
Redox Homeostasis in Pancreatic β-Cells: From Development to Failure


Abstract
1. Redox Homeostasis in β-Cell Development and Maturation
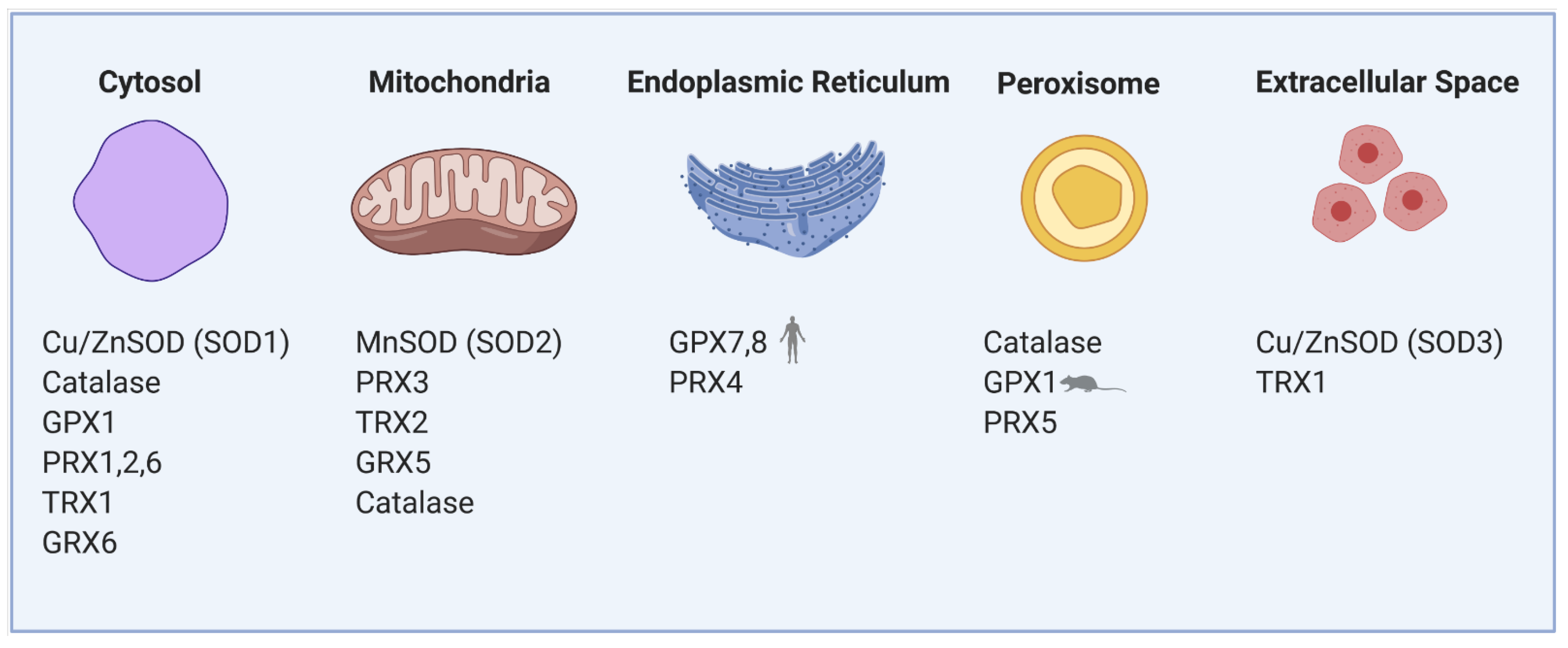
1.1. Sources of Reactive Oxygen Species and Antioxidants in β-Cells
1.1.1. Mitochondria
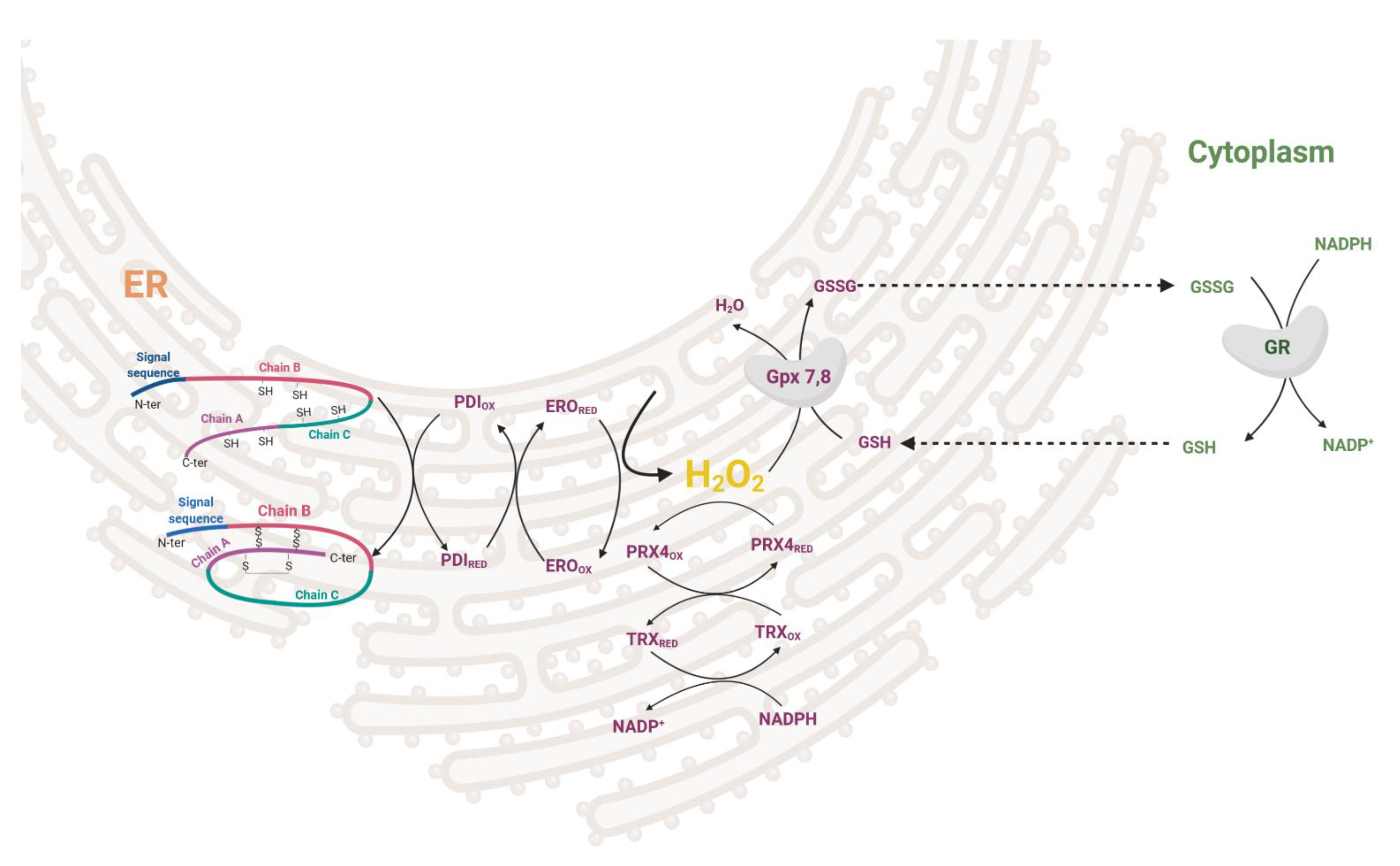
1.1.2. Endoplasmic Reticulum
1.1.3. Peroxisomes
1.1.4. Cytoplasm and Membrane Rafts
1.2. ROS Signaling Pathways and Transcription Factors in β-Cell Physiology
1.2.1. Redox Sensitive Signaling Pathways
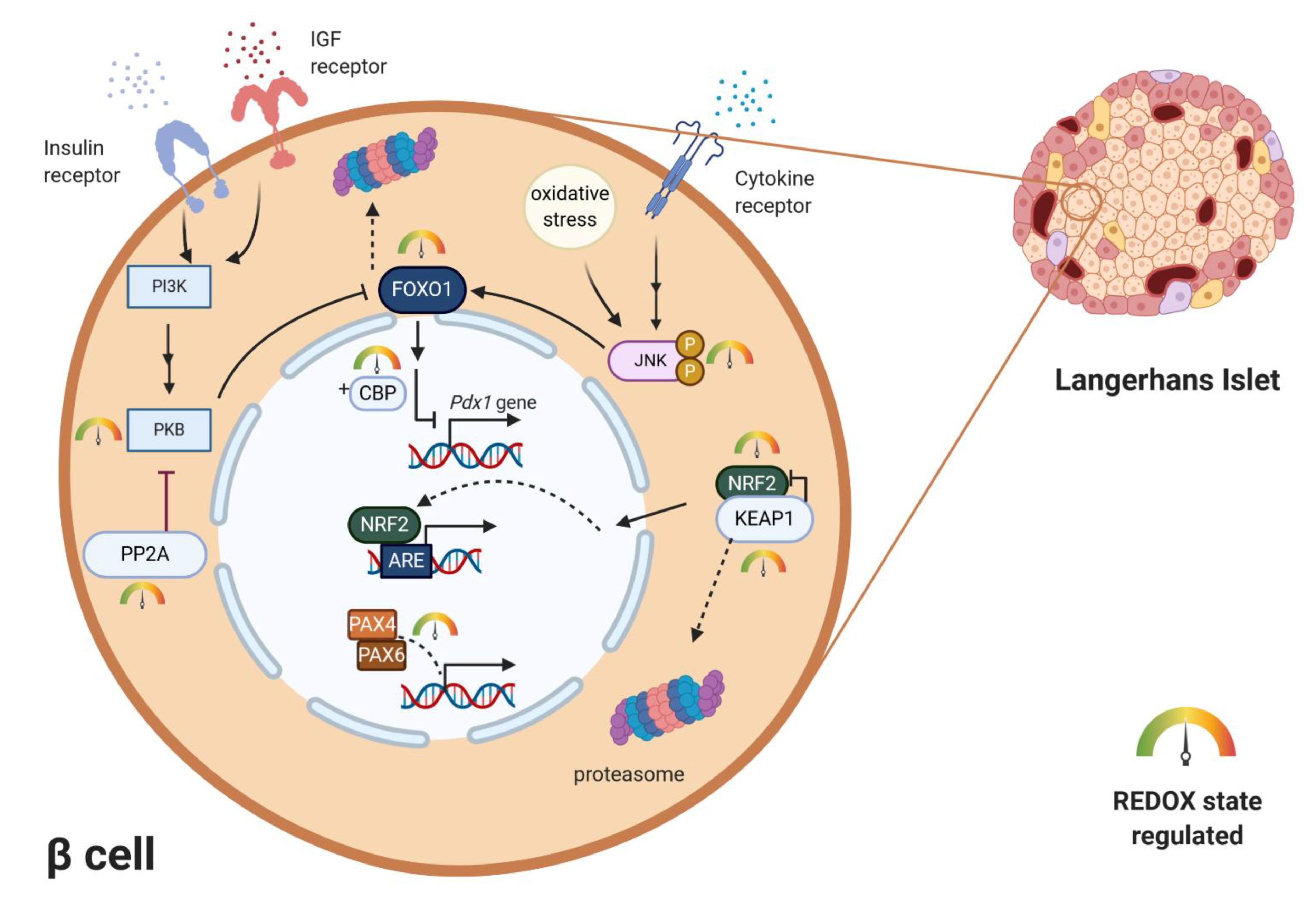
1.2.2. Redox Regulation of Transcription Factors Essential for β-Cell Maturation
1.3. Redox Regulation of β-Cell Differentiation and Proliferation
1.3.1. Redox Regulation of β-Cell Differentiation
1.3.2. Adipokines Stimulate β-Cells Proliferation through Redox Signaling
1.3.3. ROS-Mediated Ca2+ Signaling in β-Cell Proliferation
2. Redox Homeostasis in Optimum β-Cell Function
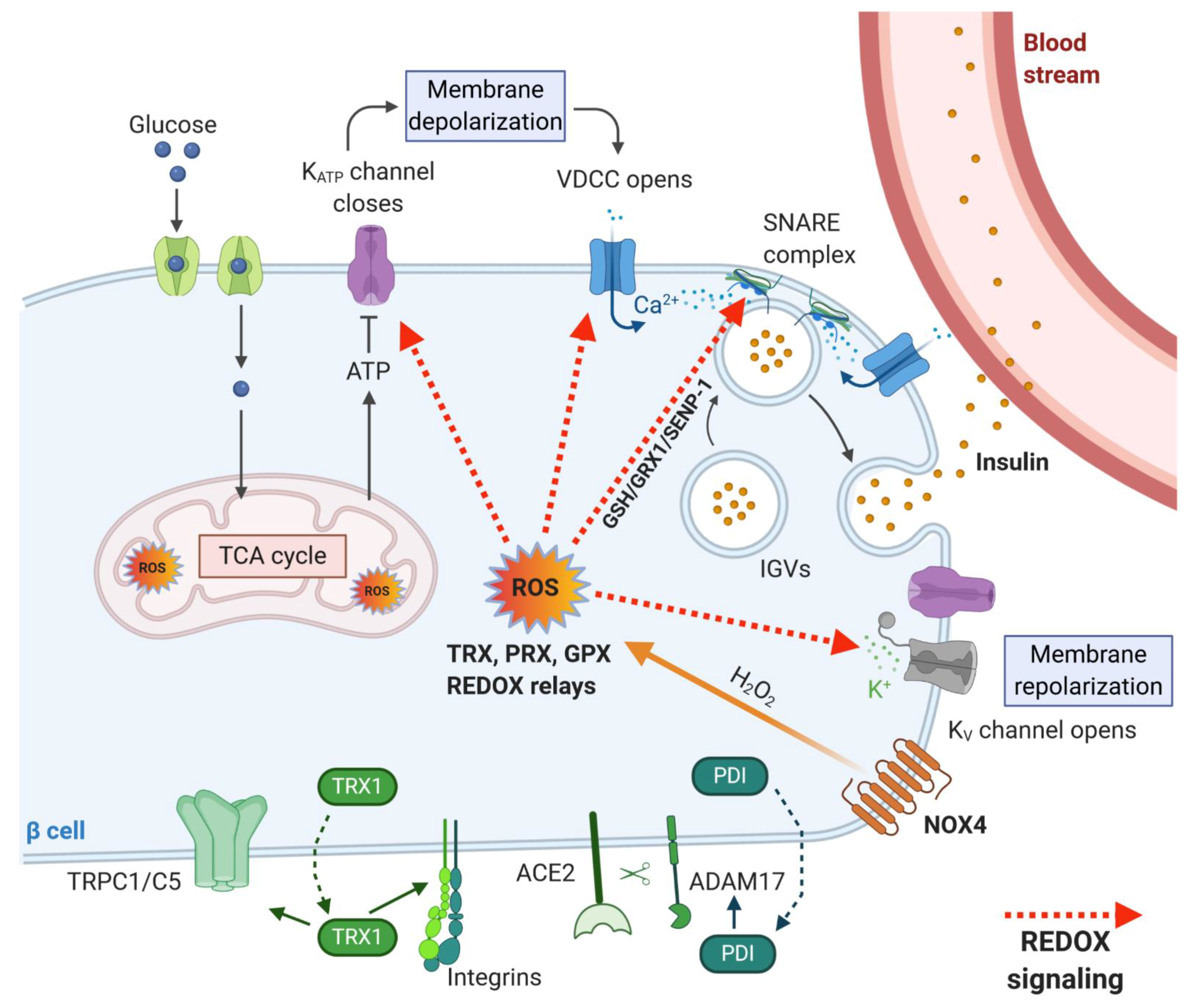
2.1. Redox Signaling in Insulin Secretory Pathway
2.2. Extracellular Redox Signaling in β-Cell Function
3. Dysbalanced Redox Homeostasis Contributing to β-Cell Dysfunction under Metabolic Stress
3.1. Redox-Linked Transition to Dysfunctional β-Cell in T2D Pathology
3.2. Affected Redox Homeostasis Impacts Insulin Biosynthesis in β-Cells
3.3. Adipose Tissue Hypertrophy-Induced β-Cell Dysfunction
3.4. Impaired Redox Homeostasis of the Immune System Leading to Β-Cell Dysfunction
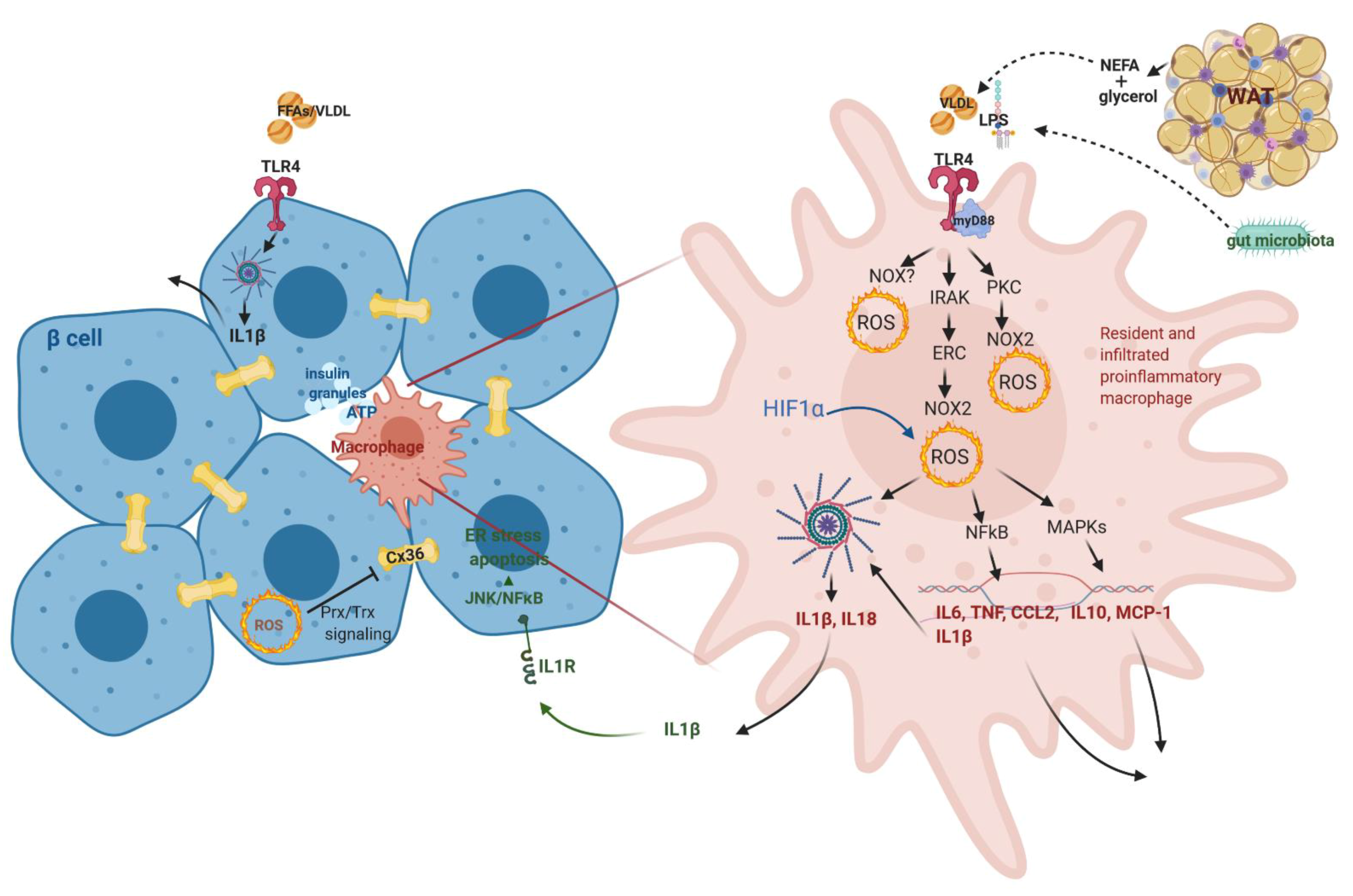
3.4.1. Macrophage Role in Langerhans Islets- from Guarding Β-Cell Activity to Its Dysfunction
3.4.2. IL1β Dual Role in β-Cell Function/Dysfunction
3.5. Gut Microbiota Supervision of the Immune System
3.6. β-Cell Dedifferentiation as the Adaptation to Metabolic and Redox Changes
3.7. β-Cell Communication in Islets under Redox Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Protein Name | Regulation | Dataset | Reference |
|---|---|---|---|---|
| TXN1 | Thioredoxin 1 | Up | GSE38642 | [237,238,239,240] |
| TXNR1 | Thioredoxin reductase 1 | Up | GSE26168 | [241] |
| TXNIP | Thioredoxin-interacting protein | Down | GSE26168 | [241] |
| PRX2 | Peroxiredoxin 2 | Up | GSE25724 | [242] |
| PRX4 | Peroxiredoxin 4 | Down | GSE25724 | [242] |
| PRX5 | Peroxiredoxin 5 | Up | GSE26168 | [241] |
| PRX6 | Peroxiredoxin 6 | Up | GSE38642 | [237,238,239,240] |
| GPX1 | Glutathione peroxidase 1 | Up | GSE26168 | [241] |
| SOD2 | Superoxide dismutase 2, mitochondrial | Up | GSE26168 | [241] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leloup, C.; Tourrel-Cuzin, C.; Magnan, C.; Karaca, M.; Castel, J.; Carneiro, L.; Colombani, A.L.; Ktorza, A.; Casteilla, L.; Penicaud, L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 2009, 58, 673–681. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Jaburek, M.; Holendova, B.; Tauber, J.; Pavluch, V.; Berkova, Z.; Cahova, M.; Schroeder, K.; Brandes, R.P.; Siemen, D.; et al. Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H. Oxidative Stress in Pancreatic Beta Cell Regeneration. Oxid. Med. Cell. Longev. 2017, 2017, 1930261. [Google Scholar] [CrossRef] [PubMed]
- Grankvist, K.; Marklund, S.L.; Taljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Miki, A.; Ricordi, C.; Sakuma, Y.; Yamamoto, T.; Misawa, R.; Mita, A.; Molano, R.D.; Vaziri, N.D.; Pileggi, A.; Ichii, H. Divergent antioxidant capacity of human islet cell subsets: A potential cause of beta-cell vulnerability in diabetes and islet transplantation. PLoS ONE 2018, 13, e0196570. [Google Scholar] [CrossRef]
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry 2008, 73, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Stancill, J.S.; Broniowska, K.A.; Oleson, B.J.; Naatz, A.; Corbett, J.A. Pancreatic beta-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J. Biol. Chem. 2019, 294, 4843–4853. [Google Scholar] [CrossRef]
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef]
- Roma, L.P.; Jonas, J.C. Nutrient Metabolism, Subcellular Redox State, and Oxidative Stress in Pancreatic Islets and beta-Cells. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef]
- Gurgul, E.; Lortz, S.; Tiedge, M.; Jorns, A.; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Engstova, H.; Jezek, J.; Holendova, B.; Tauber, J.; Petraskova, L.; Kren, V.; Jezek, P. Potential of Mitochondria-Targeted Antioxidants to Prevent Oxidative Stress in Pancreatic beta-cells. Oxid. Med. Cell. Longev. 2019, 2019, 1826303. [Google Scholar] [CrossRef] [PubMed]
- Plecita-Hlavata, L.; Engstova, H.; Holendova, B.; Tauber, J.; Spacek, T.; Petraskova, L.; Kren, V.; Spackova, J.; Gotvaldova, K.; Jezek, J.; et al. Mitochondrial Superoxide Production Decreases on Glucose-Stimulated Insulin Secretion in Pancreatic beta Cells Due to Decreasing Mitochondrial Matrix NADH/NAD(+) Ratio. Antioxid. Redox Signal. 2020, 33, 789–815. [Google Scholar] [CrossRef]
- Jezek, J.; Dlaskova, A.; Zelenka, J.; Jaburek, M.; Jezek, P. H(2)O(2)-Activated Mitochondrial Phospholipase iPLA(2)gamma Prevents Lipotoxic Oxidative Stress in Synergy with UCP2, Amplifies Signaling via G-Protein-Coupled Receptor GPR40, and Regulates Insulin Secretion in Pancreatic beta-Cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Riemer, J.; Zito, E.; Chin, K.T.; Ron, D.; Spiess, M.; Ellgaard, L. Disulphide production by Ero1alpha-PDI relay is rapid and effectively regulated. EMBO J. 2010, 29, 3318–3329. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Elsner, M.; Lenzen, S. Peroxiredoxin 4 improves insulin biosynthesis and glucose-induced insulin secretion in insulin-secreting INS-1E cells. J. Biol. Chem. 2014, 289, 26904–26913. [Google Scholar] [CrossRef]
- Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic beta-cells. Biochim Biophys Acta Gen. Subj. 2017, 1861, 1929–1942. [Google Scholar] [CrossRef]
- Tavender, T.J.; Sheppard, A.M.; Bulleid, N.J. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 2008, 411, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Hassler, J.R.; Scheuner, D.L.; Wang, S.; Han, J.; Kodali, V.K.; Li, P.; Nguyen, J.; George, J.S.; Davis, C.; Wu, S.P.; et al. The IRE1alpha/XBP1s Pathway Is Essential for the Glucose Response and Protection of beta Cells. PLoS Biol. 2015, 13, e1002277. [Google Scholar] [CrossRef] [PubMed]
- Pearse, B.R.; Hebert, D.N. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 684–693. [Google Scholar] [CrossRef]
- Plemper, R.K.; Bohmler, S.; Bordallo, J.; Sommer, T.; Wolf, D.H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 1997, 388, 891–895. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Brozzi, F.; Eizirik, D.L. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J. Med. Sci. 2016, 121, 133–139. [Google Scholar] [CrossRef]
- Baboota, R.K.; Shinde, A.B.; Lemaire, K.; Fransen, M.; Vinckier, S.; Van Veldhoven, P.P.; Schuit, F.; Baes, M. Functional peroxisomes are required for beta-cell integrity in mice. Mol. Metab. 2019, 22, 71–83. [Google Scholar] [CrossRef]
- Oliveira, H.R.; Verlengia, R.; Carvalho, C.R.; Britto, L.R.; Curi, R.; Carpinelli, A.R. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003, 52, 1457–1463. [Google Scholar] [CrossRef]
- Uchizono, Y.; Takeya, R.; Iwase, M.; Sasaki, N.; Oku, M.; Imoto, H.; Iida, M.; Sumimoto, H. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci. 2006, 80, 133–139. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Yang, L.; Chen, R.; Yang, R.; Zhang, H.; Cai, D.; Chen, H. The cytotoxic role of intermittent high glucose on apoptosis and cell viability in pancreatic beta cells. J. Diabetes Res. 2014, 2014, 712781. [Google Scholar] [CrossRef] [PubMed]
- Stancill, J.S.; Happ, J.T.; Broniowska, K.A.; Hogg, N.; Corbett, J.A. Peroxiredoxin 1 plays a primary role in protecting pancreatic β-cells from hydrogen peroxide and peroxynitrite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R1004–R1013. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef]
- Wondafrash, D.Z.; Nire’a, A.T.; Tafere, G.G.; Desta, D.M.; Berhe, D.A.; Zewdie, K.A. Thioredoxin-Interacting Protein as a Novel Potential Therapeutic Target in Diabetes Mellitus and Its Underlying Complications. Diabetes Metab. Syndr. Obes. 2020, 13, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef]
- Nishinaka, Y.; Masutani, H.; Oka, S.; Matsuo, Y.; Yamaguchi, Y.; Nishio, K.; Ishii, Y.; Yodoi, J. Importin alpha1 (Rch1) mediates nuclear translocation of thioredoxin-binding protein-2/vitamin D(3)-up-regulated protein 1. J. Biol. Chem. 2004, 279, 37559–37565. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef]
- Jing, G.; Westwell-Roper, C.; Chen, J.; Xu, G.; Verchere, C.B.; Shalev, A. Thioredoxin-interacting protein promotes islet amyloid polypeptide expression through miR-124a and FoxA2. J. Biol. Chem. 2014, 289, 11807–11815. [Google Scholar] [CrossRef]
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Collins, S.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharm. 2010, 244, 77–83. [Google Scholar] [CrossRef]
- Janjic, D.; Maechler, P.; Sekine, N.; Bartley, C.; Annen, A.S.; Wolheim, C.B. Free radical modulation of insulin release in INS-1 cells exposed to alloxan. Biochem. Pharm. 1999, 57, 639–648. [Google Scholar] [CrossRef]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem. 1999, 274, 27905–27913. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef]
- Travasso, R.D.M.; Sampaio Dos Aidos, F.; Bayani, A.; Abranches, P.; Salvador, A. Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 2017, 12, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Kontou, M.; Will, R.D.; Adelfalk, C.; Wittig, R.; Poustka, A.; Hirsch-Kauffmann, M.; Schweiger, M. Thioredoxin, a regulator of gene expression. Oncogene 2004, 23, 2146–2152. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bian, M.; Fan, R.; Zhao, S.; Liu, W. Targeting the Thioredoxin System as a Strategy for Cancer Therapy. J. Med. Chem. 2019, 62, 7309–7321. [Google Scholar] [CrossRef] [PubMed]
- Jastrząb, A.; Skrzydlewska, E. Thioredoxin-dependent system. Application of inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Reviews. Immunol. 2020. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Schultheis, J.; Beckmann, D.; Mulac, D.; Muller, L.; Esselen, M.; Dufer, M. Nrf2 Activation Protects Mouse Beta Cells from Glucolipotoxicity by Restoring Mitochondrial Function and Physiological Redox Balance. Oxid. Med. Cell. Longev. 2019, 2019, 7518510. [Google Scholar] [CrossRef]
- He, J.; Zhang, X.; Lian, C.; Wu, J.; Fang, Y.; Ye, X. KEAP1/NRF2 axis regulates H2O2-induced apoptosis of pancreatic beta-cells. Gene 2019, 691, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, A.K.; Heimberg, H.; Heremans, Y.; Leeman, R.; Kutlu, B.; Kruhoffer, M.; Orntoft, T.; Eizirik, D.L. A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J. Biol. Chem. 2001, 276, 48879–48886. [Google Scholar] [CrossRef] [PubMed]
