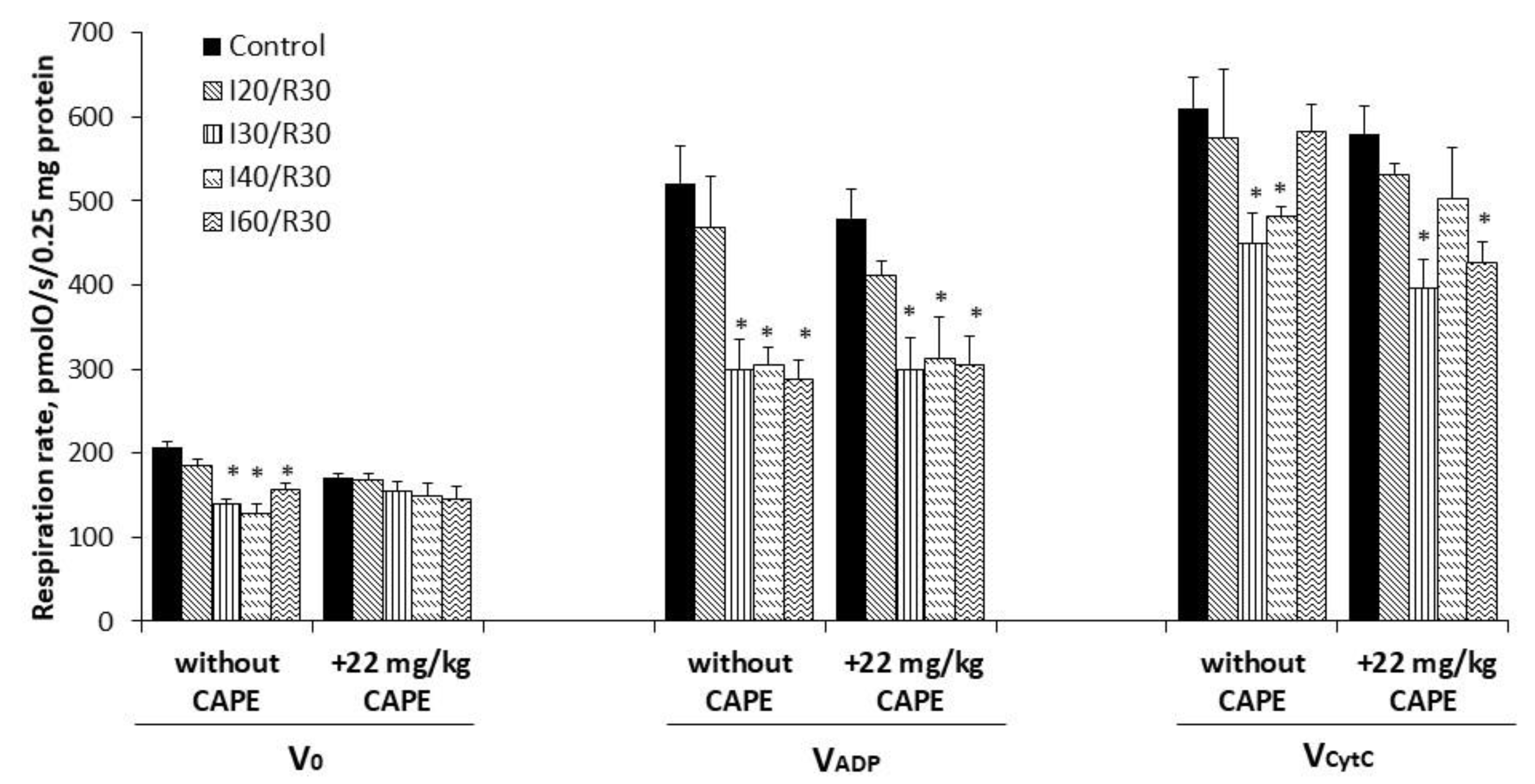
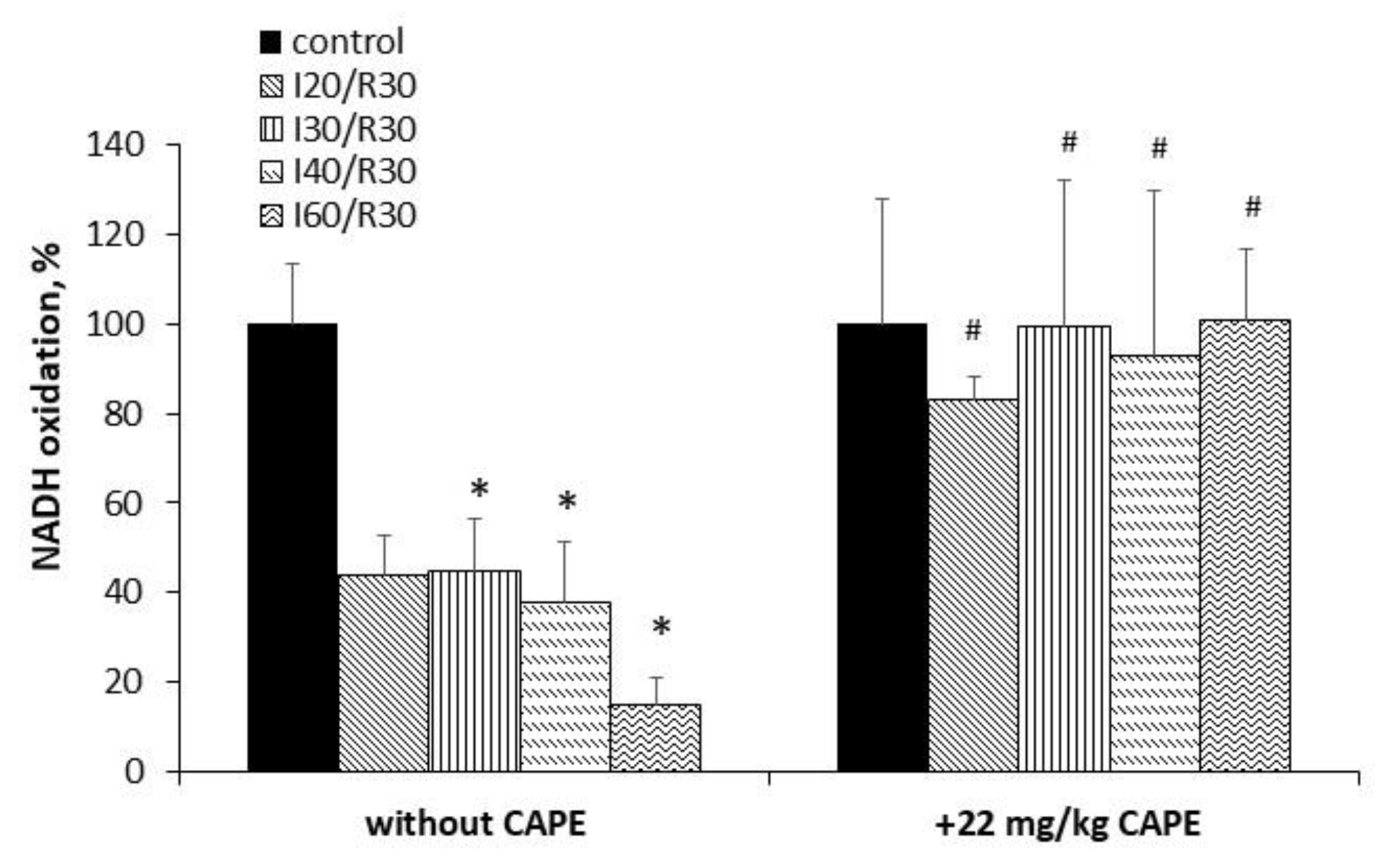
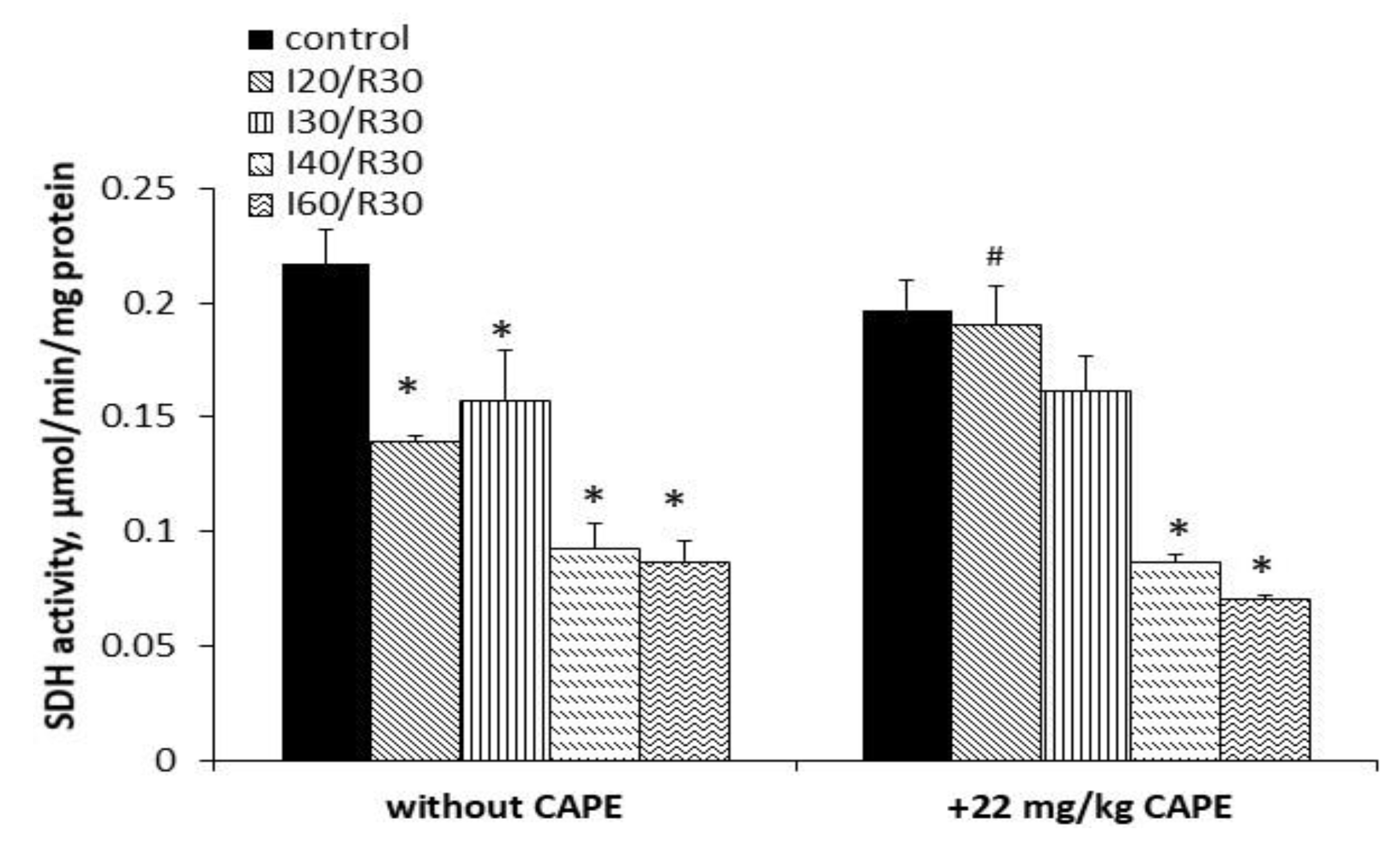
3.4. Effects of Ischemia/Reperfusion and Pretreatment with CAPE on Kidney Mitochondrial Complex I, II+III, and II (SDH) Activity
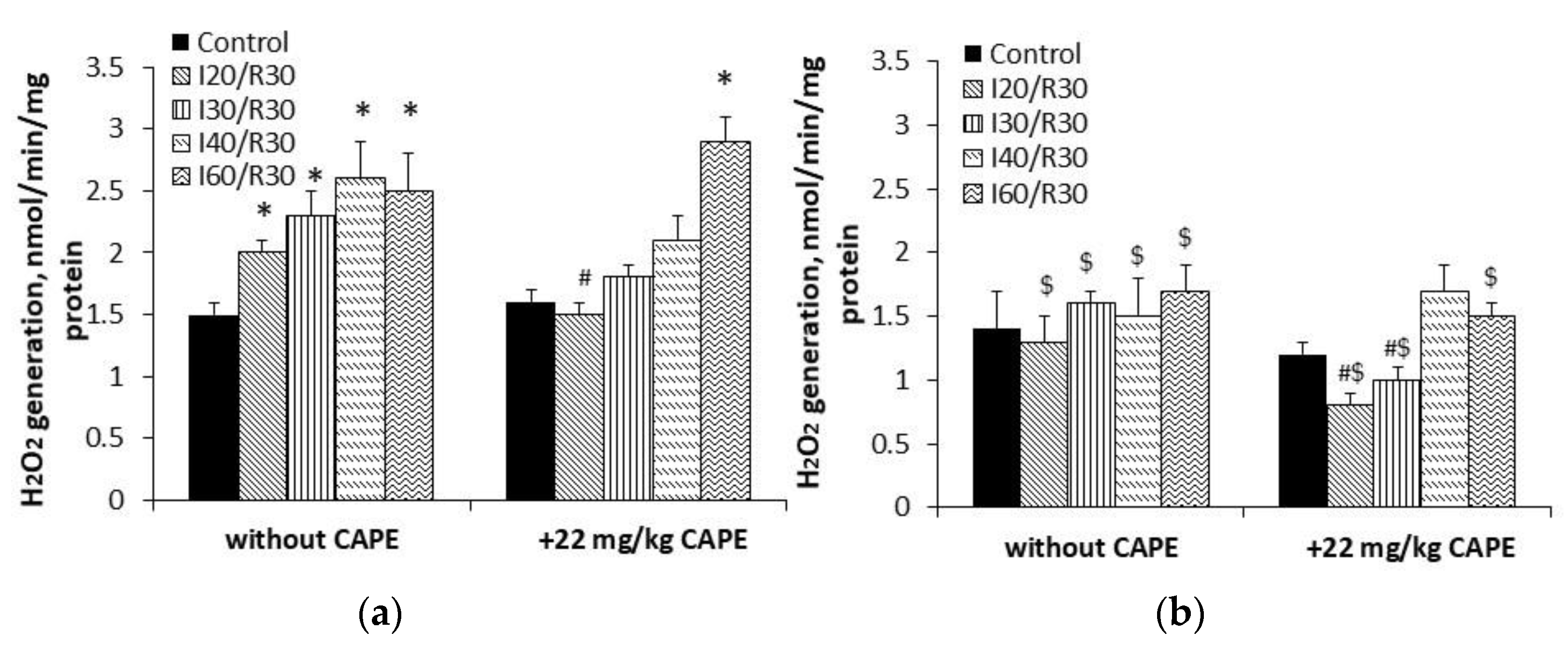
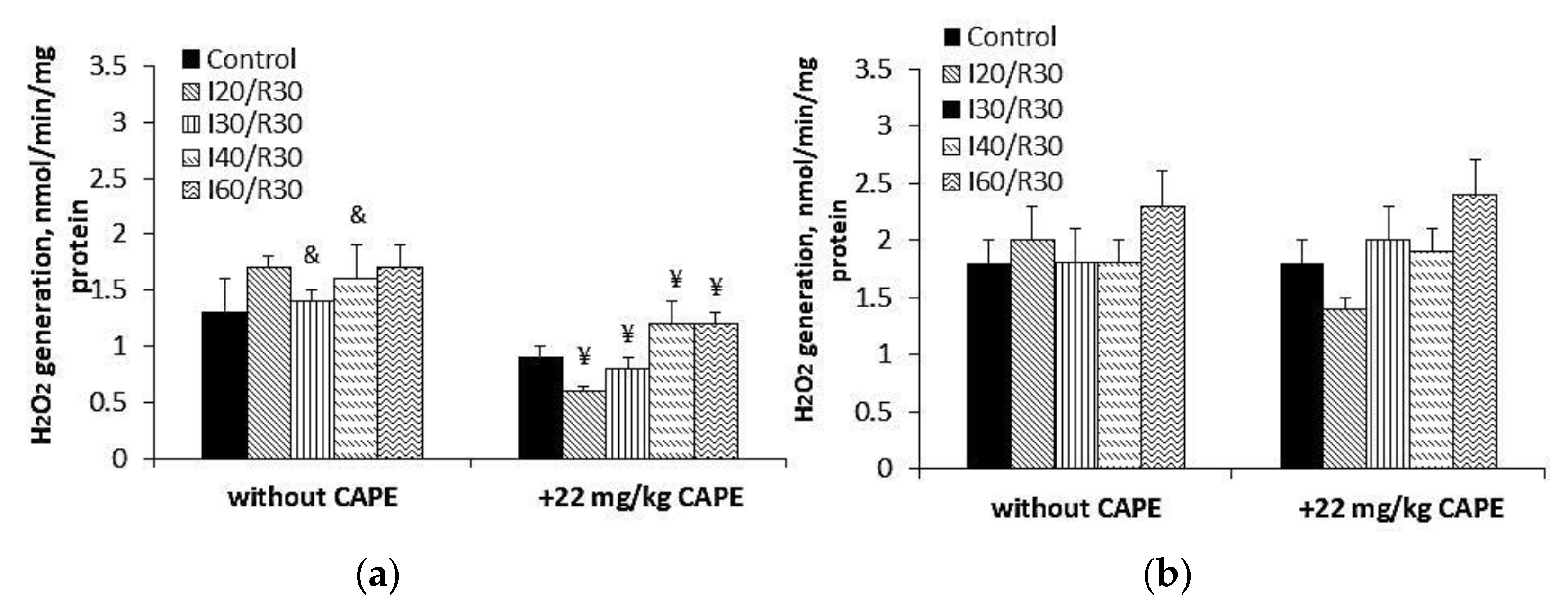
3.5. Effects of CAPE on Complex II Mediated ROS Generation in Kidney Mitochondria after Ischemia/Reperfusion
3.6. Effects of CAPE on LDH Activity after Ischemia/Reperfusion
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration Range (mM) | ABTS Method | FRAP Method | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Slope (a) | Intercept (b) | Correlation (R) | Coefficient of Determination (R2) | Slope (a) | Intercept (b) | Correlation (R) | Coefficient of Determination (R2) | ||
| Trolox | 0.25–16 | 0.0883 | +0.0248 | 0.9995 | 0.9991 | 0.1179 | +0.0164 | 0.9992 | 0.9986 |
| CAPE | 0.22–7 | 0.0753 | +0.0697 | 0.9999 | 0.9999 | 0.1443 | −0.0388 | 0.9996 | 0.9994 |
| Compound | Concentration Range (mM) | DPPH Method | CUPRAC Method | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Slope (a) | Intercept (b) | Correlation (R) | Coefficient of Determination (R2) | Slope (a) | Intercept (b) | Correlation (R) | Coefficient of Determination (R2) | ||
| Trolox | 0.25–32 | 0.0708 | +0.0223 | 0.9995 | 0.9991 | 0.04 | −0.0161 | 0.9994 | 0.999 |
| CAPE | 0.22–14 | 0.1145 | +0.0295 | 0.9996 | 0.9993 | 0.0903 | +0.0124 | 0.9992 | 0.9986 |

References
