Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress

 ,
,  and
and 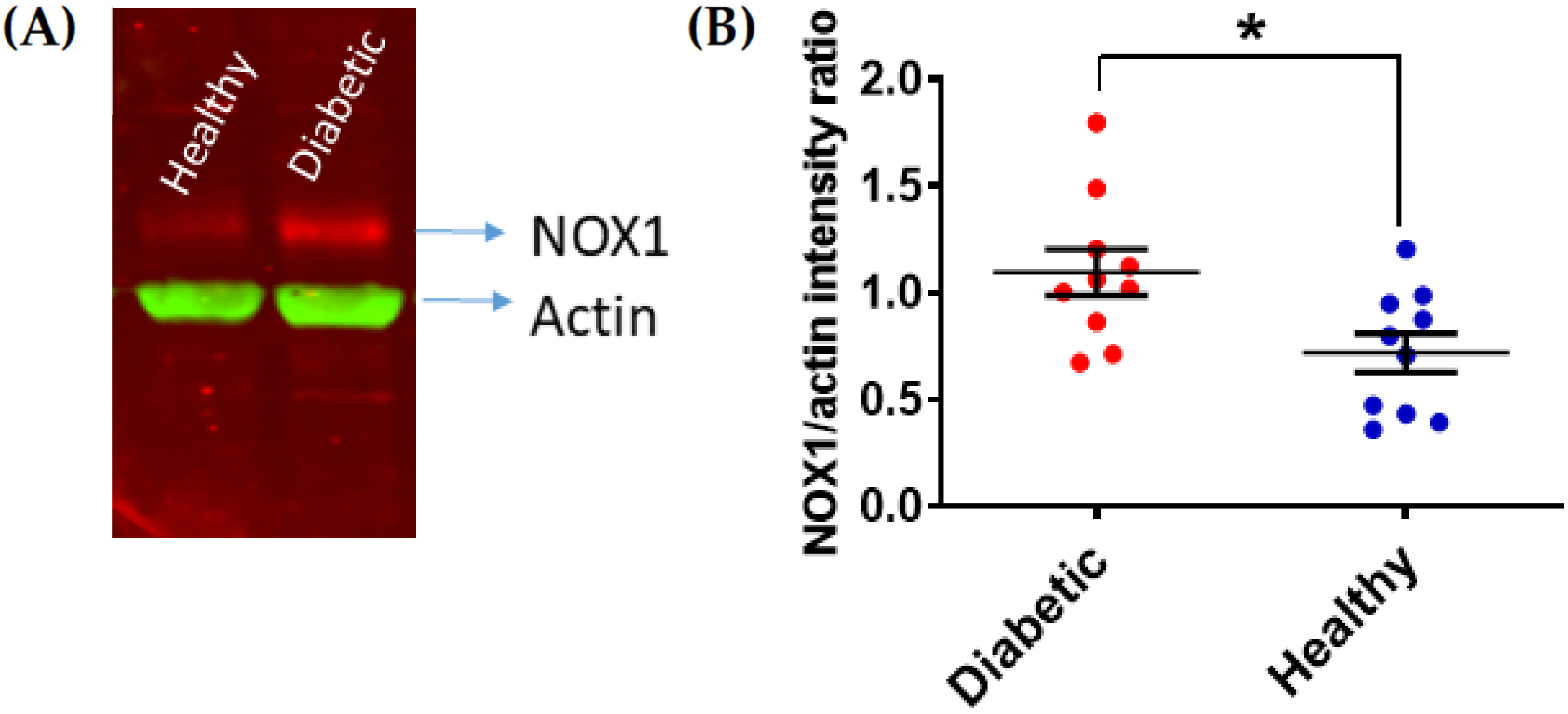{kind=link}
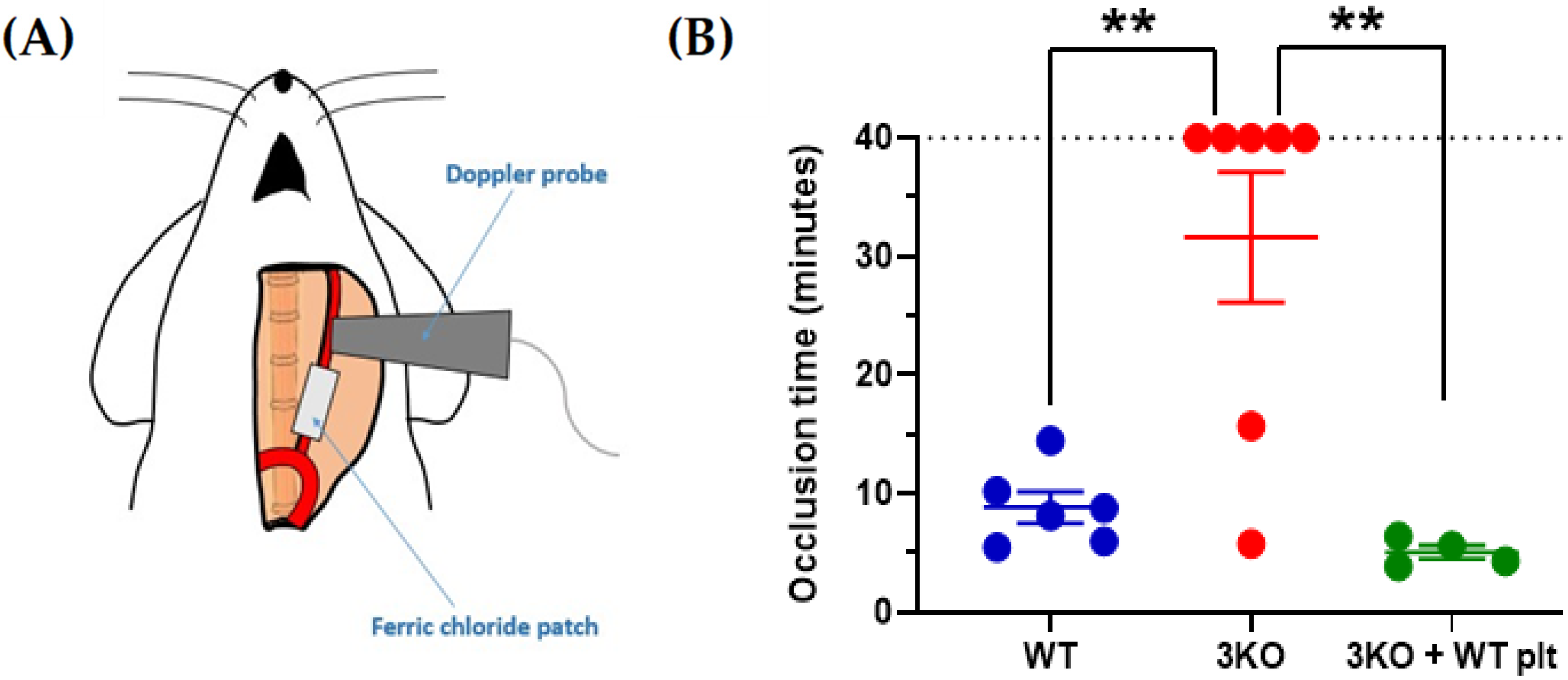{kind=link}
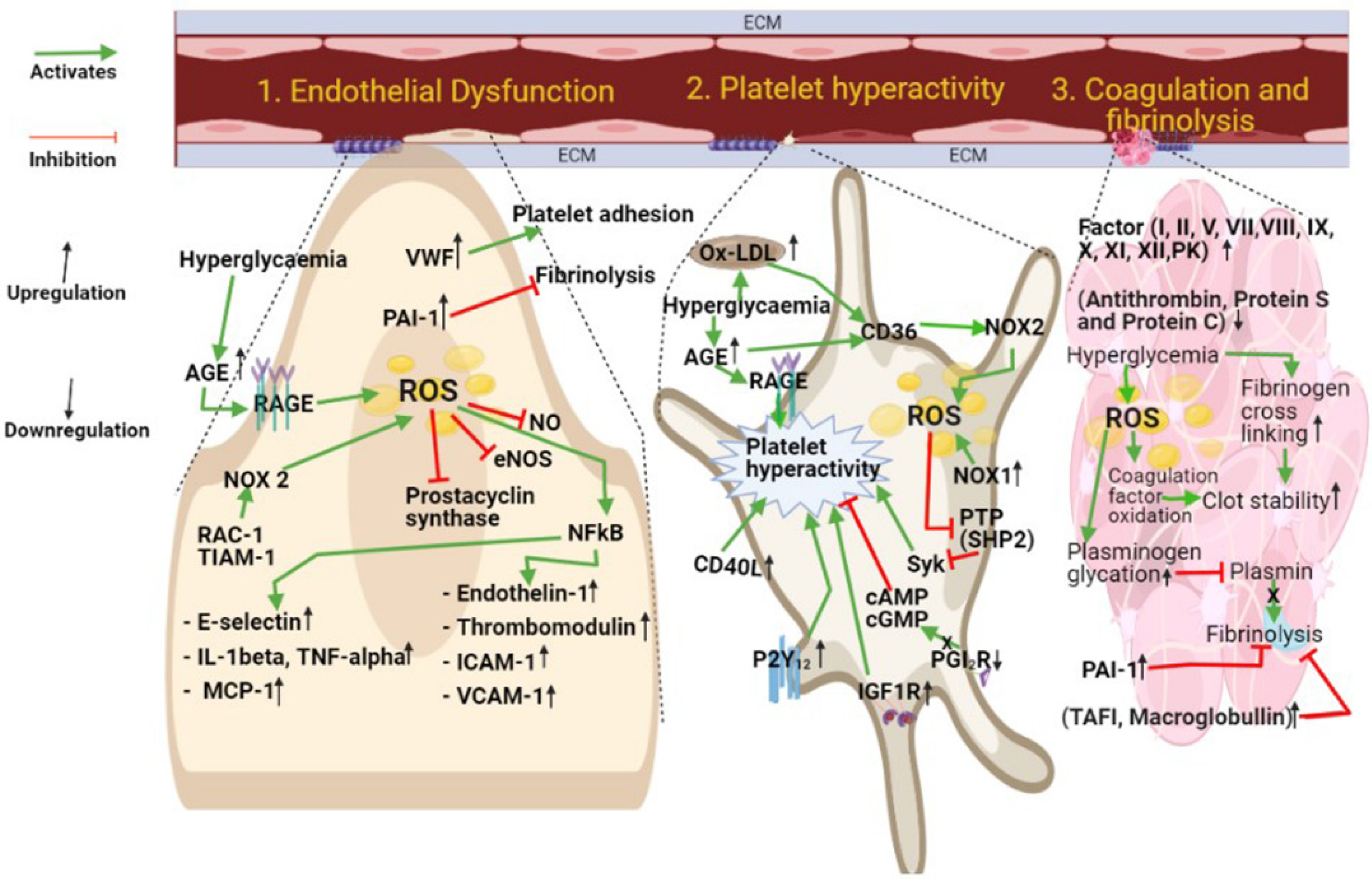{kind=link}
Abstract
1. Introduction
2. Platelet Hyperactivity in Diabetes
3. Coagulation and Fibrinolysis
4. Endothelial Cell Dysfunction
5. Therapeutic Intervention
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zinman, B. The International Diabetes Federation World Diabetes Congress 2015. Eur. Endocrinol. 2015, 11, 66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kannel, W.B.; D’Agostino, R.B.; Wilson, P.W.; Belanger, A.J.; Gagnon, D.R. Diabetes, fibrinogen, and risk of cardiovascular disease: The Framingham experience. Am. Heart J. 1990, 120, 672–676. [Google Scholar] [CrossRef]
- Laiteerapong, N.; Ham, S.A.; Gao, Y.; Moffet, H.H.; Liu, J.Y.; Huang, E.S.; Karter, A.J. The Legacy Effect in Type 2 Diabetes: Impact of Early Glycemic Control on Future Complications (The Diabetes & Aging Study). Diabetes Care 2019, 42, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Kahkoska, A.R.; Geybels, M.S.; Klein, K.R.; Kreiner, F.F.; Marx, N.; Nauck, M.A.; Pratley, R.E.; Wolthers, B.O.; Buse, J.B. Validation of distinct type 2 diabetes clusters and their association with diabetes complications in the DEVOTE, LEADER and SUSTAIN -6 cardiovascular outcomes trials. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Prospective Studies Collaboration; Asia Pacific Cohort Studies Colaboration. Sex-specific relevance of diabetes to occlusive vascular and other mortality: A collaborative meta-analysis of individual data from 980,793 adults from 68 prospective studies. Lancet Diabetes Endocrinol. 2018, 6, 538–546. [Google Scholar] [CrossRef]
- Kaseta, J.R.; Skafar, D.F.; Ram, J.L.; Jacober, S.J.; Sowers, J.R. Cardiovascular Disease in the Diabetic Woman. J. Clin. Endocrinol. Metab. 1999, 84, 1835–1838. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Gaiz, A.; Mosawy, S.; Colson, N.; Singh, I. Thrombotic and cardiovascular risks in type two diabetes; Role of platelet hyperactivity. Biomed. Pharmacother. 2017, 94, 679–686. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane Biosynthesis and Platelet Function in Type II Diabetes Mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef]
- Davì, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In Vivo Formation of 8-Iso-Prostaglandin F 2α and Platelet Activation in Diabetes Mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar] [CrossRef]
- Knebel, S.M.; Sprague, R.S.; Stephenson, A.H. Prostacyclin receptor expression on platelets of humans with type 2 diabetes is inversely correlated with hemoglobin A1c levels. Prostaglandins Other Lipid Mediat. 2015, 116, 131–135. [Google Scholar] [CrossRef]