- Meyerovich, K.; Fukaya, M.; Terra, L.F.; Ortis, F.; Eizirik, D.L.; Cardozo, A.K. The non-canonical NF-kappaB pathway is induced by cytokines in pancreatic beta cells and contributes to cell death and proinflammatory responses in vitro. Diabetologia 2016, 59, 512–521. [Google Scholar] [CrossRef]
- Meyerovich, K.; Ortis, F.; Cardozo, A.K. The non-canonical NF-kappaB pathway and its contribution to beta-cell failure in diabetes. J. Mol. Endocrinol. 2018, 61, F1–F6. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Esselman, W.J. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic. Biol. Med. 2002, 33, 1121–1132. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Flohe, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Schulze-Osthoff, K.; Beyaert, R.; Vandevoorde, V.; Haegeman, G.; Fiers, W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993, 12, 3095–3104. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Cerf, M.E. Transcription factors regulating beta-cell function. Eur. J. Endocrinol. 2006, 155, 671–679. [Google Scholar] [CrossRef]
- Jara, M.A.; Werneck-De-Castro, J.P.; Lubaczeuski, C.; Johnson, J.D.; Bernal-Mizrachi, E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to beta-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets 2020, 12, 32–40. [Google Scholar] [CrossRef]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet beta-cell function. Mol. Endocrinol. 2007, 21, 2764–2774. [Google Scholar] [CrossRef]
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem. 2005, 280, 11107–11113. [Google Scholar] [CrossRef]
- Kondo, T.; El Khattabi, I.; Nishimura, W.; Laybutt, D.R.; Geraldes, P.; Shah, S.; King, G.; Bonner-Weir, S.; Weir, G.; Sharma, A. p38 MAPK is a major regulator of MafA protein stability under oxidative stress. Mol. Endocrinol. 2009, 23, 1281–1290. [Google Scholar] [CrossRef]
- El Khattabi, I.; Sharma, A. Preventing p38 MAPK-mediated MafA degradation ameliorates beta-cell dysfunction under oxidative stress. Mol. Endocrinol. 2013, 27, 1078–1090. [Google Scholar] [CrossRef]
- Harmon, J.S.; Bogdani, M.; Parazzoli, S.D.; Mak, S.S.; Oseid, E.A.; Berghmans, M.; Leboeuf, R.C.; Robertson, R.P. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009, 150, 4855–4862. [Google Scholar] [CrossRef]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005, 16, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Smits, L.M.; van Triest, M.H.; de Keizer, P.L.; van Leenen, D.; Koerkamp, M.G.; Szypowska, A.; Meppelink, A.; Brenkman, A.B.; Yodoi, J.; et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009, 5, 664–672. [Google Scholar] [CrossRef]
- De Keizer, P.L.; Burgering, B.M.; Dansen, T.B. Forkhead box o as a sensor, mediator, and regulator of redox signaling. Antioxid. Redox Signal. 2011, 14, 1093–1106. [Google Scholar] [CrossRef]
- Burgering, B.M.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef]
- Huang, X.; Begley, M.; Morgenstern, K.A.; Gu, Y.; Rose, P.; Zhao, H.; Zhu, X. Crystal structure of an inactive Akt2 kinase domain. Structure 2003, 11, 21–30. [Google Scholar] [CrossRef]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H., 3rd; Wright, C.V.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef]
- Nishimura, W.; Kondo, T.; Salameh, T.; El Khattabi, I.; Dodge, R.; Bonner-Weir, S.; Sharma, A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev. Biol. 2006, 293, 526–539. [Google Scholar] [CrossRef]
- Cao, X.; Kambe, F.; Ohmori, S.; Seo, H. Oxidoreductive modification of two cysteine residues in paired domain by Ref-1 regulates DNA-binding activity of Pax-8. Biochem. Biophys Res. Commun 2002, 297, 288–293. [Google Scholar] [CrossRef]
- Walther, C.; Guenet, J.L.; Simon, D.; Deutsch, U.; Jostes, B.; Goulding, M.D.; Plachov, D.; Balling, R.; Gruss, P. Pax: A murine multigene family of paired box-containing genes. Genomics 1991, 11, 424–434. [Google Scholar] [CrossRef]
- Swisa, A.; Avrahami, D.; Eden, N.; Zhang, J.; Feleke, E.; Dahan, T.; Cohen-Tayar, Y.; Stolovich-Rain, M.; Kaestner, K.H.; Glaser, B.; et al. PAX6 maintains beta cell identity by repressing genes of alternative islet cell types. J. Clin. Investig. 2017, 127, 230–243. [Google Scholar] [CrossRef]
- Rieck, S.; Bankaitis, E.D.; Wright, C.V. Lineage determinants in early endocrine development. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2012; Volume 23, pp. 673–684. [Google Scholar] [CrossRef]
- Bastidas-Ponce, A.; Roscioni, S.S.; Burtscher, I.; Bader, E.; Sterr, M.; Bakhti, M.; Lickert, H. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic beta-cells. Mol. Metab. 2017, 6, 524–534. [Google Scholar] [CrossRef]
- Bensellam, M.; Jonas, J.C.; Laybutt, D.R. Mechanisms of beta-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef]
- Balakrishnan, S.; Dhavamani, S.; Prahalathan, C. beta-Cell specific transcription factors in the context of diabetes mellitus and beta-cell regeneration. Mech. Dev. 2020, 163, 103634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Chetboun, M.; Abitbol, G.; Rozenberg, K.; Rozenfeld, H.; Deutsch, A.; Sampson, S.R.; Rosenzweig, T. Maintenance of redox state and pancreatic beta-cell function: Role of leptin and adiponectin. J. Cell. Biochem. 2012, 113, 1966–1976. [Google Scholar] [CrossRef]
