
Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry

 and
and 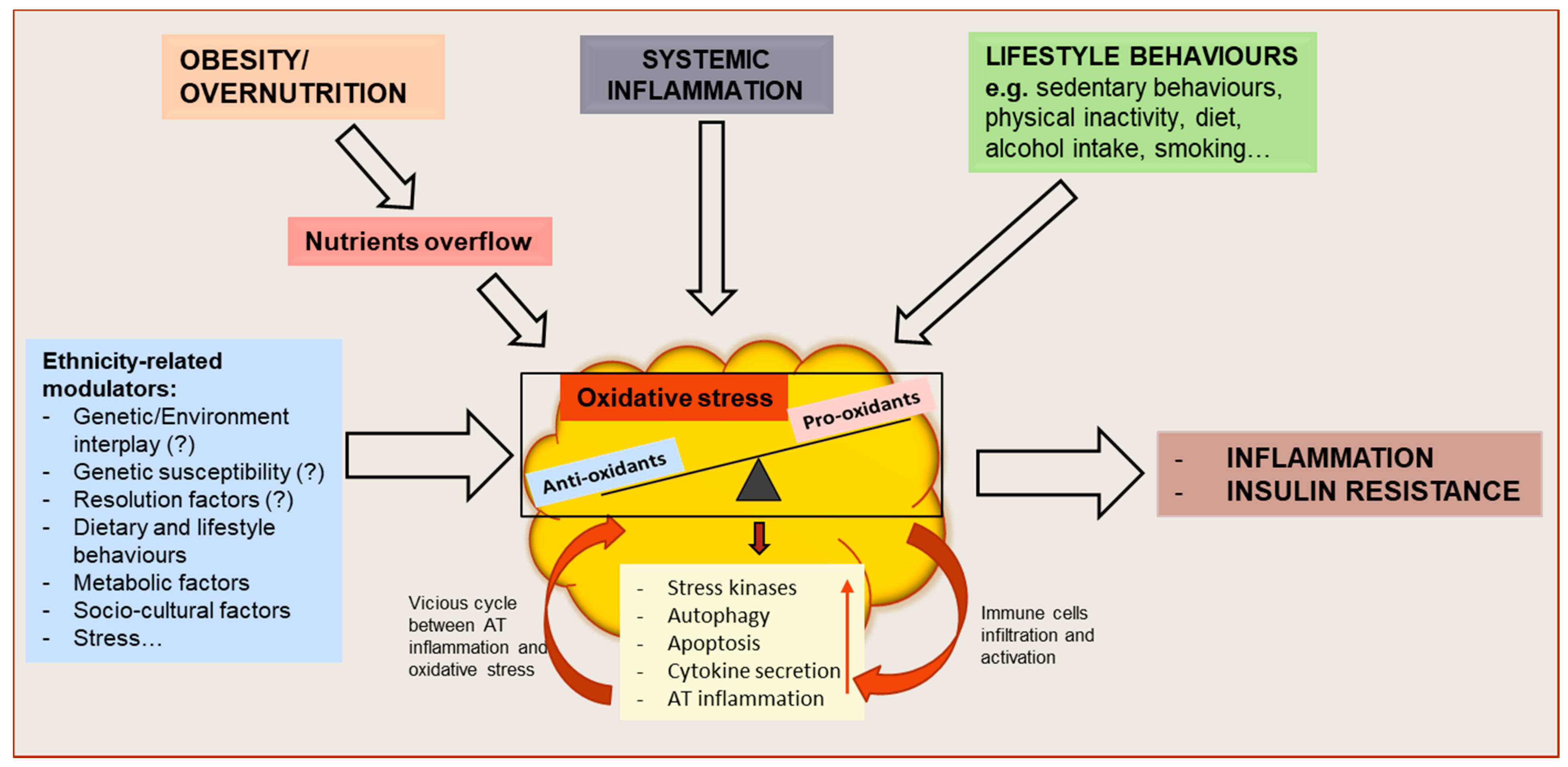{kind=link}
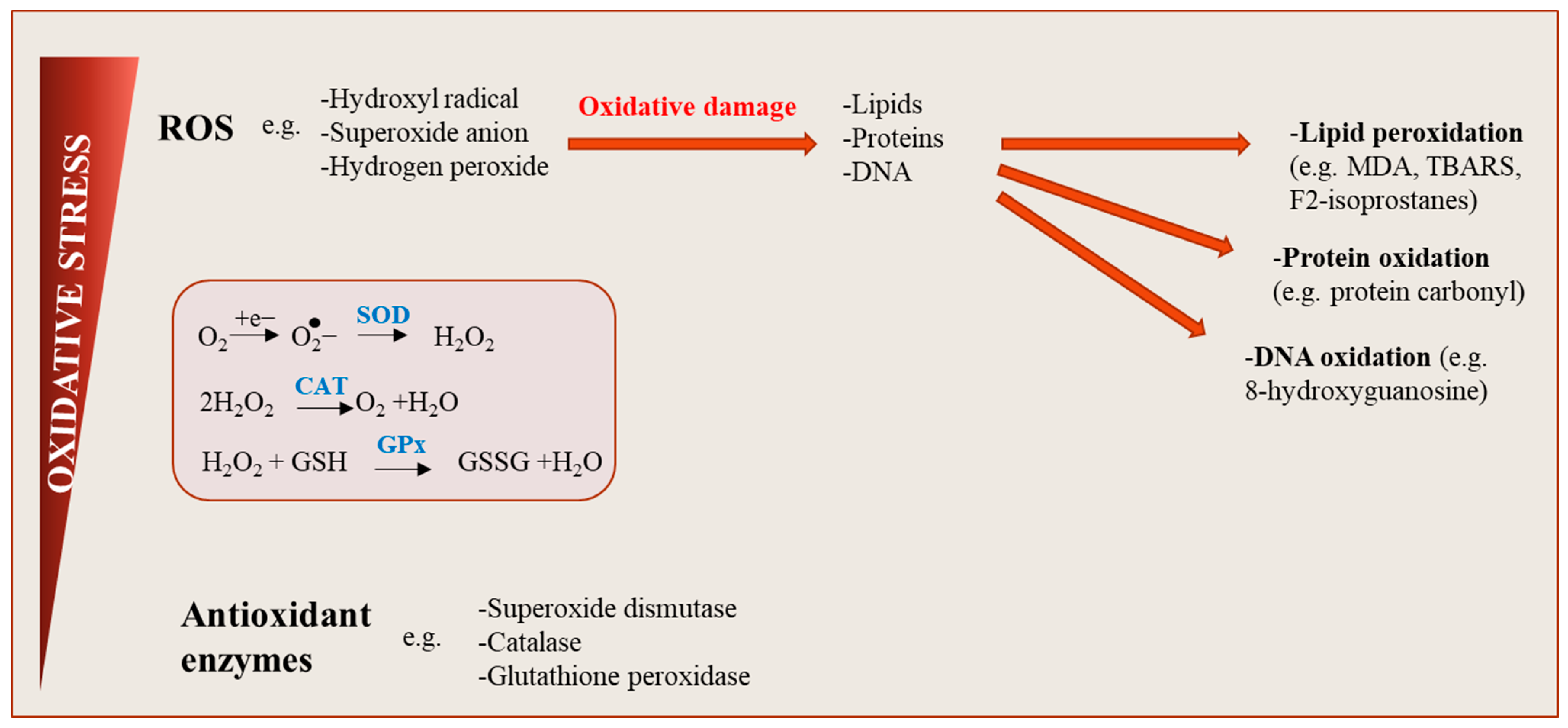{kind=link}
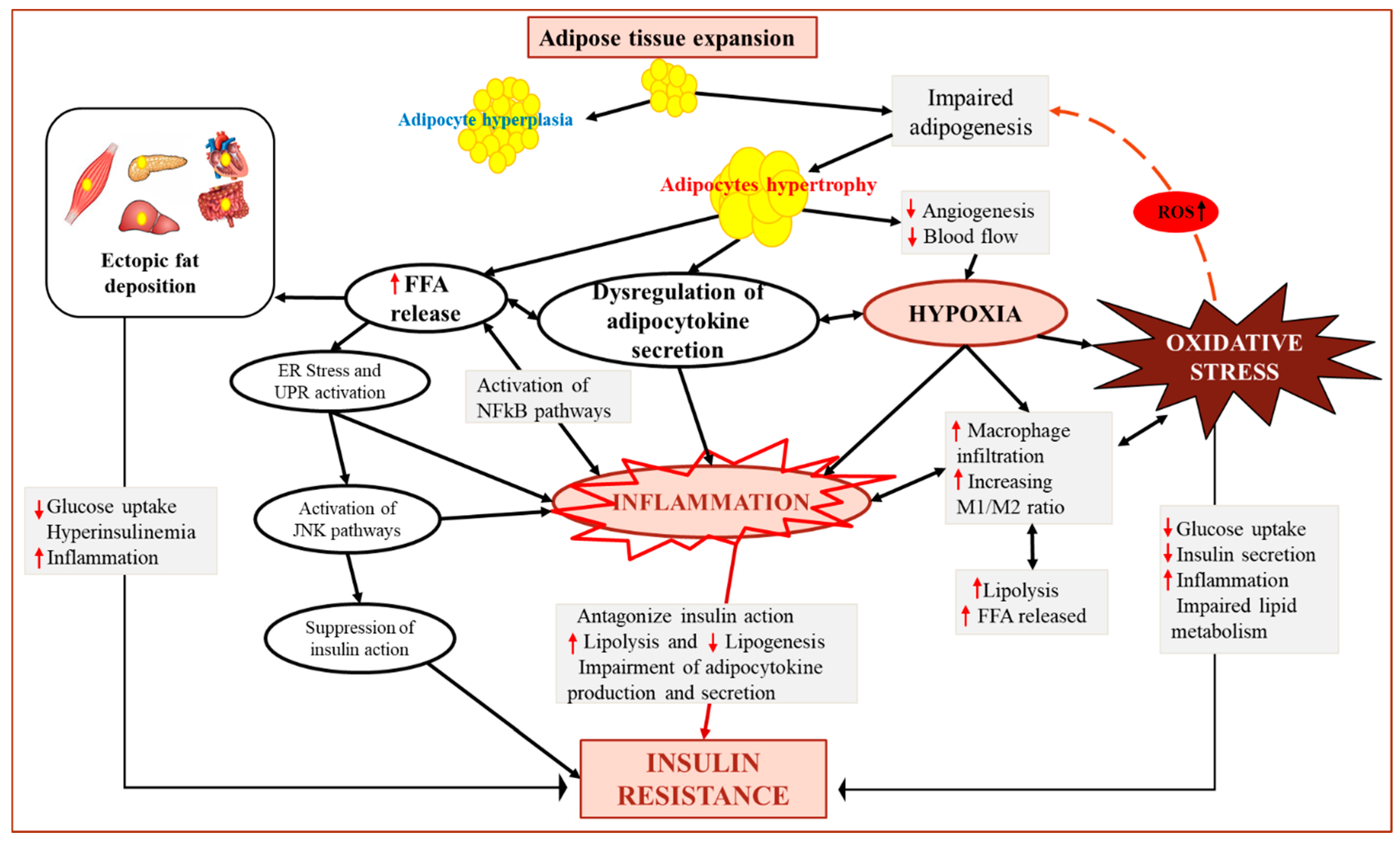{kind=link}
Abstract
1. Introduction
2. Definition of Oxidative Stress
3. Oxidative Stress in Obesity
4. Obesity-Induced AT Dysfunction and Oxidative Stress
5. Dysregulation of Adipokine Secretion in Response to AT Oxidative Stress
6. Role of Fat Distribution in Obesity-Associated Oxidative Stress
Role of Ethnicity in Oxidative Stress Regulation
7. Adipose Tissue Oxidative Stress as a Risk Factor of Metabolic Dysfunction in Africans
8. Mechanisms Contributing to Higher Measures of Oxidative Stress Markers in Africans
8.1. Hyperglycemia
8.2. Adipose Tissue Function
8.3. AT Storage and Elevated Lipid Levels
8.4. Environmental, Socioeconomic and Lifestyle Factors
8.5. Impaired Mitochondrial Function
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Asp. Med. 2013, 34, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef]
- Sacks, H.S.; Fain, J.N. Human epicardial adipose tissue: A review. Am. Heart J. 2007, 153, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Wronska, A.; Kmiec, Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol. 2012, 205, 194–208. [Google Scholar] [CrossRef]
- Lenz, M.; Arts, I.C.W.; Peeters, R.L.M.; de Kok, T.M.; Ertaylan, G. Adipose tissue in health and disease through the lens of its building blocks. Sci. Rep. 2020, 10, 10433. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Petrus, P.; Esteve, D.; Boulomie, A.; Naslund, E.; Thorell, A.; Gao, H.; Dahlman, I.; Ryden, M. Screening of potential adipokines identifies S100A4 as a marker of pernicious adipose tissue and insulin resistance. Int. J. Obes. 2018, 42, 2047–2056. [Google Scholar] [CrossRef]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Clinical relevance of adipokines. Diabetes Metab. J. 2012, 36, 317–327. [Google Scholar] [CrossRef]
- Blüher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 163–177. [Google Scholar] [CrossRef]
- Rudich, A.; Kanety, H.; Bashan, N. Adipose stress-sensing kinases: Linking obesity to malfunction. Trends Endocrinol. Metab. 2007, 18, 291–299. [Google Scholar] [CrossRef]
- Huang, C.J.; McAllister, M.J.; Slusher, A.L.; Webb, H.E.; Mock, J.T.; Acevedo, E.O. Obesity-Related Oxidative Stress: The Impact of Physical Activity and Diet Manipulation. Sports Med. Open 2015, 1, 1–12. [Google Scholar] [CrossRef]
- Bluher, M.; Bashan, N.; Shai, I.; Harman-Boehm, I.; Tarnovscki, T.; Avinaoch, E.; Stumvoll, M.; Dietrich, A.; Kloting, N.; Rudich, A. Activated Ask1-MKK4-p38MAPK/JNK stress signaling pathway in human omental fat tissue may link macrophage infiltration to whole-body Insulin sensitivity. J. Clin. Endocrinol. Metab. 2009, 94, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Haim, Y.; Bluher, M.; Konrad, D.; Goldstein, N.; Kloting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Tarnovscki, T.; Gepner, Y.; et al. ASK1 (MAP3K5) is transcriptionally upregulated by E2F1 in adipose tissue in obesity, molecularly defining a human dys-metabolic obese phenotype. Mol. Metab. 2017, 6, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Mazini, L.; Malka, G.; Zeller, M.; Cottin, Y.; Vergely, C. The Crosstalk of Adipose-Derived Stem Cells (ADSC), Oxidative Stress, and Inflammation in Protective and Adaptive Responses. Int. J. Mol. Sci. 2020, 21, 9262. [Google Scholar] [CrossRef]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef]
- Matsuoka, T.; Kajimoto, Y.; Watada, H.; Kaneto, H.; Kishimoto, M.; Umayahara, Y.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasak, Y. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J. Clin. Investig. 1997, 99, 144–150. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, M.T.; Crowther, N.J.; Schlaphoff, G.P.; Gray, I.P.; Joffe, B.I.; Lönnroth, P.N. Evidence for insulin resistance in black women from South Africa. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 1340–1346. [Google Scholar] [CrossRef]
- Hill, J.O.; Sidney, S.; Lewis, C.E.; Tolan, K.; Scherzinger, A.L.; Stamm, E.R. Racial differences in amounts of visceral adipose tissue in young adults: The CARDIA (Coronary Artery Risk Development in Young Adults) study. Am. J. Clin. Nutr. 1999, 69, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Goedecke, J.H.; Levitt, N.S.; Utzschneider, K.M.; Faulenbach, M.V.; Dave, J.A.; West, S.; Victor, H.; Evans, J.; Olsson, T.; Walker, B.R.; et al. Differential effects of abdominal adipose tissue distribution on insulin sensitivity in black and white South African women. Obesity 2009, 17, 1506–1512. [Google Scholar] [CrossRef]