- Hu, L.; Chang, L.; Zhang, Y.; Zhai, L.; Zhang, S.; Qi, Z.; Yan, H.; Yan, Y.; Luo, X.; Zhang, S.; et al. Platelets Express Activated P2Y12Receptor in Patients with Diabetes Mellitus. Circulation 2017, 136, 817–833. [Google Scholar] [CrossRef]
- Gligorijevic, N.; Robajac, D.; Nedic, O. Enhanced Platelet Sensitivity to IGF-1 in Patients with Type 2 Diabetes Mellitus. Biochemistry 2019, 84, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Hers, I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kα pathway. Blood 2007, 110, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Przygodzki, T.; Luzak, B.; Kassassir, H.; Mnich, E.; Boncler, M.; Siewiera, K.; Kosmalski, M.; Szymanski, J.; Watala, C. Diabetes and Hyperglycemia Affect Platelet GPIIIa Expression: Effects on Adhesion Potential of Blood Platelets from Diabetic Patients under In Vitro Flow Conditions. Int. J. Mol. Sci. 2020, 21, 3222. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Cifuentes-Pagano, E.; Pagano, P.J.; Pula, G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica 2019, 104, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arter. Thromb. Vasc. Biol. 2021, 41, 683–697. [Google Scholar] [CrossRef]
- Pignatelli, P.; Violi, F. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb. Haemost. 2014, 111, 817–823. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef]
- Yngen, M.; Östenson, C.-G.; Hu, H.; Li, N.; Hjemdahl, P.; Wallén, N.H. Enhanced P-selectin expression and increased soluble CD40 Ligand in patients with Type 1 diabetes mellitus and microangiopathy: Evidence for platelet hyperactivity and chronic inflammation. Diabetologia 2004, 47, 537–540. [Google Scholar] [CrossRef]
- Varo, N.; Libby, P.; Nuzzo, R.; Italiano, J.; Doria, A.; Schönbeck, U. Elevated release of sCD40L from platelets of diabetic patients by thrombin, glucose and advanced glycation end products. Diabetes Vasc. Dis. Res. 2005, 2, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Campanella, M.; Pula, G. The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br. J. Pharmacol. 2012, 168, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Kilhovd, B.K.; Berg, T.J.; Birkeland, K.I.; Thorsby, P.; Hanssen, K.F. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care 1999, 22, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.; Rojas, A.; Palomo, I. Role of multiligand/RAGE axis in platelet activation. Thromb. Res. 2014, 133, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef]
- Razmara, M.; Hjemdahl, P.; Östenson, C.; Li, N. Platelet hyperprocoagulant activity in Type 2 diabetes mellitus: Attenuation by glycoprotein IIb/IIIa inhibition. J. Thromb. Haemost. 2008, 6, 2186–2192. [Google Scholar] [CrossRef]