- Bankova, V.; Trusheva, B.; Popova, M. Caffeic acid phenethyl ester (CAPE)—Natural sources, analytical procedures and synthetic approaches. Comptes Rendus L’Acad. Bulg. Sci. 2018, 71, 1157–1169. [Google Scholar] [CrossRef]
- Trumbeckaite, S.; Pauziene, N.; Trumbeckas, D.; Jievaltas, M.; Baniene, R. Caffeic Acid Phenethyl Ester Reduces Ischemia-Induced Kidney Mitochondrial Injury in Rats. Oxid. Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 298. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–27. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Vanova, K.H.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef]
- Dröse, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef]
- Cecchini, G. Function and Structure of Complex II of the Respiratory Chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Shiao, M.-S.; Wang, S.-Y. The antioxidant caffeic acid phenethyl ester induces apoptosis associated with selective scavenging of hydrogen peroxide in human leukemic HL-60 cells. Anti-Cancer Drugs 2001, 12, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Longo, R.; Vanella, A. Antioxidant activity of propolis: Role of caffeic acid phenethyl ester and galangin. Fitoterapia 2002, 73, 21–29. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Seok, P.R.; Oh, S.J.; Choi, J.W.; Shin, J.-H. A Study on the Protective Effect of Antioxidants on Damage Induced by Liver Ischemia/Repefusion in a Rat Model. Korean J. Clin. Lab. Sci. 2019, 51, 370–378. [Google Scholar] [CrossRef]