- Després, J.-P.; Couillard, C.; Gagnon, J.; Bergeron, J.; Leon, A.S.; Rao, D.c.; Skinner, J.S.; Wilmore, J.H.; Bouchard, C. Race, Visceral Adipose Tissue, Plasma Lipids, and Lipoprotein Lipase Activity in Men and Women: The Health, Risk Factors, Exercise Training, and Genetics (HERITAGE) Family Study. Arter. Thromb. Vasc. Biol. 2000, 20, 1932–1938. [Google Scholar] [CrossRef]
- Lindquist, C.H.; Gower, B.A.; Goran, M.I. Role of dietary factors in ethnic differences in early risk of cardiovascular disease and type 2 diabetes. Am. J. Clin. Nutr. 2000, 71, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Osei, K.; Schuster, D.P.; Owusu, S.K.; Amoah, A.G. Race and ethnicity determine serum insulin and C-peptide concentrations and hepatic insulin extraction and insulin clearance: Comparative studies of three populations of West African ancestry and white Americans. Metab. Clin. Exp. 1997, 46, 53–58. [Google Scholar] [CrossRef]
- Katzmarzyk, P.T.; Bray, G.A.; Greenway, F.L.; Johnson, W.D.; Newton, R.L., Jr.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Bouchard, C. Racial differences in abdominal depot-specific adiposity in white and African American adults. Am. J. Clin. Nutr. 2010, 91, 7–15. [Google Scholar] [CrossRef]
- Stults-Kolehmainen, M.A.; Stanforth, P.R.; Bartholomew, J.B. Fat in Android, Trunk, and Peripheral Regions Varies by Ethnicity and Race in College Aged Women. Obesity 2012, 20, 660–665. [Google Scholar] [CrossRef]
- Gower, B.A.; Fowler, L.A. Obesity in African-Americans: The role of physiology. J. Intern. Med. 2020, 288, 295–304. [Google Scholar] [CrossRef]
- Evans, J.; Goedecke, J.H.; Soderstrom, I.; Buren, J.; Alvehus, M.; Blomquist, C.; Jonsson, F.; Hayes, P.M.; Adams, K.; Dave, J.A.; et al. Depot- and ethnic-specific differences in the relationship between adipose tissue inflammation and insulin sensitivity. Clin. Endocrinol. 2011, 74, 51–59. [Google Scholar] [CrossRef]
- Kotze-Horstmann, L.M.; Keswell, D.; Adams, K.; Dlamini, T.; Goedecke, J.H. Hypoxia and extra-cellular matrix gene expression in adipose tissue associates with reduced insulin sensitivity in black South African women. Endocrine 2016, 55, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Goedecke, J.H.; Levitt, N.S.; Evans, J.; Ellman, N.; Hume, D.J.; Kotze, L.; Tootla, M.; Victor, H.; Keswell, D. The role of adipose tissue in insulin resistance in women of African ancestry. J. Obes. 2013, 2013, 952916. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.; Alvarez, J.A.; Ellis, A.C.; Granger, W.M.; Ovalle, F.; Man, C.D.; Cobelli, C.; Gower, B.A. Race differences in the association of oxidative stress with insulin sensitivity in African- and European-American women. Obesity 2012, 20, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Ling, K.L.; Bekele, Y.; Gerbino, E.; Earle, K.A. Lipid hydroperoxide and markers of renal disease susceptibility in African-Caribbean and Caucasian patients with Type 2 diabetes mellitus. Diabet. Med. 2001, 18, 109–115. [Google Scholar] [CrossRef]
- Feairheller, D.L.; Park, J.-Y.; Sturgeon, K.M.; Williamson, S.T.; Diaz, K.M.; Veerabhadrappa, P.; Brown, M.D. Racial Differences in Oxidative Stress and Inflammation: In Vitro and In Vivo. Clin. Transl. Sci. 2011, 4, 32–37. [Google Scholar] [CrossRef]
- Morris, A.A.; Zhao, L.; Patel, R.S.; Jones, D.P.; Ahmed, Y.; Stoyanova, N.; Gibbons, G.H.; Vaccarino, V.; Din-Dzietham, R.; Quyyumi, A.A. Differences in Systemic Oxidative Stress Based on Race and the Metabolic Syndrome: The Morehouse and Emory Team up to Eliminate Health Disparities (META-Health) Study. Metab. Syndr. Relat. Disord. 2012, 10, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Nguemeni Tiako, M.J.; Stanford, F.C. Race, racism and disparities in obesity rates in the US. J. Intern. Med. 2020, 288, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharm. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Kaviarasan, S.; Muniandy, S.; Qvist, R.; Ismail, I. F2-Isoprostanes as Novel Biomarkers for Type 2 Diabetes: A Review. J. Clin. Biochem. Nutr. 2009, 45, 1–8. [Google Scholar] [CrossRef]
- Zarrouki, B.; Soares, A.F.; Guichardant, M.; Lagarde, M.; Géloën, A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Lett. 2007, 581, 2394–2400. [Google Scholar] [CrossRef]
