
Protein Disulphide Isomerase and NADPH Oxidase 1 Cooperate to Control Platelet Function and Are Associated with Cardiometabolic Disease Risk Factors
 ,
,  , and
, and
Abstract
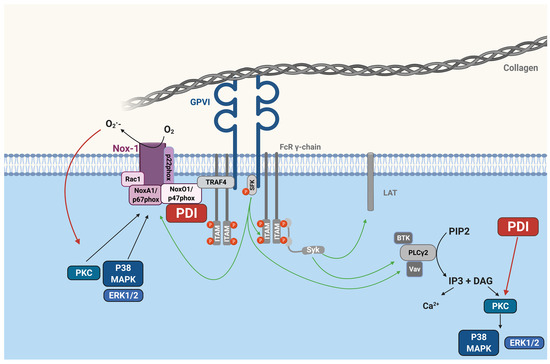
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Washed Platelets Preparation
2.2. Collection of Mouse Blood and Platelet Preparation
2.3. Immunofluorescence Microscopy
2.4. Turbidimetry and Plate-Based Platelet Aggregation
2.5. Fibrinogen Binding and P-Selectin Exposure
2.6. Calcium Measurement
2.7. Platelet Spreading
2.8. Immunoblotting
2.9. Tail Bleeding Assay
2.10. Population Study
2.11. Statistical Analysis
2.12. Study Approval
3. Results
3.1. PDI and Nox-1 Cellular Localization in Resting and Activated Platelets
3.2. Co-Inhibition of PDI and Nox-1 Results in Additive Inhibitory Effect on Platelet Aggregation Induced by Collagen and CRP
3.3. Co-Inhibition of PDI and Nox-1 Leads to an Additive Inhibitory Effect on Platelet Activation and Calcium Mobilization Induced by CRP
3.4. Co-Inhibition of PDI and Nox-1 Disrupts Collagen-Stimulated Signalling
3.5. Anti-Platelet Effects in Nox-1−/− Mice Are Increased by PDI Inhibition Without Increasing Bleeding Time
3.6. Platelet PDI and Nox-1 Protein Levels Are Upregulated in Conditions of Increased Cardiovascular Disease Risk
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | protein kinase B |
| CRP | collagen-related peptide |
| ERK | extracellular signal-regulated kinases |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GPVI | glycoprotein VI |
| NO | nitric oxide |
| Nox | nicotinamide adenine dinucleotide phosphate oxidase |
| PAPA-NONOate | propylamine propylamine NONOate |
| PDI | protein disulphide isomerase |
| PKC | protein kinase C |
| PMA | phorbol-12-myristate-13-acetate |
| PRP | platelet-rich plasma |
| ROS | reactive oxygen species |
| Src | proto-oncogene tyrosine-protein kinase |
| Syk | spleen tyrosine kinase |
| TRAP-6 | thrombin receptor activator peptide 6 |
| VASP | vasodilator-stimulated phospho-protein |
| WP | washed platele |
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Detwiler, T.C.; Essex, D.W. Characterization of protein disulphide isomerase released from activated platelets. Br. J. Haematol. 1995, 90, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Hahm, E.; Li, J.; Holbrook, L.M.; Sasikumar, P.; Stanley, R.G.; Ushio-Fukai, M.; Gibbins, J.M.; Cho, J. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood 2013, 122, 1052–1061. [Google Scholar] [CrossRef]
- Schwaller, M.; Wilkinson, B.; Gilbert, H.F. Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 2003, 278, 7154–7159. [Google Scholar] [CrossRef]
- Crescente, M.; Pluthero, F.G.; Li, L.; Lo, R.W.; Walsh, T.G.; Schenk, M.P.; Holbrook, L.M.; Louriero, S.; Ali, M.S.; Vaiyapuri, S.; et al. Intracellular Trafficking, Localization, and Mobilization of Platelet-Borne Thiol Isomerases. Arter. Thromb. Vasc. Biol. 2016, 36, 1164–1173. [Google Scholar] [CrossRef]
- Cho, J.; Kennedy, D.R.; Lin, L.; Huang, M.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of beta3 integrins. Blood 2012, 120, 647–655. [Google Scholar] [CrossRef]
- Lahav, J.; Wijnen, E.M.; Hess, O.; Hamaia, S.W.; Griffiths, D.; Makris, M.; Knight, C.G.; Essex, D.W.; Farndale, R.W. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood 2003, 102, 2085–2092. [Google Scholar] [CrossRef]