- Ferreira, I.A.; Mocking, A.I.; Feijge, M.A.; Gorter, G.; Van Haeften, T.W.; Heemskerk, J.W.; Akkerman, J.-W.N. Platelet Inhibition by Insulin Is Absent in Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 417–422. [Google Scholar] [CrossRef]
- Ravindran, R.; Krishnan, L.K. Increased Platelet Cholesterol and Decreased Percentage Volume of Platelets as a Secondary Risk Factor for Coronary Artery Disease. Pathophysiol. Haemost. Thromb. 2007, 36, 45–51. [Google Scholar] [CrossRef]
- Frostegård, J. Immune Mechanisms in Atherosclerosis, Especially in Diabetes Type 2. Front. Endocrinol. 2013, 4, 162. [Google Scholar] [CrossRef]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2–mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Edelstein, D.; Du, X.-L.; Brownlee, M. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes 2001, 50, 1491–1494. [Google Scholar] [CrossRef]
- Jang, J.Y.; Min, J.H.; Bin Wang, S.; Chae, Y.H.; Baek, J.Y.; Kim, M.; Ryu, J.-S.; Chang, T.-S. Resveratrol inhibits collagen-induced platelet stimulation through suppressing NADPH oxidase and oxidative inactivation of SH2 domain-containing protein tyrosine phosphatase-2. Free. Radic. Biol. Med. 2015, 89, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Bin Wang, S.; Park, S.J.; Oh, G.T.; Lee, S.-H.; Ho, Y.-S.; Chang, T.-S. Reactive Oxygen Species Play a Critical Role in Collagen-Induced Platelet ActivationviaSHP-2 Oxidation. Antioxid. Redox Signal. 2014, 20, 2528–2540. [Google Scholar] [CrossRef]
- Wang, S.B.; Jang, J.Y.; Chae, Y.H.; Min, J.H.; Baek, J.Y.; Kim, M.; Park, Y.; Hwang, G.S.; Ryu, J.S.; Chang, T.S. Kaempferol suppresses collagen-induced platelet activation by inhibiting NADPH oxidase and protecting SHP-2 from oxidative inactivation. Free Radic. Biol. Med. 2015, 83, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2019, 9, 3076. [Google Scholar] [CrossRef]
- Carr, M.E. Diabetes mellitus: A hypercoagulable state. J. Diabetes Complicat. 2001, 15, 44–54. [Google Scholar] [CrossRef]
- Kim, H.K.; Kim, J.E.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S. High coagulation factor levels and low protein C levels contribute to enhanced thrombin generation in patients with diabetes who do not have macrovascular complications. J. Diabetes Complicat. 2014, 28, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Sommeijer, D.W.; Hansen, H.R.; Van Oerle, R.; Hamulyák, K.; Van Zanten, A.P.; Meesters, E.; Spronk, H.M.H.; Cate, H.T. Soluble tissue factor is a candidate marker for progression of microvascular disease in patients with Type 2 diabetes. J. Thromb. Haemost. 2006, 4, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.W.; Kohler, H.P.; Ariens, R.A.; McCormack, L.J.; Grant, P.J. Circulating levels of coagulation factor XIII in subjects with type 2 diabetes and in their first-degree relatives. Diabetes Care 2000, 23, 703–705. [Google Scholar] [CrossRef][Green Version]