- Ovadia, H.; Haim, Y.; Nov, O.; Almog, O.; Kovsan, J.; Bashan, N.; Benhar, M.; Rudich, A. Increased Adipocyte S-Nitrosylation Targets Anti-lipolytic Action of Insulin. J. Biol. Chem. 2011, 286, 30433–30443. [Google Scholar] [CrossRef]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2015, 20, 709–711. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lucchini, F.C.; Wueest, S.; Challa, T.D.; Item, F.; Modica, S.; Borsigova, M.; Haim, Y.; Wolfrum, C.; Rudich, A.; Konrad, D. ASK1 inhibits browning of white adipose tissue in obesity. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Haim, Y.; Blüher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Maixner, N.; Bechor, S.; Vershinin, Z.; Pecht, T.; Goldstein, N.; Haim, Y.; Rudich, A. Transcriptional Dysregulation of Adipose Tissue Autophagy in Obesity. Physiology 2016, 31, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.K.; Gowder, S.J.T. Members of Antioxidant Machinery and Their Functions. In Basic Principles and Clinical Significance of Oxidative Stress; IntechOpen Limited. 5 Princes Gate Court: London, UK, 2015. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2019, 54, 287–293. [Google Scholar] [CrossRef]
- Niedowicz, D.M.; Daleke, D.L. The Role of Oxidative Stress in Diabetic Complications. Cell Biochem. Biophisics 2005, 43, 289–330. [Google Scholar]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J.; et al. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham Study. Arter. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef]
- Warolin, J.; Coenen, K.R.; Kantor, J.L.; Whitaker, L.E.; Wang, L.; Acra, S.A.; Roberts, L.J., 2nd; Buchowski, M.S. The relationship of oxidative stress, adiposity and metabolic risk factors in healthy Black and White American youth. Pediatr. Obes. 2014, 9, 43–52. [Google Scholar] [CrossRef]
- Elrayess, M.A.; Almuraikhy, S.; Kafienah, W.; Al-Menhali, A.; Al-Khelaifi, F.; Bashah, M.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Alsayrafi, M.; et al. 4-hydroxynonenal causes impairment of human subcutaneous adipogenesis and induction of adipocyte insulin resistance. Free Radic. Biol. Med. 2017, 104, 129–137. [Google Scholar] [CrossRef]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulere, L.; Lagarde, M.; Soulage, C.O. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef]
- Montes-Nieto, R.; Insenser, M.; Murri, M.; Fernández-Durán, E.; Ojeda-Ojeda, M.; Martínez-García, M.Á.; Luque-Ramírez, M.; Escobar-Morreale, H.F. Plasma thiobarbituric acid reactive substances (TBARS) in young adults: Obesity increases fasting levels only in men whereas glucose ingestion, and not protein or lipid intake, increases postprandial concentrations regardless of sex and obesity. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Kar, K.; Bhattacharyya, A.; Paria, B. Elevated MDA Level Correlates with Insulin Resistance in Prediabetes. J. Clin. Diagn. Res. 2018. [Google Scholar] [CrossRef]
- Tinahones, F.J.; Murri-Pierri, M.; Garrido-Sanchez, L.; Garcia-Almeida, J.M.; Garcia-Serrano, S.; Garcia-Arnes, J.; Garcia-Fuentes, E. Oxidative stress in severely obese persons is greater in those with insulin resistance. Obesity 2009, 17, 240–246. [Google Scholar] [CrossRef]
- Vincent, H.K.; Taylor, A.G. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int. J. Obes. 2006, 30, 400–418. [Google Scholar] [CrossRef]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Skoumas, I.; Papademetriou, L.; Economou, M.; Stefanadis, C. The implication of obesity on total antioxidant capacity in apparently healthy men and women: The ATTICA study. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 590–597. [Google Scholar] [CrossRef]
- Heit, C.; Marshall, S.; Singh, S.; Yu, X.; Charkoftaki, G.; Zhao, H.; Orlicky, D.J.; Fritz, K.S.; Thompson, D.C.; Vasiliou, V. Catalase deletion promotes prediabetic phenotype in mice. Free Radic. Biol. Med. 2017, 103, 48–56. [Google Scholar] [CrossRef]
- Shin, S.K.; Cho, H.W.; Song, S.E.; Im, S.S.; Bae, J.H.; Song, D.K. Oxidative stress resulting from the removal of endogenous catalase induces obesity by promoting hyperplasia and hypertrophy of white adipocytes. Redox Biol. 2020, 37, 101749. [Google Scholar] [CrossRef]
- Jankovic, A.; Korac, A.; Srdic-Galic, B.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Vucetic, M.; Markelic, M.; Velickovic, K.; Golic, I.; et al. Differences in the redox status of human visceral and subcutaneous adipose tissues--relationships to obesity and metabolic risk. Metabolism 2014, 63, 661–671. [Google Scholar] [CrossRef]
- Fukai, T.; Folz, R.J.; Landmesser, U.; Harrison, D.G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc. Res. 2002, 55, 239–249. [Google Scholar] [CrossRef]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef]