- Gornall, A.G.; Bardawill, C.J.; David, M.M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949, 177, 751–766. [Google Scholar] [CrossRef]
- Hs, Y.; Fy, C.; Ct, T.; Yc, N.; Cw, H. Antioxidant Activities and Total Phenolic Content of Aqueous Extract of Pleurotus ostreatus (Cultivated Oyster Mushroom). Malays. J. Nutr. 2010, 16, 281–291. [Google Scholar]
- Raudone, L.; Vilkickyte, G.; Pitkauskaite, L.; Raudonis, R.; Vainoriene, R.; Motiekaityte, V. Antioxidant Activities of Vaccinium vitis-idaea L. Leaves within Cultivars and Their Phenolic Compounds. Molecules 2019, 24, 844. [Google Scholar] [CrossRef] [PubMed]
- Apak, R.; Güçlü, K.; Özyürek, M.; Çelik, S.E. Mechanism of antioxidant capacity assays and the CUPRAC (cupric ion reducing antioxidant capacity) assay. Microchim. Acta 2007, 160, 413–419. [Google Scholar] [CrossRef]
- Koleva, I.I.; Niederländer, H.A.G.; Van Beek, T.A. Application of ABTS Radical Cation for Selective On-Line Detection of Radical Scavengers in HPLC Eluates. Anal. Chem. 2001, 73, 3373–3381. [Google Scholar] [CrossRef]
- Tisdale, H.D. Preparation and properties of succinic–cytochrome c reductase (complex II-III). Methods Enzymol. 1967, 10, 213–215. [Google Scholar] [CrossRef]
- Baliutyte, G.; Baniene, R.; Trumbeckaite, S.; Borutaite, V.; Toleikis, A. Effects of Ginkgo biloba extract on heart and liver mitochondrial functions: Mechanism(s) of action. J. Bioenerg. Biomembr. 2010, 42, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Meiliana, A.; Dewi, N.M.; Wijaya, A. Mitochondria in Health and Disease. Indones. Biomed. J. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Cui, H.; Ma, Z.; Liu, X.; Yang, L. Recent progresses in the pharmacological activities of caffeic acid phenethyl ester. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 1–13. [Google Scholar] [CrossRef]
- Wei, X.; Zhao, L.; Ma, Z.; Holtzman, D.M.; Yan, C.; Dodel, R.C.; Hampel, H.; Oertel, W.; Farlow, M.R.; Du, Y. Caffeic acid phenethyl ester prevents neonatal hypoxic-ischaemic brain injury. Brain 2004, 127, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Elango, C.; Ansari, M.A.; Singh, I.; Singh, A.K. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. J. Neurochem. 2007, 102, 365–377. [Google Scholar] [CrossRef]
- Yildiz, Y.; Serter, M.; Ek, R.O.; Ergin, K.; Cecen, S.; Demir, E.M.; Yenisey, C. Protective Effects of Caffeic Acid Phenethyl Ester on Intestinal Ischemia-Reperfusion Injury. Dig. Dis. Sci. 2008, 54, 738–744. [Google Scholar] [CrossRef]
- Akyol, S.; Ginis, Z.; Armutcu, F.; Ozturk, G.; Yigitoglu, M.R.; Akyol, O. The potential usage of caffeic acid phenethyl ester (CAPE) against chemotherapy-induced and radiotherapy-induced toxicity. Cell Biochem. Funct. 2012, 30, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Göçer, H.; Gülçin, I. Caffeic acid phenethyl ester (CAPE): Correlation of structure and antioxidant properties. Int. J. Food Sci. Nutr. 2011, 62, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tang, Y.; Li, N.-G.; Zhu, Y.; Duan, J.-A. Bioactivity and Chemical Synthesis of Caffeic Acid Phenethyl Ester and Its Derivatives. Molecules 2014, 19, 16458–16476. [Google Scholar] [CrossRef]