- Burgess, J.K.; Hotchkiss, K.A.; Suter, C.; Dudman, N.P.; Szollosi, J.; Chesterman, C.N.; Chong, B.H.; Hogg, P.J. Physical proximity and functional association of glycoprotein 1balpha and protein-disulfide isomerase on the platelet plasma membrane. J. Biol Chem. 2000, 275, 9758–9766. [Google Scholar] [CrossRef]
- Gimenez, M.; Verissimo-Filho, S.; Wittig, I.; Schickling, B.M.; Hahner, F.; Schurmann, C.; Netto, L.E.S.; Rosa, J.C.; Brandes, R.P.; Sartoretto, S.; et al. Redox Activation of Nox1 (NADPH Oxidase 1) Involves an Intermolecular Disulfide Bond Between Protein Disulfide Isomerase and p47(phox) in Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2019, 39, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.G.; Berndt, M.C.; Carrim, N.; Cowman, J.; Kenny, D.; Metharom, P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014, 2, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Bayraktutan, U.; Blayney, L.; Shah, A.M. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arter. Thromb. Vasc. Biol. 2000, 20, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef]
- Seno, T.; Inoue, N.; Gao, D.; Okuda, M.; Sumi, Y.; Matsui, K.; Yamada, S.; Hirata, K.I.; Kawashima, S.; Tawa, R.; et al. Involvement of NADH/NADPH oxidase in human platelet ROS production. Thromb. Res. 2001, 103, 399–409. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arter. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef]
- Vara, D.; Campanella, M.; Pula, G. The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br. J. Pharm. 2013, 168, 212–224. [Google Scholar] [CrossRef]
- Vara, D.; Cifuentes-Pagano, E.; Pagano, P.J.; Pula, G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica 2019, 104, 1879–1891. [Google Scholar] [CrossRef]
- Gaspar, R.S.; Trostchansky, A.; Paes, A.M. Potential Role of Protein Disulfide Isomerase in Metabolic Syndrome-Derived Platelet Hyperactivity. Oxidative Med. Cell. Longev. 2016, 2016, 2423547. [Google Scholar] [CrossRef]
- Qiu, S.; Mintz, J.D.; Salet, C.D.; Han, W.; Giannis, A.; Chen, F.; Yu, Y.; Su, Y.; Fulton, D.J.; Stepp, D.W. Increasing muscle mass improves vascular function in obese (db/db) mice. J. Am. Heart Assoc. 2014, 3, e000854. [Google Scholar] [CrossRef]
- Gaspar, R.S.; da Silva, S.A.; Stapleton, J.; Fontelles, J.L.L.; Sousa, H.R.; Chagas, V.T.; Alsufyani, S.; Trostchansky, A.; Gibbins, J.M.; Paes, A.M.A. Myricetin, the Main Flavonoid in Syzygium cumini Leaf, Is a Novel Inhibitor of Platelet Thiol Isomerases PDI and ERp5. Front. Pharm. 2019, 10, 1678. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, G.; Banfi, B.; Deffert, C.; Fiette, L.; Schappi, M.; Herrmann, F.; Krause, K.H. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006, 580, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Bye, A.P.; Unsworth, A.J.; Desborough, M.J.; Hildyard, C.A.T.; Appleby, N.; Bruce, D.; Kriek, N.; Nock, S.H.; Sage, T.; Hughes, C.E.; et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017, 1, 2610–2623. [Google Scholar] [CrossRef]
- Morton, L.; Hargreaves, P.; Farndale, R.; Young, R.; Barnes, M. Integrin α 2 β 1-independent activation of platelets by simple collagen-like peptides: Collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for α 2 β 1-independent platelet reactivity. Biochem. J. 1995, 306, 337–344. [Google Scholar] [CrossRef]
- Bekendam, R.; Bendapudi, P.; Lin, L.; Nag, P.P.; Pu, J.; Kennedy, D.R.; Feldenzer, A.; Chiu, J.; Cook, K.M.; Furie, B.; et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat. Commun. 2016, 7, 12579. [Google Scholar] [CrossRef]