- László, G.; Rass, P.; Páy, A. Catalase Enzyme Mutations and their Association with Diseases. Mol. Diagn. 2004, 8, 141–149. [Google Scholar]
- Shin, S.K.; Cho, H.W.; Song, S.E.; Bae, J.H.; Im, S.S.; Hwang, I.; Ha, H.; Song, D.K. Ablation of catalase promotes non-alcoholic fatty liver via oxidative stress and mitochondrial dysfunction in diet-induced obese mice. Pflug. Arch. 2019, 471, 829–843. [Google Scholar] [CrossRef]
- García-Sánchez, A.; Gámez-Nava, J.I.; Díaz-de la Cruz, E.N.; Cardona-Muñoz, E.G.; Becerra-Alvarado, I.N.; Aceves-Aceves, J.A.; Sánchez-Rodríguez, E.N.; Miranda-Díaz, A.G. The Effect of Visceral Abdominal Fat Volume on Oxidative Stress and Proinflammatory Cytokines in Subjects with Normal Weight, Overweight and Obesity. DiabetesMetab. Syndr. Obes. Targets Ther. 2020, 13, 1077–1087. [Google Scholar] [CrossRef]
- Kelli, H.M.; Corrigan, F.E., 3rd; Heinl, R.E.; Dhindsa, D.S.; Hammadah, M.; Samman-Tahhan, A.; Sandesara, P.; O’Neal, W.T.; Al Mheid, I.; Ko, Y.A.; et al. Relation of Changes in Body Fat Distribution to Oxidative Stress. Am. J. Cardiol. 2017, 120, 2289–2293. [Google Scholar] [CrossRef]
- Evans, J.L.; Maddux, B.A.; Goldfine, I. The Molecular Basis for Oxidative Stress-Induced Insulin Resistance. Antioxid. Redox. Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Billeter, A.T.; Vittas, S.; Israel, B.; Scheurlen, K.M.; Hidmark, A.; Fleming, T.H.; Kopf, S.; Buchler, M.W.; Muller-Stich, B.P. Gastric bypass simultaneously improves adipose tissue function and insulin-dependent type 2 diabetes mellitus. Langenbecks Arch. Surg. 2017, 402, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Miljkovic, M.; Stefanovic, A.; Simic-Ogrizovic, S.; Vekic, J.; Bogavac-Stanojevic, N.; Cerne, D.; Kocbek, P.; Marc, J.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; et al. Association of Dyslipidemia, Oxidative Stress, and Inflammation With Redox Status in VLDL, LDL, and HDL Lipoproteins in Patients With Renal Disease. Angiology 2018, 69, 861–870. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arter. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef]
- Han, C.Y.; Umemoto, T.; Omer, M.; Den Hartigh, L.J.; Chiba, T.; LeBoeuf, R.; Buller, C.L.; Sweet, I.R.; Pennathur, S.; Abel, E.D.; et al. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 2012, 287, 10379–10393. [Google Scholar] [CrossRef]
- Ronquillo, M.D.; Mellnyk, A.; Cardenas-Rodriguez, N.; Martinez, E.; Comoto, D.A.; Carmona-Aparicio, L.; Herrera, N.E.; Lara, E.; Pereyra, A.; Floriano-Sanchez, E. Different gene expression profiles in subcutaneous & visceral adipose tissues from Mexican patients with obesity. Indian J. Med. Res. 2019, 149, 616–626. [Google Scholar] [CrossRef]
- Akl, M.G.; Fawzy, E.; Deif, M.; Farouk, A.; Elshorbagy, A.K. Perturbed adipose tissue hydrogen peroxide metabolism in centrally obese men: Association with insulin resistance. PLoS ONE 2017, 12, e0177268. [Google Scholar] [CrossRef]
- Preis, S.R.; Massaro, J.M.; Robins, S.J.; Hoffmann, U.; Vasan, R.S.; Irlbeck, T.; Meigs, J.B.; Sutherland, P.; D’Agostino, R.B., Sr.; O’Donnell, C.J.; et al. Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity 2010, 18, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Krishnaswami, S.; Harris, T.B.; Katsiaras, A.; Kritchevsky, S.B.; Simonsick, E.M.; Nevitt, M.; Holvoet, P.; Newman, A.B. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Arch. Intern. Med. 2005, 165, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Desprès, J.P. Abdominal obesity as important component of insulin-resistance syndrome. Nutrition 1993, 9, 452–459. [Google Scholar] [PubMed]
- Patel, P.; Abate, N. Body fat distribution and insulin resistance. Nutrients 2013, 5, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.L.; Parkinson, J.R.; Frost, G.S.; Goldstone, A.P.; Dore, C.J.; McCarthy, J.P.; Collins, A.L.; Fitzpatrick, J.A.; Durighel, G.; Taylor-Robinson, S.D.; et al. The missing risk: MRI and MRS phenotyping of abdominal adiposity and ectopic fat. Obesity 2012, 20, 76–87. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H Oxidase Homolog Nox4 Modulates Insulin-Stimulated Generation of H2O2 and Plays an Integral Role in Insulin Signal Transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef]
- Sakurai, T.; Ogasawara, J.; Shirato, K.; Izawa, T.; Oh-ishi, S.; Ishibashi, Y.; Radák, Z.; Ohno, H.; Kizaki, T. Exercise Training Attenuates the Dysregulated Expression of Adipokines and Oxidative Stress in White Adipose Tissue. Oxidative Med. Cell. Longev. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.-i.; et al. Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 2008, 57, 1071–1077. [Google Scholar] [CrossRef]