- Ahmed Alfar, E.; Kirova, D.; Konantz, J.; Birke, S.; Mansfeld, J.; Ninov, N. Distinct Levels of Reactive Oxygen Species Coordinate Metabolic Activity with Beta-cell Mass Plasticity. Sci. Rep. 2017, 7, 3994. [Google Scholar] [CrossRef]
- Liang, J.; Wu, S.Y.; Zhang, D.; Wang, L.; Leung, K.K.; Leung, P.S. NADPH Oxidase-Dependent Reactive Oxygen Species Stimulate beta-Cell Regeneration Through Differentiation of Endocrine Progenitors in Murine Pancreas. Antioxid. Redox Signal. 2016, 24, 419–433. [Google Scholar] [CrossRef]
- Hoarau, E.; Chandra, V.; Rustin, P.; Scharfmann, R.; Duvillie, B. Pro-oxidant/antioxidant balance controls pancreatic beta-cell differentiation through the ERK1/2 pathway. Cell Death Dis. 2014, 5, e1487. [Google Scholar] [CrossRef]
- Costes, S.; Broca, C.; Bertrand, G.; Lajoix, A.D.; Bataille, D.; Bockaert, J.; Dalle, S. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: A key role in glucose-mediated pancreatic beta-cell survival. Diabetes 2006, 55, 2220–2230. [Google Scholar] [CrossRef]
- Hussain, M.A.; Porras, D.L.; Rowe, M.H.; West, J.R.; Song, W.J.; Schreiber, W.E.; Wondisford, F.E. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Mol. Cell. Biol. 2006, 26, 7747–7759. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Hawkins, D.F.; Whitecavage, M.K.; Colter, D.C.; Stokes, D.G.; Jimenez, S.A. Regulation of the human SOX9 promoter by Sp1 and CREB. Exp. Cell Res. 2007, 313, 1069–1079. [Google Scholar] [CrossRef]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef]
- Fruhbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef]
- Lee, Y.H.; Magkos, F.; Mantzoros, C.S.; Kang, E.S. Effects of leptin and adiponectin on pancreatic beta-cell function. Metabolism 2011, 60, 1664–1672. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Hurley, J.D.; Smith, D.M.; Ghatei, M.A.; Withers, D.J.; Gardiner, J.V.; Bailey, C.J.; Bloom, S.R. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Investig. 1997, 100, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.J.; Heller, R.S.; Leech, C.A.; Holz, G.G.; Habener, J.F. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 1997, 46, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Kuehnen, P.; Laubner, K.; Raile, K.; Schofl, C.; Jakob, F.; Pilz, I.; Path, G.; Seufert, J. Protein phosphatase 1 (PP-1)-dependent inhibition of insulin secretion by leptin in INS-1 pancreatic beta-cells and human pancreatic islets. Endocrinology 2011, 152, 1800–1808. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sim, A.T.; Baldwin, M.L.; Rostas, J.A.; Holst, J.; Ludowyke, R.I. The role of serine/threonine protein phosphatases in exocytosis. Biochem. J. 2003, 373, 641–659. [Google Scholar] [CrossRef]
- Staiger, K.; Stefan, N.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Machicao, F.; Kellerer, M.; Stumvoll, M.; Fritsche, A.; et al. Adiponectin is functionally active in human islets but does not affect insulin secretory function or beta-cell lipoapoptosis. J. Clin. Endocrinol. Metab. 2005, 90, 6707–6713. [Google Scholar] [CrossRef]
- Llanos, P.; Contreras-Ferrat, A.; Barrientos, G.; Valencia, M.; Mears, D.; Hidalgo, C. Glucose-Dependent Insulin Secretion in Pancreatic beta-Cell Islets from Male Rats Requires Ca2+ Release via ROS-Stimulated Ryanodine Receptors. PLoS ONE 2015, 10, e0129238. [Google Scholar] [CrossRef]
- Jansson, D.; Ng, A.C.; Fu, A.; Depatie, C.; Al Azzabi, M.; Screaton, R.A. Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc. Natl. Acad. Sci. USA 2008, 105, 10161–10166. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocana, A. Human beta-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef]
- Sato, Y.; Endo, H.; Okuyama, H.; Takeda, T.; Iwahashi, H.; Imagawa, A.; Yamagata, K.; Shimomura, I.; Inoue, M. Cellular hypoxia of pancreatic beta-cells due to high levels of oxygen consumption for insulin secretion in vitro. J. Biol. Chem. 2011, 286, 12524–12532. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, A.V.; Ward, M.W.; Prehn, J.H.; Papkovsky, D.B. Dynamics of intracellular oxygen in PC12 Cells upon stimulation of neurotransmission. J. Biol. Chem. 2008, 283, 5650–5661. [Google Scholar] [CrossRef]
- O’Hagan, K.A.; Cocchiglia, S.; Zhdanov, A.V.; Tambuwala, M.M.; Cummins, E.P.; Monfared, M.; Agbor, T.A.; Garvey, J.F.; Papkovsky, D.B.; Taylor, C.T.; et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2188–2193. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef]
- Olsson, R.; Carlsson, P.O. A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes 2011, 60, 2068–2075. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef]
- Yasui, S.; Mawatari, K.; Morizumi, R.; Furukawa, H.; Shimohata, T.; Harada, N.; Takahashi, A.; Nakaya, Y. Hydrogen peroxide inhibits insulin-induced ATP-sensitive potassium channel activation independent of insulin signaling pathway in cultured vascular smooth muscle cells. J. Med. Investig. 2012, 59, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, R.; Mori, Y. Transient receptor potential (TRP) channels: Biosensors for redox environmental stimuli and cellular status. Free Radic. Biol. Med. 2020, 146, 36–44. [Google Scholar] [CrossRef]
- Finol-Urdaneta, R.K.; Remedi, M.S.; Raasch, W.; Becker, S.; Clark, R.B.; Struver, N.; Pavlov, E.; Nichols, C.G.; French, R.J.; Terlau, H. Block of Kv1.7 potassium currents increases glucose-stimulated insulin secretion. EMBO Mol. Med. 2012, 4, 424–434. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Salapatek, A.M.; Wheeler, M.B. Temperature and redox state dependence of native Kv2.1 currents in rat pancreatic beta-cells. J. Physiol. 2003, 546, 647–653. [Google Scholar] [CrossRef]