- Vara, D.; Tarafdar, A.; Celikag, M.; Patinha, D.; Gulacsy, C.E.; Hounslea, E.; Warren, Z.; Ferreira, B.; Koeners, M.P.; Caggiano, L.; et al. NADPH oxidase 1 is a novel pharmacological target for the development of an antiplatelet drug without bleeding side effects. FASEB J. 2020, 34, 13959–13977. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Taulet, N.; Zhang, H.; DerMardirossian, C.; Kister, J.; Martinez, L.; Roush, W.R.; Brown, S.J.; Bokoch, G.M.; Rosen, H.A. Novel and Specific NADPH Oxidase-1 (Nox1) Small-Molecule Inhibitor Blocks the Formation of Functional Invadopodia in Human Colon Cancer Cells. ACS Chem. Biol. 2010, 10, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension global hypertension practice guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef]
- Santilli, F.; Vazzana, N.; Liani, R.; Guagnano, M.; Davì, G. Platelet activation in obesity and metabolic syndrome. Obes. Rev. 2012, 13, 27–42. [Google Scholar] [CrossRef]
- El Haouari, M.; Rosado, J.A. Platelet function in hypertension. Blood Cells Mol. Dis. 2009, 42, 38–43. [Google Scholar] [CrossRef]
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): A prospective cohort study. Lancet 2020, 395, 795–808. [Google Scholar] [CrossRef]
- Fernandes, D.C.; Manoel, A.H.; Wosniak, J., Jr.; Laurindo, F.R. Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: Effects of nitrosothiol exposure. Arch. Biochem. Biophys. 2009, 484, 197–204. [Google Scholar] [CrossRef]
- de A Paes, A.M.; Veríssimo-Filho, S.; Guimarães, L.L.; Silva, A.C.; Takiuti, J.T.; Santos, C.X.; Janiszewski, M.; Laurindo, F.R.; Lopes, L.R. Protein disulfide isomerase redox-dependent association with p47(phox): Evidence for an organizer role in leukocyte NADPH oxidase activation. J. Leukoc. Biol. 2011, 90, 799–810. [Google Scholar] [CrossRef]
- Matsumoto, M.; Katsuyama, M.; Iwata, K.; Ibi, M.; Zhang, J.; Zhu, K.; Nauseef, W.M.; Yabe-Nishimura, C. Characterization of N-glycosylation sites on the extracellular domain of NOX1/NADPH oxidase. Free. Radic. Biol. Med. 2014, 68, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Bendapudi, P.; Sharda, A.; Chen, V.; Bellido-Martin, L.; Jasuja, R.; Furie, B.C.; Flaumenhaft, R.; Furie, B. Extracellular Thiol Isomerases and Their Role in Thrombus Formation. Antioxid. Redox Signal 2016, 24, 1–15. [Google Scholar] [CrossRef]
- Chen, W.; Shang, W.H.; Adachi, Y.; Hirose, K.; Ferrari, D.M.; Kamata, T. A possible biochemical link between NADPH oxidase (Nox) 1 redox-signalling and ERp72. Biochem. J. 2008, 416, 55–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, W.J.; Li, J.Y.; Chen, R.J.; Huang, L.T.; Lee, T.Y.; Lin, K.H. VAS2870 and VAS3947 attenuate platelet activation and thrombus formation via a NOX-independent pathway downstream of PKC. Sci. Rep. 2019, 9, 18852. [Google Scholar] [CrossRef] [PubMed]
- Gilio, K.; Munnix, I.C.; Mangin, P.; Cosemans, J.M.; Feijge, M.A.; Van der Meijden, P.E.; Olieslagers, S.; Chrzanowska-Wodnicka, M.B.; Lillian, R.; Schoenwaelder, S. Non-redundant roles of phosphoinositide 3-kinase isoforms α and β in glycoprotein VI-induced platelet signaling and thrombus formation. J. Biol. Chem. 2009, 284, 33750–33762. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; De, S.; Damron, D.S.; Chen, W.S.; Hay, N.; Byzova, T.V. Impaired platelet responses to thrombin and collagen in AKT-1–deficient mice. Blood 2004, 104, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Pravin, P.; Ulhas, P.N. Platelet MAPKs a 20+ year History: What Do We Really Know? J. Thromb. Haemost. 2020, 18, 2087–2102. [Google Scholar]