- Fernandez-Sanchez, A.; Madrigal-Santillan, E.; Bautista, M.; Esquivel-Soto, J.; Morales-Gonzalez, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sanchez-Rivera, G.; Valadez-Vega, C.; Morales-Gonzalez, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Gao, C.L.; Zhu, C.; Zhao, Y.P.; Chen, X.; Ji, C.; Zahang, C.; Zhu, J.; Xia, Z.; Tomng, M.; Guo, X. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3- L1 adipocytes. Mol. Cell. Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wang, C.C.; Huang, H.C.; Wei, Y.H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS production and oxidative damage in subcutaneous white adipose tissue mitochondria in obese and type 2 diabetes subjects. Mol. Cell Biochem. 2015, 399, 95–103. [Google Scholar] [CrossRef]
- Goossens, G.H. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol. Behav. 2008, 94, 206–218. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef]
- Gealekman, O.; Guseva, N.; Hartigan, C.; Apotheker, S.; Gorgoglione, M.; Gurav, K.; Tran, K.V.; Straubhaar, J.; Nicoloro, S.; Czech, M.P.; et al. Depot-specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation 2011, 123, 186–194. [Google Scholar] [CrossRef]
- Lacobini, C.; Pugliese, G.; Blasetti Fantauzzi, C.; Federici, M.; Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metabolism 2019, 92, 51–60. [Google Scholar] [CrossRef]
- Pasarica, M.; Sereda, O.R.; Redman, L.M.; Albarado, D.C.; Hymel, D.T.; Roan, L.E.; Rood, J.C.; Burk, D.H.; Smith, S.R. Reduced adipose tissue oxygenation in human obesity: Evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 2009, 58, 718–725. [Google Scholar] [CrossRef]
- Trayhurn, P. Hypoxia and adipocyte physiology: Implications for adipose tissue dysfunction in obesity. Annu. Rev. Nutr. 2014, 34, 207–236. [Google Scholar] [CrossRef]
- Trayhurn, P.; Alomar, S.Y. Oxygen Deprivation and the Cellular Response to Hypoxia in Adipocytes - Perspectives on White and Brown Adipose Tissues in Obesity. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; González-Campoy, J.M.; Bray, G.A. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev. Cardiovasc. 2008, 6, 343–368. [Google Scholar] [CrossRef] [PubMed]
- Devries, M.C.; Hamadeh, M.J.; Glover, A.W.; Raha, S.; Samjoo, I.A.; Tarnopolsky, M.A. Endurance training without weight loss lowers systemic, but not muscle, oxidative stress with no effect on inflammation in lean and obese women. Free Radic. Biol. Med. 2008, 45, 503–511. [Google Scholar] [CrossRef]
- Han, C.Y. Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue. Diabetes Metab. J. 2016, 40, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Fukuhara, A.; Hashimoto, E.; Kobayashi, H.; Kobayashi, S.; Otsuki, M.; Shimomura, I. Oxidative Stress Inhibits Healthy Adipose Expansion Through Suppression of SREBF1-Mediated Lipogenic Pathway. Diabetes 2018, 67, 1113–1127. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.; Bloomer, R.J. Acute exercise and oxidative stress: A 30 year history. Dyn. Med. 2009, 8, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, Y.J.; Choi, H.; Ko, E.H.; Kim, J.W. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 2009, 284, 10601–10609. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Dusting, G.J.; Peshavariya, H.; Jiang, F.; Hsiao, S.T.; Chan, E.C.; Liu, G.S. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead box O1 mediated upregulation of antioxidant enzymes. Stem. Cells Dev. 2013, 22, 878–888. [Google Scholar] [CrossRef]
- Mouche, S.; Mkaddem, S.B.; Wang, W.; Katic, M.; Tseng, Y.-H.; Carnesecchi, S.; Steger, K.; Foti, M.; Meier, C.A.; Muzzin, P.; et al. Reduced expression of the NADPH oxidase NOX4 is a hallmark of adipocyte differentiation. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1015–1027. [Google Scholar] [CrossRef]
- Schroder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arter. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef]
- Hutchings, G.; Janowicz, K.; Moncrieff, L.; Dompe, C.; Strauss, E.; Kocherova, I.; Nawrocki, M.J.; Kruszyna, L.; Wasiatycz, G.; Antosik, P.; et al. The Proliferation and Differentiation of Adipose-Derived Stem Cells in Neovascularization and Angiogenesis. Int. J. Mol. Sci. 2020, 21, 3790. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Maslov, L.N.; Naryzhnaya, N.V.; Boshchenko, A.A.; Popov, S.V.; Ivanov, V.V.; Oeltgen, P.R. Is oxidative stress of adipocytes a cause or a consequence of the metabolic syndrome? J. Clin. Transl. Endocrinol. 2019, 15, 1–5. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Marra, F. Adipokines and redox signaling: Impact on fatty liver disease. Antioxid. Redox Signal. 2011, 15, 461–483. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Edelstein, D.; Du, X.L.; Kaneda, Y.; Guzman, M.; Brownlee, M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J. Biol. Chem. 2001, 276, 25096–25100. [Google Scholar] [CrossRef]