- Tolba, M.F.; Omar, H.A.; Azab, S.S.; Khalifa, A.E.; Abdel-Naim, A.B.; Abdel-Rahman, S.Z. Caffeic Acid Phenethyl Ester: A Review of Its Antioxidant Activity, Protective Effects against Ischemia-reperfusion Injury and Drug Adverse Reactions. Crit. Rev. Food Sci. Nutr. 2016, 56, 2183–2190. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, H.; Zheng, J.; Liang, G. Structure-Thermodynamics-Antioxidant Activity Relationships of Selected Natural Phenolic Acids and Derivatives: An Experimental and Theoretical Evaluation. PLoS ONE 2015, 10, e0121276. [Google Scholar] [CrossRef]
- Shi, Y.; Wu, X.; Gong, Y.; Qiu, Y.; Zhang, H.; Huang, Z.; Su, K. Protective Effects of Caffeic Acid Phenethyl Ester on Retinal Ischemia/Reperfusion Injury in Rats. Curr. Eye Res. 2010, 35, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Celik, O.; Turkoz, Y.; Hascalik, S.; Hascalik, M.; Cigremis, Y.; Mizrak, B.; Yologlu, S. The protective effect of caffeic acid phenethyl ester on ischemia-reperfusion injury in rat ovary. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 117, 183–188. [Google Scholar] [CrossRef]
- Irmak, M.K.; Koltuksuz, U.; Kutlu, N.O.; Yağmurca, M.; Özyurt, H.; Karaman, A.; Akyol, Ö. The effect of caffeic acid phenethyl ester on ischemia-reperfusion injury in comparison with α-tocopherol in rat kidneys. Urol. Res. 2001, 29, 190–193. [Google Scholar] [CrossRef]