- Mazharian, A.; Roger, S.; Maurice, P.; Berrou, E.; Popoff, M.R.; Hoylaerts, M.F.; Fauvel-Lafeve, F.; Bonnefoy, A.; Bryckaert, M. Differential involvement of ERK2 and p38 in platelet adhesion to collagen. J. Biol. Chem. 2005, 280, 26002–26010. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jin, J.; Kunapuli, S.P. Akt activation in platelets depends on Gi signaling pathways. J. Biol. Chem. 2004, 279, 4186–4195. [Google Scholar] [CrossRef]
- Lopes Pires, M.E.; Antunes Naime, A.C.; Oliveira, J.G.F.; Anhe, G.F.; Garraud, O.; Cognasse, F.; Antunes, E.; Marcondes, S. Signalling pathways involved in p47(phox) -dependent reactive oxygen species in platelets of endotoxemic rats. Basic Clin. Pharm. Toxicol. 2019, 124, 394–403. [Google Scholar] [CrossRef]
- Androwiki, A.C.; de Camargo, L.L.; Sartoretto, S.; Couto, G.K.; Ribeiro, I.M.; Verissimo-Filho, S.; Rossoni, L.V.; Lopes, L.R. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Front. Chem. 2015, 3, 24. [Google Scholar] [CrossRef]
- DeVallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709. [Google Scholar] [CrossRef]
- Chien, C.Y.; Hung, Y.J.; Shieh, Y.S.; Hsieh, C.H.; Lu, C.H.; Lin, F.H.; Su, S.C.; Lee, C.H. A novel potential biomarker for metabolic syndrome in Chinese adults: Circulating protein disulfide isomerase family A, member 4. PLoS ONE 2017, 12, e0179963. [Google Scholar] [CrossRef]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef]
- Camargo, L.L.; Harvey, A.P.; Rios, F.J.; Tsiropoulou, S.; Da Silva, R.d.N.O.; Cao, Z.; Graham, D.; McMaster, C.; Burchmore, R.J.; Hartley, R.C. Vascular Nox (NADPH oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension 2018, 72, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kranz, P.; Neumann, F.; Wolf, A.; Classen, F.; Pompsch, M.; Ocklenburg, T.; Baumann, J.; Janke, K.; Baumann, M.; Goepelt, K.; et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. 2017, 8, e2986. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaspar, R.S.; Sage, T.; Little, G.; Kriek, N.; Pula, G.; Gibbins, J.M. Protein Disulphide Isomerase and NADPH Oxidase 1 Cooperate to Control Platelet Function and Are Associated with Cardiometabolic Disease Risk Factors. Antioxidants 2021, 10, 497. https://doi.org/10.3390/antiox10030497
Gaspar RS, Sage T, Little G, Kriek N, Pula G, Gibbins JM. Protein Disulphide Isomerase and NADPH Oxidase 1 Cooperate to Control Platelet Function and Are Associated with Cardiometabolic Disease Risk Factors. Antioxidants. 2021; 10(3):497. https://doi.org/10.3390/antiox10030497
Chicago/Turabian StyleGaspar, Renato Simões, Tanya Sage, Gemma Little, Neline Kriek, Giordano Pula, and Jonathan M. Gibbins. 2021. "Protein Disulphide Isomerase and NADPH Oxidase 1 Cooperate to Control Platelet Function and Are Associated with Cardiometabolic Disease Risk Factors" Antioxidants 10, no. 3: 497. https://doi.org/10.3390/antiox10030497
APA StyleGaspar, R. S., Sage, T., Little, G., Kriek, N., Pula, G., & Gibbins, J. M. (2021). Protein Disulphide Isomerase and NADPH Oxidase 1 Cooperate to Control Platelet Function and Are Associated with Cardiometabolic Disease Risk Factors. Antioxidants, 10(3), 497. https://doi.org/10.3390/antiox10030497