- Fortuno, A.; Bidegain, J.; Baltanas, A.; Moreno, M.U.; Montero, L.; Landecho, M.F.; Beloqui, O.; Diez, J.; Zalba, G. Is leptin involved in phagocytic NADPH oxidase overactivity in obesity? Potential clinical implications. J. Hypertens. 2010, 28, 1944–1950. [Google Scholar] [CrossRef]
- Zhang, H.; Park, Y.; Wu, J.; Chen, X.; Lee, S.; Yang, J.; Dellsperger, K.C.; Zhang, C. Role of TNF-alpha in vascular dysfunction. Clin. Sci. 2009, 116, 219–230. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, X.; Zheng, H.; Hu, D.; Zhang, Y.; Guan, Q.; Liu, L.; Ding, Q.; Li, Y. Clematichinenoside inhibits VCAM-1 and ICAM-1 expression in TNF-alpha-treated endothelial cells via NADPH oxidase-dependent IkappaB kinase/NF-kappaB pathway. Free Radic. Biol. Med. 2015, 78, 190–201. [Google Scholar] [CrossRef]
- Shen, H.-M.; Pervaiz, S. TNF receptor superfamily-induced cell death: Redox-dependent execution. FASEB J. 2006, 20, 1589–1598. [Google Scholar] [CrossRef]
- Skalicky, J.; Muzakova, V.; Kandar, R.; Meloun, M.; Rousar, T.; Palicka, V. Evaluation of oxidative stress and inflammation in obese adults with metabolic syndrome. Clin. Chem. Lab. Med. 2008, 46, 499–505. [Google Scholar] [CrossRef]
- Hulsmans, M.; Holvoet, P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell Mol. Med. 2010, 14, 70–78. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Murr, C.; Schroecksnadel, K.; Winkler, C.; Ledochowski, M.; Fuchs, D. Antioxidants may increase the probability of developing allergic diseases and asthma. Med. Hypotheses 2005, 64, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Dommel, S.; Bluher, M. Does C-C Motif Chemokine Ligand 2 (CCL2) Link Obesity to a Pro-Inflammatory State? Int. J. Mol. Sci. 2021, 22, 1500. [Google Scholar] [CrossRef]
- Stults Bashan, N.; Kovsan, J.; Kachko, I.; Ovadia, H.; Rudich, A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev. 2009, 89, 27–71. [Google Scholar] [CrossRef]
- Bloch-Damti, A.; Bashan, N. Proposed Mechanisms for the Induction of Insulin Resistance by Oxidative Stress. Antioxid. Redox Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Goedecke, J.H.; Evans, J.; Keswell, D.; Stimson, R.H.; Livingstone, D.E.; Hayes, P.; Adams, K.; Dave, J.A.; Victor, H.; Levitt, N.S.; et al. Reduced gluteal expression of adipogenic and lipogenic genes in Black South African women is associated with obesity-related insulin resistance. J. Clin. Endocrinol. Metab. 2011, 96, E2029–E2033. [Google Scholar] [CrossRef]
- Shay, C.M.; Carnethon, M.R.; Church, T.R.; Hankinson, A.L.; Chan, C.; Jacobs, D.R., Jr.; Lewis, C.E.; Schreiner, P.J.; Sternfeld, B.; Sidney, S. Lower extremity fat mass is associated with insulin resistance in overweight and obese individuals: The CARDIA study. Obesity 2011, 19, 2248–2253. [Google Scholar] [CrossRef]
- Keswell, D.; Tootla, M.; Goedecke, J.H. Associations between body fat distribution, insulin resistance and dyslipidaemia in black and white South African women. Cardiovasc. J. Afr. 2016, 27, 177–183. [Google Scholar] [CrossRef]
- Rush, E.C.; Goedecke, J.H.; Jennings, C.; Micklesfield, L.; Dugas, L.; Lambert, E.V.; Plank, L.D. BMI, fat and muscle differences in urban women of five ethnicities from two countries. Int. J. Obes. 2007, 31, 1232–1239. [Google Scholar] [CrossRef]
- Pou, K.M.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; Maurovich-Horvat, P.; Larson, M.G.; Keaney, J.F., Jr.; Meigs, J.B.; Lipinska, I.; Kathiresan, S.; et al. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: The Framingham Heart Study. Circulation 2007, 116, 1234–1241. [Google Scholar] [CrossRef]
- Sriram, S.; Yuan, C.; Chakraborty, S.; Tay, W.; Park, M.; Shabbir, A.; Toh, S.-A.; Han, W.; Sugii, S. Oxidative stress mediates depot-specific functional differences of human adipose-derived stem cells. Stem. Cell Res. Ther. 2019, 10. [Google Scholar] [CrossRef]
- Rydén, M.; Elizalde, M.; van Harmelen, V.; Öhlund, A.; Hoffstedt, J.; Bringman, S.; Andersson, K. Increased expression of eNOS protein in omental versus subcutaneous adipose tissue in obese human subjects. Int. J. Obes. 2001, 25, 811–815. [Google Scholar] [CrossRef]