- Teke, Z.; Bostanci, E.B.; Yenisey, C.; Sacar, M.; Simsek, N.G.; Akoglu, M. Caffeic Acid Phenethyl Ester Alleviates Mesenteric Ischemia/Reperfusion Injury. J. Investig. Surg. 2012, 25, 354–365. [Google Scholar] [CrossRef]
- Siebels, I.; Dröse, S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1827, 1156–1164. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Kozlovsky, V.S.; Vinogradov, A.D. Respiratory complex II: ROS production and the kinetics of ubiquinone reduction. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, Subunit of Complex II Triggers Reactive Oxygen Species-Dependent Hypoxia-Inducible Factor Activation and Tumorigenesis. Mol. Cell. Biol. 2007, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Na, U.; Yu, W.; Cox, J.; Bricker, D.K.; Brockmann, K.; Rutter, J.; Thummel, C.S.; Winge, D.R. The LYR Factors SDHAF1 and SDHAF3 Mediate Maturation of the Iron-Sulfur Subunit of Succinate Dehydrogenase. Cell Metab. 2014, 20, 253–266. [Google Scholar] [CrossRef]
- Maklashina, E.; Rajagukguk, S.; Iverson, T.M.; Cecchini, G. The unassembled flavoprotein subunits of human and bacterial complex II have impaired catalytic activity and generate only minor amounts of ROS. J. Biol. Chem. 2018, 293, 7754–7765. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Brookes, P.S. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 2010, 104, 121–129. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Ruiz-Meana, M.; Rodríguez-Sinovas, A.; Garcia-Dorado, D. Selective Inhibition of Succinate Dehydrogenase in Reperfused Myocardium with Intracoronary Malonate Reduces Infarct Size. Sci. Rep. 2018, 8, 2442. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-P.; Wang, F.; Jiang, W.; Liu, J.; Liu, B.-L.; Qi, L.-W.; Zhou, W. A mitochondrion-targeting tanshinone IIA derivative attenuates myocardial hypoxia reoxygenation injury through a SDH-dependent antioxidant mechanism. J. Drug Target. 2019, 27, 896–902. [Google Scholar] [CrossRef]
- Wang, F.; Peng, Q.; Liu, J.; Alolga, R.N.; Zhou, W. A novel ferulic acid derivative attenuates myocardial cell hypoxia reoxygenation injury through a succinate dehydrogenase dependent antioxidant mechanism. Eur. J. Pharmacol. 2019, 856, 172417. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Regulation of apoptosis by the redox state of cytochrome c. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 877–881. [Google Scholar] [CrossRef]
- Skemiene, K.; Pampuscenko, K.; Rekuviene, E.; Borutaite, V. Protective effects of anthocyanins against brain ischemic damage. J. Bioenerg. Biomembr. 2020, 52, 71–82. [Google Scholar] [CrossRef]
- Jussupow, A.; Di Luca, A.; Kaila, V.R.I. How cardiolipin modulates the dynamics of respiratory complex I. Sci. Adv. 2019, 5, eaav1850. [Google Scholar] [CrossRef]
- Feng, Y.; Lu, Y.-W.; Xu, P.-H.; Long, Y.; Wu, W.-M.; Li, W.; Wang, R. Caffeic acid phenethyl ester and its related compounds limit the functional alterations of the isolated mouse brain and liver mitochondria submitted to in vitro anoxia–reoxygenation: Relationship to their antioxidant activities. Biochim. Biophys. Acta (BBA) Gen. Subj. 2008, 1780, 659–672. [Google Scholar] [CrossRef]