- Song, D.Y.; Gu, J.-Y.; Yoo, H.J.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Activation of Factor XII and Kallikrein-kinin System Combined with Neutrophil Extracellular Trap Formation in Diabetic Retinopathy. Exp. Clin. Endocrinol. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Boden, G.; Rao, A.K. Tissue factor and Toll-like receptor (TLR)4 in hyperglycaemia-hyperinsulinaemia. Effects in healthy subjects, and type 1 and type 2 diabetes mellitus. Thromb. Haemost. 2015, 113, 750–758. [Google Scholar] [CrossRef]
- Yasuma, T.; Yano, Y.; Alessandro-Gabazza, C.N.D.; Toda, M.; Gil-Bernabe, P.; Kobayashi, T.; Nishihama, K.; Hinneh, J.A.; Mifuji-Moroka, R.; Roeen, Z.; et al. Amelioration of Diabetes Mellitus by Protein S. Diabetes 2016, 65, 1940–1951. [Google Scholar] [CrossRef]
- Wieczór, R.; Wieczór, A.M.; Kulwas, A.; Rość, D. Type 2 Diabetes and Cardiovascular Factors Contrasted with Fibrinolysis Disorders in the Blood of Patients with Peripheral Arterial Disease. Medicina 2019, 55, 395. [Google Scholar] [CrossRef]
- Bryk, A.H.; Konieczynska, M.; Rostoff, P.; Broniatowska, E.; Hohendorff, J.; Malecki, M.T.; Undas, A. Plasma Protein Oxidation as a Determinant of Impaired Fibrinolysis in Type 2 Diabetes. Thromb. Haemost. 2019, 119, 213–222. [Google Scholar] [CrossRef]
- Maatman, B.T.; Schmeisser, G.; Kreutz, R.P. Fibrin Clot Strength in Patients with Diabetes Mellitus Measured by Thrombelastography. J. Diabetes Res. 2018, 2018, 4543065. [Google Scholar] [CrossRef]
- Bryk, A.H.; Konieczyńska, M.; Polak, M.; Plicner, D.; Bochenek, M.; Undas, A. Plasma fibrin clot properties and cardiovascular mortality in patients with type 2 diabetes: A long-term follow-up study. Cardiovasc. Diabetol. 2021, 20, 47. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Gamlen, T.; Standeven, K.F.; Mughal, S.; Hess, K.; Smith, K.A.; Dunn, E.J.; Anwar, M.M.; Rabbani, N.; Thornalley, P.J.; et al. Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood 2013, 122, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Konieczynska, M.; Fil, K.; Bazanek, M.; Undas, A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb. Haemost. 2014, 111, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Aso, Y.; Okumura, K.-I.; Yoshida, N.; Tayama, K.; Takemura, Y.; Inukai, T. Enhancement of Fibrinolysis in Poorly Controlled, Hospitalized Type 2 Diabetic Patients by Short-Term Metabolic Control: Association with a Decrease in Plasminogen Activator Inhibitor 1. Exp. Clin. Endocrinol. Diabetes 2004, 112, 175–180. [Google Scholar] [CrossRef]