- Mittal, M.; Gu, X.Q.; Pak, O.; Pamenter, M.E.; Haag, D.; Fuchs, D.B.; Schermuly, R.T.; Ghofrani, H.A.; Brandes, R.P.; Seeger, W.; et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2012, 52, 1033–1042. [Google Scholar] [CrossRef]
- Gerst, J.E. SNARE regulators: Matchmakers and matchbreakers. Biochim Biophys Acta 2003, 1641, 99–110. [Google Scholar] [CrossRef]
- Ivarsson, R.; Quintens, R.; Dejonghe, S.; Tsukamoto, K.; Renstrom, E.; Schuit, F.C. Redox control of exocytosis: Regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 2005, 54, 2132–2142. [Google Scholar] [CrossRef]
- Reinbothe, T.M.; Ivarsson, R.; Li, D.Q.; Niazi, O.; Jing, X.; Zhang, E.; Stenson, L.; Bryborn, U.; Renstrom, E. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol. Endocrinol. 2009, 23, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Ferdaoussi, M.; Dai, X.; Jensen, M.V.; Wang, R.; Peterson, B.S.; Huang, C.; Ilkayeva, O.; Smith, N.; Miller, N.; Hajmrle, C.; et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional beta cells. J. Clin. Investig. 2015, 125, 3847–3860. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lam, L.S.; Lam, L.H.; Chau, S.F.; Ng, T.B.; Au, S.W. Molecular basis of the redox regulation of SUMO proteases: A protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008, 22, 127–137. [Google Scholar] [CrossRef]
- Ferdaoussi, M.; MacDonald, P.E. Toward Connecting Metabolism to the Exocytotic Site. Trends Cell Biol. 2017, 27, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.M. Thiol switches in membrane proteins - Extracellular redox regulation in cell biology. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Lundberg, M.; Fernandes, A.P.; Kumar, S.; Holmgren, A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem. Biophys. Res. Commun. 2004, 319, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Jha, V.; Min, J.K.; Cho, J. Protein disulfide isomerase in cardiovascular disease. Exp. Mol. Med. 2020, 52, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine Oxidation Targets Peroxiredoxins 1 and 2 for Exosomal Release through a Novel Mechanism of Redox-Dependent Secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of β-cell function by thioredoxin. Redox Biol. 2020, 34, 101570. [Google Scholar] [CrossRef] [PubMed]
- Jikimoto, T.; Nishikubo, Y.; Koshiba, M.; Kanagawa, S.; Morinobu, S.; Morinobu, A.; Saura, R.; Mizuno, K.; Kondo, S.; Toyokuni, S.; et al. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol. Immunol. 2002, 38, 765–772. [Google Scholar] [CrossRef]
- Kakisaka, Y.; Nakashima, T.; Sumida, Y.; Yoh, T.; Nakamura, H.; Yodoi, J.; Senmaru, H. Elevation of serum thioredoxin levels in patients with type 2 diabetes. Horm. Metab. Res. 2002, 34, 160–164. [Google Scholar] [CrossRef]
- Asami, K.; Inagaki, A.; Imura, T.; Sekiguchi, S.; Fujimori, K.; Masutani, H.; Yodoi, J.; Satomi, S.; Ohuchi, N.; Goto, M. Thioredoxin-1 attenuates early graft loss after intraportal islet transplantation in mice. PLoS ONE 2013, 8, e70259. [Google Scholar] [CrossRef]
- Willems, S.H.; Tape, C.J.; Stanley, P.L.; Taylor, N.A.; Mills, I.G.; Neal, D.E.; McCafferty, J.; Murphy, G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem. J. 2010, 428, 439–450. [Google Scholar] [CrossRef]
- Bass, R.; Edwards, D.R. ADAMs and protein disulfide isomerase: The key to regulated cell-surface protein ectodomain shedding? Biochem. J. 2010, 428, e3–e5. [Google Scholar] [CrossRef]
- Düsterhöft, S.; Jung, S.; Hung, C.W.; Tholey, A.; Sönnichsen, F.D.; Grötzinger, J.; Lorenzen, I. Membrane-proximal domain of a disintegrin and metalloprotease-17 represents the putative molecular switch of its shedding activity operated by protein-disulfide isomerase. J. Am. Chem. Soc. 2013, 135, 5776–5781. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Chodavarapu, H.; Porretta, C.; Robinson, L.K.; Lazartigues, E. Dynamics of ADAM17-Mediated Shedding of ACE2 Applied to Pancreatic Islets of Male db/db Mice. Endocrinology 2015, 156, 4411–4425. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, K.H.; Chodavarapu, H.; Lazartigues, E. Angiotensin converting enzyme 2: A new important player in the regulation of glycemia. IUBMB Life 2013, 65, 731–738. [Google Scholar] [CrossRef]
- Bergerhausen, L.; Grosche, J.; Meißner, J.; Hecker, C.; Caliandro, M.F.; Westerhausen, C.; Kamenac, A.; Rezaei, M.; Mörgelin, M.; Poschmann, G.; et al. Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants 2020, 9, 227. [Google Scholar] [CrossRef]
- Passam, F.; Chiu, J.; Ju, L.; Pijning, A.; Jahan, Z.; Mor-Cohen, R.; Yeheskel, A.; Kolšek, K.; Thärichen, L.; Aponte-Santamaría, C.; et al. Mechano-redox control of integrin de-adhesion. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.E.; Gannon, M. Extracellular Matrix-Associated Factors Play Critical Roles in Regulating Pancreatic β-Cell Proliferation and Survival. Endocrinology 2019, 160, 1885–1894. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Alam, N.; Goel, H.L.; Zarif, M.J.; Butterfield, J.E.; Perkins, H.M.; Sansoucy, B.G.; Sawyer, T.K.; Languino, L.R. The integrin-growth factor receptor duet. J. Cell. Physiol. 2007, 213, 649–653. [Google Scholar] [CrossRef]
