
NAC and Vitamin D Restore CNS Glutathione in Endotoxin-Sensitized Neonatal Hypoxic-Ischemic Rats

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
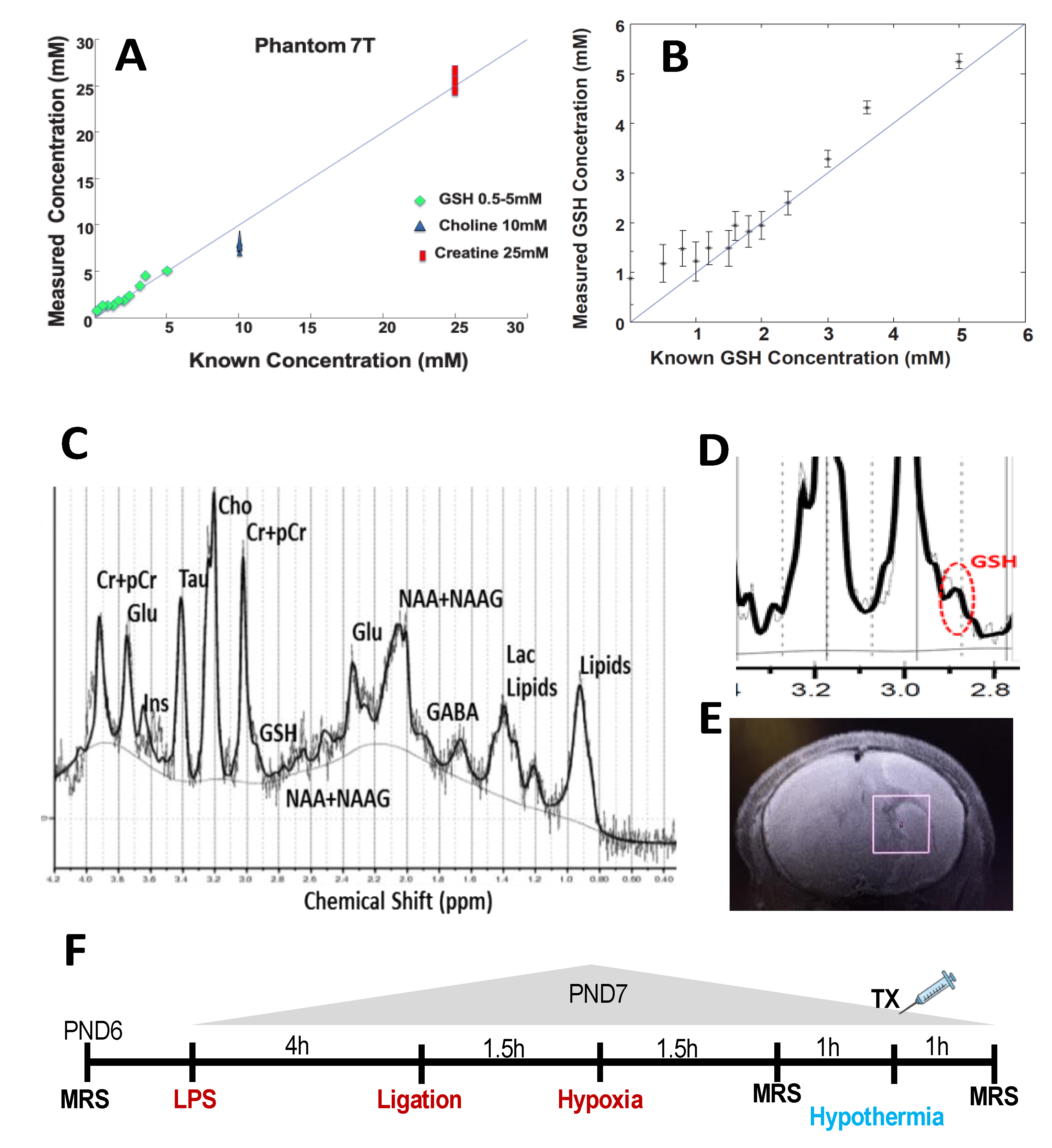
2.1. Validation of GSH by MRS