- Verkleij, C.J.N.; De Bruijn, R.E.; Meesters, E.W.; Gerdes, V.E.; Meijers, J.C.M.; Marx, P.F. The Hemostatic System in Patients with Type 2 Diabetes with and Without Cardiovascular Disease. Clin. Appl. Thromb. 2010, 17, E57–E63. [Google Scholar] [CrossRef] [PubMed]
- Sherif, E.M.; Elbarbary, N.S.; Al Aziz, M.M.A.; Mohamed, S.F. Plasma thrombin-activatable fibrinolysis inhibitor levels in children and adolescents with type 1 diabetes mellitus: Possible relation to diabetic microvascular complications. Blood Coagul. Fibrinolysis 2014, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular form and concentration of serum alpha2-macroglobulin in diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef] [PubMed]
- Fattah, M.A.; Shaheen, M.H.; Mahfouz, M.H. Disturbances of Haemostasis in Diabetes Mellitus. Dis. Markers 2004, 19, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Erem, C.; Hacıhasanoğlu, A.; Çelik, Ş.; Ovalı, E.; Ersöz, H.Ö.; Ukinç, K.; Deger, O.; Telatar, M. Coagulation and Fibrinolysis Parameters in Type 2 Diabetic Patients with and without Diabetic Vascular Complications. Med. Princ. Pract. 2005, 14, 22–30. [Google Scholar] [CrossRef]
- Wu, M.D.; Atkinson, T.M.; Lindner, J.R. Platelets and von Willebrand factor in atherogenesis. Blood 2017, 129, 1415–1419. [Google Scholar] [CrossRef]
- Urano, T.; Castellino, F.J.; Suzuki, Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018. [Google Scholar] [CrossRef]
- Peng, X.; Wang, X.; Fan, M.; Zhao, J.; Lin, L.; Liu, J. Plasma levels of von Willebrand factor in type 2 diabetes patients with and without cardiovascular diseases: A meta-analysis. Diabetes Metab. Res. Rev. 2020, 36, e3193. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.; Chow, W.-S.; Ai, V.H.; Metz, C.; Bucala, R.; Lam, K.S. Advanced glycation end products and endothelial dysfunction in type 2 diabetes. Diabetes Care 2002, 25, 1055–1059. [Google Scholar] [CrossRef]
- Kemeny, S.F.; Figueroa, D.S.; Andrews, A.M.; Barbee, K.A.; Clyne, A.M. Glycated collagen alters endothelial cell actin alignment and nitric oxide release in response to fluid shear stress. J. Biomech. 2011, 44, 1927–1935. [Google Scholar] [CrossRef]
- Khan, G.; Aftab, M.F.; Bano, B.; Khan, K.M.; Murtaza, M.; Siddiqui, S.; Rehman, M.H.; Waraich, R.S. A new indanedione derivative alleviates symptoms of diabetes by modulating RAGE-NF-kappaB pathway in db/db mice. Biochem. Biophys. Res. Commun. 2018, 501, 863–870. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1–RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, N.; Ranalli, P.; Cuccurullo, C.; Davì, G. Diabetes mellitus and thrombosis. Thromb. Res. 2012, 129, 371–377. [Google Scholar] [CrossRef]