- Xu, S.Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC channel activation by extracellular thioredoxin. Nature 2008, 451, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S. Molecular Regulations and Functions of the Transient Receptor Potential Channels of the Islets of Langerhans and Insulinoma Cells. Cells 2020, 9, 685. [Google Scholar] [CrossRef]
- Bensellam, M.; Van Lommel, L.; Overbergh, L.; Schuit, F.C.; Jonas, J.C. Cluster analysis of rat pancreatic islet gene mRNA levels after culture in low-, intermediate- and high-glucose concentrations. Diabetologia 2009, 52, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Corkey, B.E.; Deeney, J.T. The Redox Communication Network as a Regulator of Metabolism. Front. Physiol. 2020, 11, 567796. [Google Scholar] [CrossRef] [PubMed]
- Corkey, B.E.; Shirihai, O. Metabolic master regulators: Sharing information among multiple systems. Trends Endocrinol. Metab. 2012, 23, 594–601. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27S, S60–S68. [Google Scholar] [CrossRef] [PubMed]
- Herbert, T.P.; Laybutt, D.R. A Reevaluation of the Role of the Unfolded Protein Response in Islet Dysfunction: Maladaptation or a Failure to Adapt? Diabetes 2016, 65, 1472–1480. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Banuls, C.; Diaz-Morales, N.; Iannantuoni, F.; Rovira-Llopis, S.; Gomis, R.; Rocha, M.; Hernandez-Mijares, A.; Murphy, M.P.; Victor, V.M. The Mitochondria-Targeted Antioxidant MitoQ Modulates Mitochondrial Function and Endoplasmic Reticulum Stress in Pancreatic beta Cells Exposed to Hyperglycaemia. Cell Physiol. Biochem. 2019, 52, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Yuan, Q.; Tang, W.; Zhang, X.; Hinson, J.A.; Liu, C.; Osei, K.; Wang, J. Proinsulin atypical maturation and disposal induces extensive defects in mouse Ins2+/Akita beta-cells. PLoS ONE 2012, 7, e35098. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef]
- Vasiljevic, J.; Torkko, J.M.; Knoch, K.P.; Solimena, M. The making of insulin in health and disease. Diabetologia 2020, 63, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Gutierrez, T.; Paredes, F.; Gatica, D.; Rodriguez, A.E.; Pedrozo, Z.; Chiong, M.; Parra, V.; Quest, A.F.; Rothermel, B.A.; et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 2012, 44, 16–20. [Google Scholar] [CrossRef]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Nadler, J.L. Islet inflammation: A unifying target for diabetes treatment? Trends Endocrinol. Metab. 2013, 24, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Kim, J.J.; Sears, D.D. TLR4 and Insulin Resistance. Gastroenterol. Res. Pract. 2010, 2010. [Google Scholar] [CrossRef]
- Singh, A.; Singh, V.; Tiwari, R.L.; Chandra, T.; Kumar, A.; Dikshit, M.; Barthwal, M.K. The IRAK-ERK-p67phox-Nox-2 axis mediates TLR4, 2-induced ROS production for IL-1beta transcription and processing in monocytes. Cell. Mol. Immunol. 2016, 13, 745–763. [Google Scholar] [CrossRef]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef]
- Han, C.Y. Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue. Diabetes Metab. J. 2016, 40, 272–279. [Google Scholar] [CrossRef]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lieb, D.C.; Brotman, J.J.; Hatcher, M.A.; Aye, M.S.; Cole, B.K.; Haynes, B.A.; Wohlgemuth, S.D.; Fontana, M.A.; Beydoun, H.; Nadler, J.L.; et al. Adipose tissue 12/15 lipoxygenase pathway in human obesity and diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E1713–E1720. [Google Scholar] [CrossRef]
- Dunmore, S.J.; Brown, J.E. The role of adipokines in beta-cell failure of type 2 diabetes. J. Endocrinol. 2013, 216, T37–T45. [Google Scholar] [CrossRef]
- Tushuizen, M.E.; Bunck, M.C.; Pouwels, P.J.; Bontemps, S.; van Waesberghe, J.H.; Schindhelm, R.K.; Mari, A.; Heine, R.J.; Diamant, M. Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care 2007, 30, 2916–2921. [Google Scholar] [CrossRef]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e455. [Google Scholar] [CrossRef]
- Weitz, J.R.; Makhmutova, M.; Almaca, J.; Stertmann, J.; Aamodt, K.; Brissova, M.; Speier, S.; Rodriguez-Diaz, R.; Caicedo, A. Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia 2018, 61, 182–192. [Google Scholar] [CrossRef]
- Jacques-Silva, M.C.; Correa-Medina, M.; Cabrera, O.; Rodriguez-Diaz, R.; Makeeva, N.; Fachado, A.; Diez, J.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; et al. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc. Natl. Acad. Sci. USA 2010, 107, 6465–6470. [Google Scholar] [CrossRef]
- Almaca, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.O.; Caicedo, A. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.G.; Pasek, R.C.; Maulis, M.F.; Dunn, J.C.; Bolus, W.R.; Kendall, P.L.; Hasty, A.H.; Gannon, M. Macrophages are essential for CTGF-mediated adult beta-cell proliferation after injury. Mol. Metab. 2015, 4, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and beta-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.C.; Hwang, I.; Choe, S.S.; Park, J.; Ji, Y.; Kim, J.I.; Lee, G.Y.; Choi, S.H.; Ching, J.; Kovalik, J.P.; et al. Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat. Commun. 2017, 8, 1087. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Thorne, J.; Parnaud, G.; Marselli, L.; Ehses, J.A.; Kerr-Conte, J.; Pattou, F.; Halban, P.A.; Weir, G.C.; Donath, M.Y. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 2008, 93, 4065–4074. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.; Denroche, H.C.; Ehses, J.A.; Verchere, C.B. Differential Activation of Innate Immune Pathways by Distinct Islet Amyloid Polypeptide (IAPP) Aggregates. J. Biol. Chem. 2016, 291, 8908–8917. [Google Scholar] [CrossRef]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.A.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 2006, 281, 1091–1098. [Google Scholar] [CrossRef]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Martin, T.M.; Mendoza, T.; Stadler, K.; Alami, W.; Karlstad, M.D.; Robson, M.J.; Blakely, R.D.; et al. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet beta-cell de-differentiation. Mol. Metab. 2018, 14, 95–107. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Boni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in beta-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.; Zhao, B.; Fei, H. IL-1beta caused pancreatic beta-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol. Cell. Biochem. 2009, 324, 183–190. [Google Scholar] [CrossRef]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef]
- Herder, C.; Dalmas, E.; Boni-Schnetzler, M.; Donath, M.Y. The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications. Trends Endocrinol. Metab. 2015, 26, 551–563. [Google Scholar] [CrossRef]
- Sokolova, M.; Sahraoui, A.; Hoyem, M.; Ogaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Fei, N.; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013, 7, 880–884. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef]
- Vezza, T.; Abad-Jimenez, Z.; Marti-Cabrera, M.; Rocha, M.; Victor, V.M. Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes. Antioxidants 2020, 9, 848. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Segovia, I.; Yuan, X.L.; Gao, Z.H. Controversial Roles of Gut Microbiota-Derived Short-Chain Fatty Acids (SCFAs) on Pancreatic beta-Cell Growth and Insulin Secretion. Int. J. Mol. Sci. 2020, 21, 910. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Dahllof, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermudez-Humaran, L.G.; Smirnova, N.; Berge, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, C.M.; Panzer, J.K.; Drotar, D.M.; Enos, S.J.; Kipke, N.; Chen, C.; Bozsak, R.; Schoniger, E.; Ehehalt, F.; Distler, M.; et al. Dysfunction of Persisting beta Cells Is a Key Feature of Early Type 2 Diabetes Pathogenesis. Cell Rep. 2020, 31, 107469. [Google Scholar] [CrossRef]
- Da Silva Xavier, G.; Rutter, G.A. Metabolic and Functional Heterogeneity in Pancreatic beta Cells. J. Mol. Biol. 2020, 432, 1395–1406. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.S. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic. Biol. Med. 2006, 41, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, J.; Parazzoli, S.; Oseid, E.; Hertzel, A.V.; Bernlohr, D.A.; Vallerie, S.N.; Liu, C.Q.; Lopez, M.; Harmon, J.S.; Robertson, R.P. Ebselen treatment prevents islet apoptosis, maintains intranuclear Pdx-1 and MafA levels, and preserves beta-cell mass and function in ZDF rats. Diabetes 2013, 62, 3582–3588. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, D.; Kajimoto, Y.; Kaneto, H.; Umayahara, Y.; Fujitani, Y.; Miyatsuka, T.; Watada, H.; Leibiger, I.B.; Yamasaki, Y.; Hori, M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes 2003, 52, 2896–2904. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Kim-Muller, J.Y.; Zhao, S.; Srivastava, S.; Mugabo, Y.; Noh, H.L.; Kim, Y.R.; Madiraju, S.R.; Ferrante, A.W.; Skolnik, E.Y.; Prentki, M.; et al. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 2014, 20, 593–602. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of beta cells. Nat. Commun. 2016, 7, 11756. [Google Scholar] [CrossRef]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef]
- Sasaki, M.; Fujimoto, S.; Sato, Y.; Nishi, Y.; Mukai, E.; Yamano, G.; Sato, H.; Tahara, Y.; Ogura, K.; Nagashima, K.; et al. Reduction of reactive oxygen species ameliorates metabolism-secretion coupling in islets of diabetic GK rats by suppressing lactate overproduction. Diabetes 2013, 62, 1996–2003. [Google Scholar] [CrossRef]
- Bensellam, M.; Duvillie, B.; Rybachuk, G.; Laybutt, D.R.; Magnan, C.; Guiot, Y.; Pouyssegur, J.; Jonas, J.C. Glucose-induced O(2) consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS ONE 2012, 7, e29807. [Google Scholar] [CrossRef]
- Sato, Y.; Inoue, M.; Yoshizawa, T.; Yamagata, K. Moderate hypoxia induces β-cell dysfunction with HIF-1-independent gene expression changes. PLoS ONE 2014, 9, e114868. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Meshinchi, S.; Dias-Leme, C.; Raffin, D.; Johnson, J.D.; Treutelaar, M.K.; Burant, C.F. Islet microvasculature in islet hyperplasia and failure in a model of type 2 diabetes. Diabetes 2006, 55, 2965–2973. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Science’s STKE: Signal transduction knowledge environment. Sci. Stke 2007, 2007, cm8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Liu, N.; Cai, X.; Liu, T.; Zou, J.; Wang, L.; Wang, G.; Liu, Y.; Ding, X.; Zhang, B.; Sun, P.; et al. Hypoxia-inducible factor-1α mediates the expression of mature β cell-disallowed genes in hypoxia-induced β cell dedifferentiation. Biochem. Biophys. Res. Commun. 2020, 523, 382–388. [Google Scholar] [CrossRef]