- Galvin, V.B.; Barakat, H.; Kemeny, G.; Macdonald, K.G.; Pories, W.J.; Hickner, R.C. Endothelial nitric oxide synthase content in adipose tissue from obese and lean African American and white American women. Metabolism 2005, 54, 1368–1373. [Google Scholar] [CrossRef]
- Carroll, J.F.; Chiapa, A.L.; Rodriquez, M.; Phelps, D.R.; Cardarelli, K.M.; Vishwanatha, J.K.; Bae, S.; Cardarelli, R. Visceral fat, waist circumference, and BMI: Impact of race/ethnicity. Obesity 2008, 16, 600–607. [Google Scholar] [CrossRef]
- Carroll, J.F.; Fulda, K.G.; Chiapa, A.L.; Rodriquez, M.; Phelps, D.R.; Cardarelli, K.M.; Vishwanatha, J.K.; Cardarelli, R. Impact of race/ethnicity on the relationship between visceral fat and inflammatory biomarkers. Obesity 2009, 17, 1420–1427. [Google Scholar] [CrossRef]
- Van der Merwe, M.T.; Crowther, N.J.; Schlaphoff, G.P.; Boyd, I.H.; Gray, I.P.; Joffe, B.I.; Lönnroth, P.N. Lactate and glycerol release from the subcutaneous adipose tissue of obese urban women from South Africa; important metabolic implications. J. Clin. Endocrinol. Metab. 1998, 83, 4084–4091. [Google Scholar] [CrossRef]
- Lovejoy, J.C.; de la Bretonne, J.A.; Klemperer, M.; Tulley, R. Abdominal fat distribution and metabolic risk factors: Effects of race. Metabolism 1996, 145, 1119–1124. [Google Scholar] [CrossRef]
- Goedecke, J.H.; Dave, J.A.; Faulenbach, M.V.; Utzschneider, K.M.; Lambert, E.V.; West, S.; Collins, M.; Olsson, T.; Walker, B.R.; Seckl, J.R.; et al. Insulin response in relation to insulin sensitivity: An appropriate beta-cell response in black South African women. Diabetes Care 2009, 32, 860–865. [Google Scholar] [CrossRef]
- Goedecke, J.H.; Mtintsilana, A.; Dlamini, S.N.; Kengne, A.P. Type 2 diabetes mellitus in African women. Diabetes Res. Clin. Pr. 2017, 123, 87–96. [Google Scholar] [CrossRef]
- Elizalde, M.; Rydén, M.; van Harmelen, V.; Eneroth, P.; Gyllenhammar, H.; Holm, C.; Ramel, S.; Ölund, A.; Arner, P.; Andersson, K. Expression of nitric oxide synthases in subcutaneous adipose tissue of nonobese and obese humans. J. Lipid. Res. 2000, 41, 1244–1251. [Google Scholar] [CrossRef]
- Nono Nankam, P.A.; Mendham, A.; De Smidt, M.; Keswell, D.; Olsson, T.; Bluher, M.; Goedecke, J. Changes in systemic and subcutaneous adipose tissue inflammation and oxidative stress in response to exercise training in obese black African women. J. Physiol. 2020, 598, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Liu, B.; Sun, Y.; Du, Y.; Snetselaar, L.G.; Hu, F.B.; Bao, W. Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: Population based study. BMJ 2018, 362, k1497. [Google Scholar] [CrossRef]
- Duke, C.M.P.; Plowden, T.C.; Armstrong, A.Y. Disparate Cardiovascular and Diabetic Outcomes in Young Adult Black Women: Studies from across the Globe. Curr. Cardiovasc. Risk Rep. 2012, 6, 251–258. [Google Scholar] [CrossRef]
- Wright, E.; Scism-Bacon, J.L.; Glass, L.C. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pr. 2006, 60, 308–314. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Aronson, D.; Rayfield, E.J. How hyperglycemia promotes atherosclerosis: Molecular mechanisms. Cardiovasc. Diabetol 2002, 1, 1–10. [Google Scholar] [CrossRef]
- Smith, L.M.; Yao-Borengasser, A.; Starks, T.; Tripputi, M.; Kern, P.A.; Rasouli, N. Insulin resistance in African-American and Caucasian women: Differences in lipotoxicity, adipokines, and gene expression in adipose tissue and muscle. J. Clin. Endocrinol. Metab. 2010, 95, 4441–4448. [Google Scholar] [CrossRef]
- Kim, C.X.; Bailey, K.R.; Klee, G.G.; Ellington, A.A.; Liu, G.; Mosley, T.H., Jr.; Rehman, H.; Kullo, I.J. Sex and ethnic differences in 47 candidate proteomic markers of cardiovascular disease: The Mayo Clinic proteomic markers of arteriosclerosis study. PLoS ONE 2010, 5, e9065. [Google Scholar] [CrossRef]
- Festa, A.; D’Agostino, R., Jr.; Rich, S.S.; Jenny, N.S.; Tracy, R.P.; Haffner, S.M. Promoter (4G/5G) plasminogen activator inhibitor-1 genotype and plasminogen activator inhibitor-1 levels in blacks, Hispanics, and non-Hispanic whites: The Insulin Resistance Atherosclerosis Study. Circulation 2003, 107, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Lopes, H.; Morrow, J.D.; Stoijiljkovic, M.P.; Goodfriend, T.L.; Egan, B.M. Acute Hyperlipidemia Increases Oxidative Stress More in African Americans Than in White Americans. Am. J. Hypertens. 2003, 16, 331–336. [Google Scholar] [CrossRef]
- Aseervatham, G.S.; Sivasudha, T.; Jeyadevi, R.; Arul Ananth, D. Environmental factors and unhealthy lifestyle influence oxidative stress in humans--an overview. Environ. Sci. Pollut. Res. Int. 2013, 20, 4356–4369. [Google Scholar] [CrossRef] [PubMed]