- Folli, F.; Corradi, D.; Fanti, P.; Davalli, A.; Paez, A.; Giaccari, A.; Perego, C.; Muscogiuri, G. The Role of Oxidative Stress in the Pathogenesis of Type 2 Diabetes Mellitus Micro- and Macrovascular Complications: Avenues for a Mechanistic-Based Therapeutic Approach. Curr. Diabetes Rev. 2011, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kosugi, S.; Sano, F. Improvement of High Serum Levels of Biomarkers of Endothelial Injury (Vascular Cell Adhesion Molecule-1) and Inflammation (Tumor Necrosis Factor Receptor Type I) after Allogeneic Hematopoietic Stem Cell Transplantation with Sinusoidal Obstruction Syndrome Using Defibrotide. Am. J. Ther. 2020. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.-L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef]
- Elmariah, S.; Mauri, L.; Doros, G.; Galper, B.Z.; O’Neill, K.E.; Steg, P.G.; Kereiakes, D.J.; Yeh, R.W. Extended duration dual antiplatelet therapy and mortality: A systematic review and meta-analysis. Lancet 2015, 385, 792–798. [Google Scholar] [CrossRef]
- The ASCEND Study Collaborative Group; Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Seidu, S.; Kunutsor, S.K.; Sesso, H.D.; Gaziano, J.M.; Buring, J.E.; Roncaglioni, M.C.; Khunti, K. Aspirin has potential benefits for primary prevention of cardiovascular outcomes in diabetes: Updated literature-based and individual participant data meta-analyses of randomized controlled trials. Cardiovasc. Diabetol. 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Rivas Rios, J.R.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Diabetes and antiplatelet therapy: From bench to bedside. Cardiovasc. Diagn. Ther. 2018, 8, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Sharma, A.N.; Deyell, J.S.; Sharma, S.N.; Barseghian, A. Role of and Recent Evidence for Antiplatelet Therapy in Prevention of Cardiovascular Disease in Diabetes. Curr. Cardiol. Rep. 2019, 21, 78. [Google Scholar] [CrossRef]
- Meredith, I.T.; Tanguay, J.-F.; Kereiakes, D.J.; Cutlip, D.E.; Yeh, R.W.; Garratt, K.N.; Lee, D.P.; Steg, P.G.; Weaver, W.D.; Holmes, D.R., Jr.; et al. Diabetes Mellitus and Prevention of Late Myocardial Infarction After Coronary Stenting in the Randomized Dual Antiplatelet Therapy Study. Circulation 2016, 133, 1772–1782. [Google Scholar] [CrossRef]
- Calderone, D.; Capodanno, D.; Angiolillo, D.J. An updated drug profile of ticagrelor with considerations on the treatment of patients with coronary artery disease and diabetes mellitus. Expert Rev. Cardiovasc. Ther. 2020, 18, 449–464. [Google Scholar] [CrossRef]
- Neri Serneri, G.G.; Coccheri, S.; Marubini, E.; Violi, F.; Drug Evaluation in Atherosclerotic Vascular Disease in Diabetics (DAVID) Study Group. Picotamide, a combined inhibitor of thromboxane A2 synthase and receptor, reduces 2-year mortality in diabetics with peripheral arterial disease: The DAVID study. Eur. Heart J. 2004, 25, 1845–1852. [Google Scholar] [CrossRef]