- Giuliani, M.; Moritz, W.; Bodmer, E.; Dindo, D.; Kugelmeier, P.; Lehmann, R.; Gassmann, M.; Groscurth, P.; Weber, M. Central necrosis in isolated hypoxic human pancreatic islets: Evidence for postisolation ischemia. Cell Transplant. 2005, 14, 67–76. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, Q.; Tan, J.; Li, L.; An, X.; Lei, P. Intermittent hypoxia-induced rat pancreatic β-cell apoptosis and protective effects of antioxidant intervention. Nutr. Diabetes 2014, 4, e131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, X.; Wang, X.; Ma, Z.; Gupta Sunkari, V.; Botusan, I.; Takeda, T.; Björklund, A.; Inoue, M.; Catrina, S.B.; et al. Acute hypoxia induces apoptosis of pancreatic β-cell by activation of the unfolded protein response and upregulation of CHOP. Cell Death Dis. 2012, 3, e322. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Montgomery, M.K.; Luzuriaga, J.; Chan, J.Y.; Laybutt, D.R. Inhibitor of differentiation proteins protect against oxidative stress by regulating the antioxidant-mitochondrial response in mouse beta cells. Diabetologia 2015, 58, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Schug, J.; Won, K.J.; Liu, C.; Naji, A.; Avrahami, D.; Golson, M.L.; Kaestner, K.H. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes 2016, 65, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kim, D.H.; Xiao, X.; Lee, S.; Gong, Z.; Muzumdar, R.; Calabuig-Navarro, V.; Yamauchi, J.; Harashima, H.; Wang, R.; et al. FoxO1 Plays an Important Role in Regulating beta-Cell Compensation for Insulin Resistance in Male Mice. Endocrinology 2016, 157, 1055–1070. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J. Biol. Chem. 1993, 268, 22265–22268. [Google Scholar] [CrossRef]
- Jacob, S.; Kohler, M.; Troster, P.; Visa, M.; Garcia-Prieto, C.F.; Alanentalo, T.; Moede, T.; Leibiger, B.; Leibiger, I.B.; Berggren, P.O. In vivo Ca(2+) dynamics in single pancreatic beta cells. FASEB J. 2020, 34, 945–959. [Google Scholar] [CrossRef]
- Frank, J.A.; Broichhagen, J.; Yushchenko, D.A.; Trauner, D.; Schultz, C.; Hodson, D.J. Optical tools for understanding the complexity of beta-cell signalling and insulin release. Nat. Rev. Endocrinol. 2018, 14, 721–737. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef]
- Salem, V.; Silva, L.D.; Suba, K.; Georgiadou, E.; Neda Mousavy Gharavy, S.; Akhtar, N.; Martin-Alonso, A.; Gaboriau, D.C.A.; Rothery, S.M.; Stylianides, T.; et al. Leader beta-cells coordinate Ca(2+) dynamics across pancreatic islets in vivo. Nat. Metab. 2019, 1, 615–629. [Google Scholar] [CrossRef]
- Benninger, R.K.; Zhang, M.; Head, W.S.; Satin, L.S.; Piston, D.W. Gap junction coupling and calcium waves in the pancreatic islet. Biophys. J. 2008, 95, 5048–5061. [Google Scholar] [CrossRef] [PubMed]
- Head, W.S.; Orseth, M.L.; Nunemaker, C.S.; Satin, L.S.; Piston, D.W.; Benninger, R.K. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 2012, 61, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Corezola do Amaral, M.E.; Kravets, V.; Dwulet, J.M.; Farnsworth, N.L.; Piscopio, R.; Schleicher, W.E.; Miranda, J.G.; Benninger, R.K.P. Caloric restriction recovers impaired beta-cell-beta-cell gap junction coupling, calcium oscillation coordination, and insulin secretion in prediabetic mice. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E709–E720. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Garcia, I.E.; Pinto, B.I.; Pupo, A.; Baez, D.; Stehberg, J.; Del Rio, R.; Gonzalez, C. Extracellular Cysteine in Connexins: Role as Redox Sensors. Front. Physiol. 2016, 7, 1. [Google Scholar] [CrossRef]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef]
- Taneera, J.; Fadista, J.; Ahlqvist, E.; Zhang, M.; Wierup, N.; Renström, E.; Groop, L. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol. Cell. Endocrinol. 2013, 375, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Kanatsuna, N.; Taneera, J.; Vaziri-Sani, F.; Wierup, N.; Larsson, H.E.; Delli, A.; Skärstrand, H.; Balhuizen, A.; Bennet, H.; Steiner, D.F.; et al. Autoimmunity against INS-IGF2 protein expressed in human pancreatic islets. J. Biol. Chem. 2013, 288, 29013–29023. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Wang, J.; Güney, E.; Tang, Y.; Zhang, E.; Axelsson, A.S.; Nenonen, H.; Salehi, A.S.; Wollheim, C.B.; Zetterberg, E.; et al. Thrombin stimulates insulin secretion via protease-activated receptor-3. Islets 2015, 7, e1118195. [Google Scholar] [CrossRef]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Dominguez, V.; Raimondi, C.; Somanath, S.; Bugliani, M.; Loder, M.K.; Edling, C.E.; Divecha, N.; da Silva-Xavier, G.; Marselli, L.; Persaud, S.J.; et al. Class II phosphoinositide 3-kinase regulates exocytosis of insulin granules in pancreatic beta cells. J. Biol. Chem. 2011, 286, 4216–4225. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benáková, Š.; Holendová, B.; Plecitá-Hlavatá, L. Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants 2021, 10, 526. https://doi.org/10.3390/antiox10040526
Benáková Š, Holendová B, Plecitá-Hlavatá L. Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants. 2021; 10(4):526. https://doi.org/10.3390/antiox10040526
Chicago/Turabian StyleBenáková, Štěpánka, Blanka Holendová, and Lydie Plecitá-Hlavatá. 2021. "Redox Homeostasis in Pancreatic β-Cells: From Development to Failure" Antioxidants 10, no. 4: 526. https://doi.org/10.3390/antiox10040526
APA StyleBenáková, Š., Holendová, B., & Plecitá-Hlavatá, L. (2021). Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants, 10(4), 526. https://doi.org/10.3390/antiox10040526