- Rolle-Kampczyk, U.; Gebauer, S.; Haange, S.B.; Schubert, K.; Kern, M.; Moulla, Y.; Dietrich, A.; Schon, M.R.; Kloting, N.; von Bergen, M.; et al. Accumulation of distinct persistent organic pollutants is associated with adipose tissue inflammation. Sci. Total Environ. 2020, 748, 142458. [Google Scholar] [CrossRef] [PubMed]
- Janicki-Deverts, D.; Cohen, S.; Matthews, K.A.; Gross, M.D.; Jacobs, D.R., Jr. Socioeconomic status, antioxidant micronutrients, and correlates of oxidative damage: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Psychosom. Med. 2009, 71, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Seyedsadjadi, N.; Berg, J.; Bilgin, A.A.; Tung, C.; Grant, R. Significant relationships between a simple marker of redox balance and lifestyle behaviours; Relevance to the Framingham risk score. PLoS ONE 2017, 12, e0187713. [Google Scholar] [CrossRef] [PubMed]
- Cruz dos Santos, R.R.; Rosa, C.; Gris, E.F.; Ferreira, E.A.; Rosa, T.; Vieira de Andrade, R.; Amato, A. Association between Lifestyle and Oxidative Stress Markers in Young Active Men. J. Exerc. Physiol. Online 2019, 22, 75–86. [Google Scholar]
- Goedecke, J.H. Addressing the problem of obesity and associated cardiometabolic risk in black South African women-time for action! Glob. Health Action 2017, 10, 1366165. [Google Scholar] [CrossRef]
- Eick, S.M.; Meeker, J.D.; Brown, P.; Swartzendruber, A.; Rios-McConnell, R.; Shen, Y.; Milne, G.L.; Vélez Vega, C.; Rosario, Z.; Alshawabkeh, A.; et al. Associations between socioeconomic status, psychosocial stress, and urinary levels of 8-iso-prostaglandin-F2α during pregnancy in Puerto Rico. Free Radic. Biol. Med. 2019, 143, 95–100. [Google Scholar] [CrossRef]
- Bell, C.N.; Kerr, J.; Young, J.L. Associations between Obesity, Obesogenic Environments, and Structural Racism Vary by County-Level Racial Composition. Int. J. Environ. Res. Public Health 2019, 16, 861. [Google Scholar] [CrossRef]
- Cozier, Y.C.; Yu, J.; Coogan, P.F.; Bethea, T.N.; Rosenberg, L.; Palmer, J.R. Racism, segregation, and risk of obesity in the Black Women’s Health Study. Am. J. Epidemiol. 2014, 179, 875–883. [Google Scholar] [CrossRef]
- Patel, C.; Ghanim, H.; Ravishankar, S.; Sia, C.L.; Viswanathan, P.; Mohanty, P.; Dandona, P. Prolonged reactive oxygen species generation and nuclear factor-kappaB activation after a high-fat, high-carbohydrate meal in the obese. J. Clin. Endocrinol. Metab. 2007, 92, 4476–4479. [Google Scholar] [CrossRef]
- Vincent, H.K.; Powers, S.K.; Dirks, A.J.; Scarpace, P.J. Mechanism for obesity-induced increase in myocardial lipid peroxidation. Int. J. Obes. 2001, 25, 378–388. [Google Scholar] [CrossRef]
- Beltowski, J.; Wojcicka, G.; Gorny, D.; Marciniak, A. The effect of dietary-induced obesity on lipid peroxidation, antioxidant enzymes and total plasma antioxidant capacity. J. Physiol. Pharm. 2000, 51, 883–896. [Google Scholar]
- Nono Nankam, P.A.; Jaarsveld, P.J.V.; Chorell, E.; Smidt, M.C.F.; Adams, K.; Bluher, M.; Olsson, T.; Mendham, A.E.; Goedecke, J.H. Circulating and Adipose Tissue Fatty Acid Composition in Black South African Women with Obesity: A Cross-Sectional Study. Nutrients 2020, 12, 1619. [Google Scholar] [CrossRef]
- MacIntyre, U.; Kruger, H.; Venter, C.; Vorster, H. Dietary intakes of an African population in different stages of transition in the North West Province, South Africa: The THUSA study. Nutr. Res. 2002, 22, 239–256. [Google Scholar] [CrossRef]
- Mendham, A.E.; Larsen, S.; George, C.; Adams, K.; Hauksson, J.; Olsson, T.; Fortuin-de Smidt, M.C.; Nono Nankam, P.A.; Hakim, O.; Goff, L.M.; et al. Exercise training results in depot-specific adaptations to adipose tissue mitochondrial function. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nono Nankam, P.A.; Nguelefack, T.B.; Goedecke, J.H.; Blüher, M. Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry. Antioxidants 2021, 10, 622. https://doi.org/10.3390/antiox10040622
Nono Nankam PA, Nguelefack TB, Goedecke JH, Blüher M. Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry. Antioxidants. 2021; 10(4):622. https://doi.org/10.3390/antiox10040622
Chicago/Turabian StyleNono Nankam, Pamela A., Télesphore B. Nguelefack, Julia H. Goedecke, and Matthias Blüher. 2021. "Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry" Antioxidants 10, no. 4: 622. https://doi.org/10.3390/antiox10040622
APA StyleNono Nankam, P. A., Nguelefack, T. B., Goedecke, J. H., & Blüher, M. (2021). Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry. Antioxidants, 10(4), 622. https://doi.org/10.3390/antiox10040622