- Stone, G.W.; Bertrand, M.; Colombo, A.; Dangas, G.; Farkouh, M.E.; Feit, F.; Lansky, A.J.; Lincoff, A.; Mehran, R.; Moses, J.W.; et al. Acute Catheterization and Urgent Intervention Triage strategY (ACUITY) trial: Study design and rationale. Am. Heart J. 2004, 148, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, D.; Xanthopoulou, I.; Mavronasiou, E.; Stavrou, K.; Siapika, A.; Tsoni, E.; Davlouros, P. Randomized Assessment of Ticagrelor Versus Prasugrel Antiplatelet Effects in Patients with Diabetes. Diabetes Care 2013, 36, 2211–2216. [Google Scholar] [CrossRef]
- Ahmad, A.; Nawaz, M.I.; Siddiquei, M.M.; Abu El-Asrar, A.M. Apocynin ameliorates NADPH oxidase 4 (NOX4) induced oxidative damage in the hypoxic human retinal Müller cells and diabetic rat retina. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Kwon, G.; Uddin, M.J.; Lee, G.; Jiang, S.; Cho, A.; Lee, J.H.; Lee, S.R.; Bae, Y.S.; Moon, S.H.; Lee, S.J.; et al. A novel pan-Nox inhibitor, APX-115, protects kidney injury in streptozotocin-induced diabetic mice: Possible role of peroxisomal and mitochondrial biogenesis. Oncotarget 2017, 8, 74217–74232. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kim, H.M.; Lee, S.H.; Ha, K.B.; Bae, Y.S.; Lee, S.J.; Moon, S.H.; Lee, E.Y.; Lee, J.H.; Chung, C.H. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic. Biol. Med. 2020, 161, 92–101. [Google Scholar] [CrossRef]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; De Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus–Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; Van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef]
- De Livera, A.M.; Reutens, A.; Cooper, M.; Thomas, M.; Jandeleit-Dahm, K.; Shaw, J.E.; Salim, A. Evaluating the efficacy and safety of GKT137831 in adults with type 1 diabetes and persistently elevated urinary albumin excretion: A statistical analysis plan. Trials 2020, 21, 459. [Google Scholar] [CrossRef]
- Carnevale, R.; Loffredo, L.; Pignatelli, P.; Nocella, C.; Bartimoccia, S.; Di Santo, S.; Martino, F.; Catasca, E.; Perri, L.; Violi, F. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. J. Thromb. Haemost. 2012, 10, 125–132. [Google Scholar] [CrossRef]
- Wang, Y.; Chun, O.K.; Song, W.O. Plasma and Dietary Antioxidant Status as Cardiovascular Disease Risk Factors: A Review of Human Studies. Nutrients 2013, 5, 2969–3004. [Google Scholar] [CrossRef]