| Method | TEAC |
|---|---|
| ABTS | 0.85 ± 0.07 |
| DPPH | 1.65 ± 0.09 |
| FRAP | 1.22 ± 0.03 |
| CUPRAC | 2.24 ± 0.05 |
| Without CAPE | +22 mg/kg CAPE | |
|---|---|---|
| Control | 0.42 ± 0.01 | 0.44 ± 0.01 |
| I20/R30 | 0.29 ± 0.03 * | 0.39 ± 0.01 # |
| I30/R30 | 0.36 ± 0.02 * | 0.42 ± 0.01 # |
| I40/R30 | 0.30 ± 0.01 * | 0.41 ± 0.02 # |
| I60/R30 | 0.38 ± 0.02 | 0.37 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamarauskaite, J.; Baniene, R.; Trumbeckas, D.; Strazdauskas, A.; Trumbeckaite, S. Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model. Antioxidants 2021, 10, 747. https://doi.org/10.3390/antiox10050747
Kamarauskaite J, Baniene R, Trumbeckas D, Strazdauskas A, Trumbeckaite S. Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model. Antioxidants. 2021; 10(5):747. https://doi.org/10.3390/antiox10050747
Chicago/Turabian StyleKamarauskaite, Justina, Rasa Baniene, Darius Trumbeckas, Arvydas Strazdauskas, and Sonata Trumbeckaite. 2021. "Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model" Antioxidants 10, no. 5: 747. https://doi.org/10.3390/antiox10050747
APA StyleKamarauskaite, J., Baniene, R., Trumbeckas, D., Strazdauskas, A., & Trumbeckaite, S. (2021). Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model. Antioxidants, 10(5), 747. https://doi.org/10.3390/antiox10050747