- Mega, J.L.; Braunwald, E.; Wiviott, S.D.; Bassand, J.-P.; Bhatt, D.L.; Bode, C.; Burton, P.; Cohen, M.; Cook-Bruns, N.; Fox, K.A.; et al. Rivaroxaban in Patients with a Recent Acute Coronary Syndrome. N. Engl. J. Med. 2012, 366, 9–19. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Calkins, H.; Chen, L.Y.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; Furie, K.L.; et al. 2019 AHA/ACC/HRS Focused Update of the 2014 AHA/ACC/HRS Guideline for the Management of Patients with Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society in Collaboration with the Society of Thoracic Surgeons. Circulation 2019, 140, e125–e151. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-H.; Lee, H.-F.; Li, P.-R.; Liu, J.-R.; Chao, T.-F.; Wu, L.-S.; Chang, S.-H.; Yeh, Y.-H.; Kuo, C.-T.; See, L.-C.; et al. Effectiveness, safety, and major adverse limb events in atrial fibrillation patients with concomitant diabetes mellitus treated with non-vitamin K antagonist oral anticoagulants. Cardiovasc. Diabetol. 2020, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Ezekowitz, J.A.; Lewis, B.S.; Lopes, R.D.; Wojdyla, D.M.; McMurray, J.J.; Hanna, M.; Atar, D.; Bahit, M.C.; Keltai, M.; Lopez-Sendon, J.L.; et al. Clinical outcomes of patients with diabetes and atrial fibrillation treated with apixaban: Results from the ARISTOTLE trial. Eur. Heart J. Cardiovasc. Pharmacother. 2015, 1, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Brambatti, M.; Darius, H.; Oldgren, J.; Clemens, A.; Noack, H.H.; Brueckmann, M.; Yusuf, S.; Wallentin, L.; Ezekowitz, M.D.; Connolly, S.J.; et al. Comparison of dabigatran versus warfarin in diabetic patients with atrial fibrillation: Results from the RE-LY trial. Int. J. Cardiol. 2015, 196, 127–131. [Google Scholar] [CrossRef]
- De Caterina, R.; Patti, G.; Westerbergh, J.; Horowitz, J.; Ezekowitz, J.A.; Lewis, B.S.; Lopes, R.D.; McMurray, J.J.V.; Atar, D.; Bahit, M.C.; et al. Heterogeneity of diabetes as a risk factor for major adverse cardiovascular events in anticoagulated patients with atrial fibrillation: An analysis of the ARISTOTLE trial. Eur. Heart J. Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaidya, A.R.; Wolska, N.; Vara, D.; Mailer, R.K.; Schröder, K.; Pula, G. Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants 2021, 10, 706. https://doi.org/10.3390/antiox10050706
Vaidya AR, Wolska N, Vara D, Mailer RK, Schröder K, Pula G. Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants. 2021; 10(5):706. https://doi.org/10.3390/antiox10050706
Chicago/Turabian StyleVaidya, Aishwarya R., Nina Wolska, Dina Vara, Reiner K. Mailer, Katrin Schröder, and Giordano Pula. 2021. "Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress" Antioxidants 10, no. 5: 706. https://doi.org/10.3390/antiox10050706
APA StyleVaidya, A. R., Wolska, N., Vara, D., Mailer, R. K., Schröder, K., & Pula, G. (2021). Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants, 10(5), 706. https://doi.org/10.3390/antiox10050706