2.2. Animals and Reagents
2.3. Endotoxin Sensitized Hypoxic Ischemic Injury Animal Model
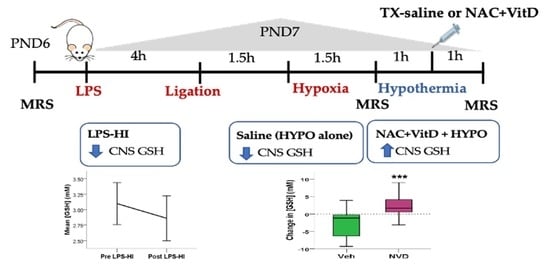
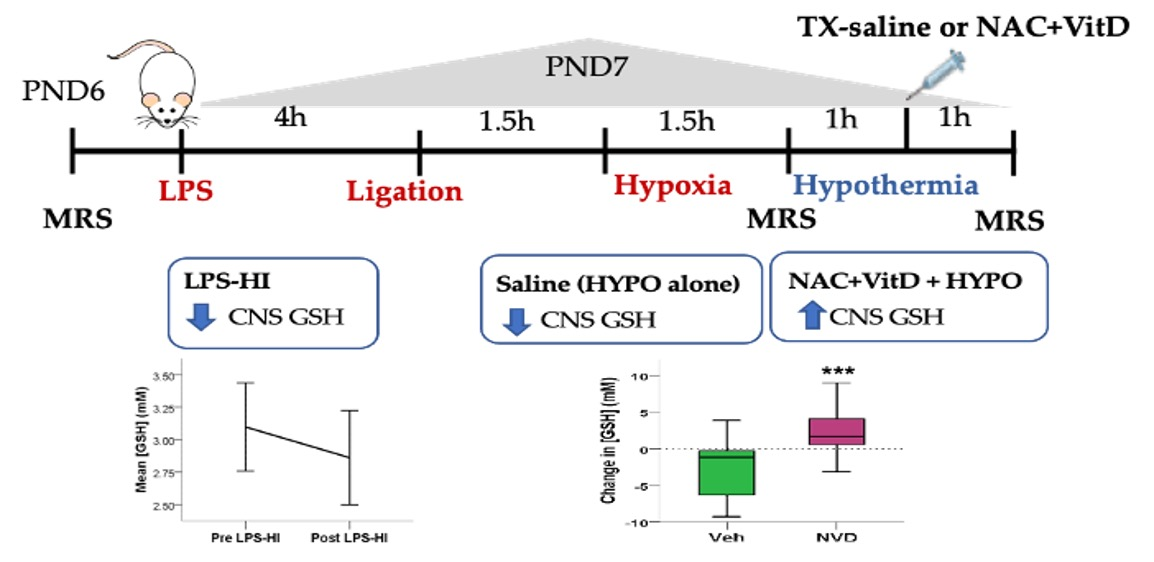
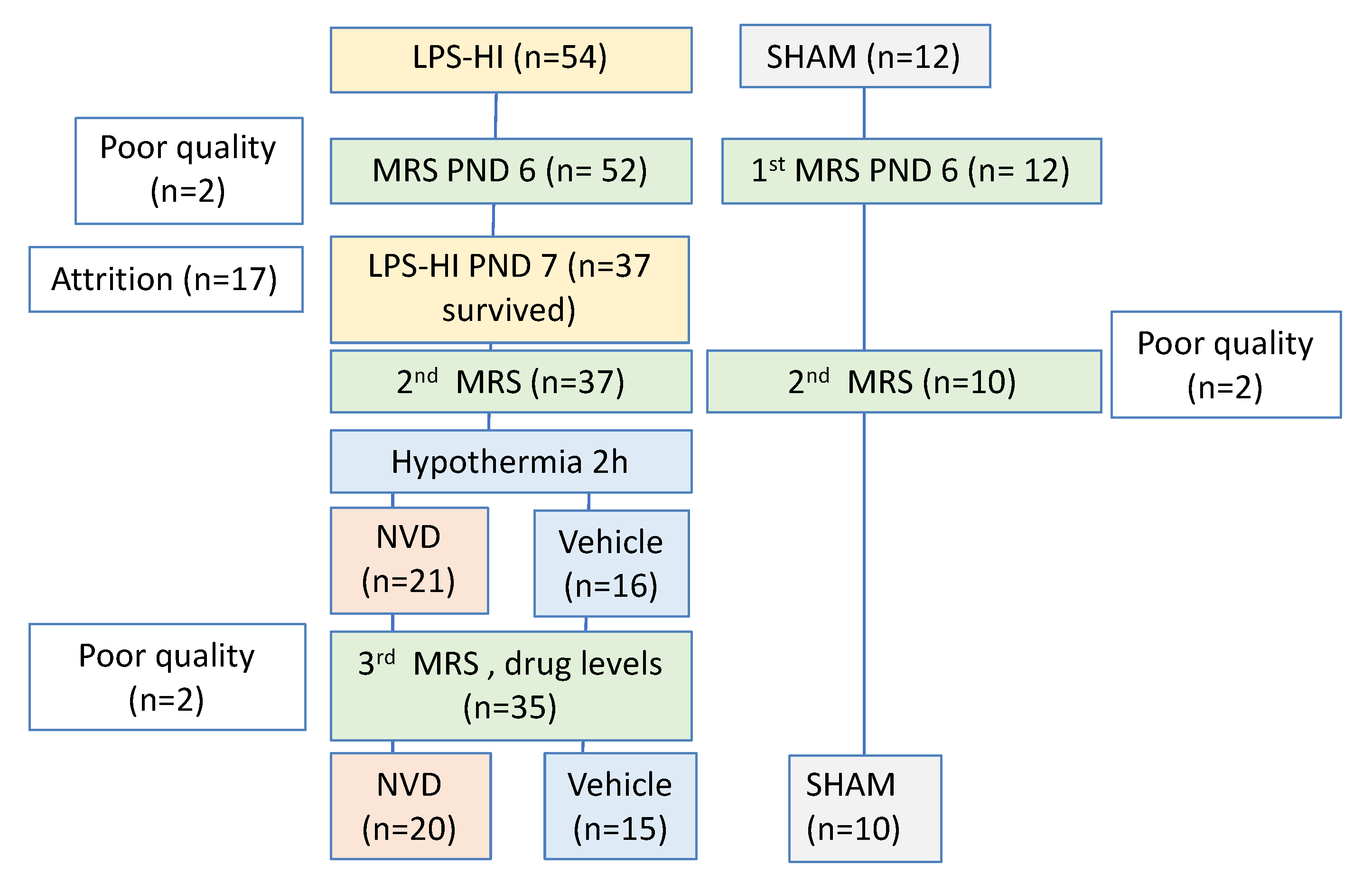
2.4. Experimental Design
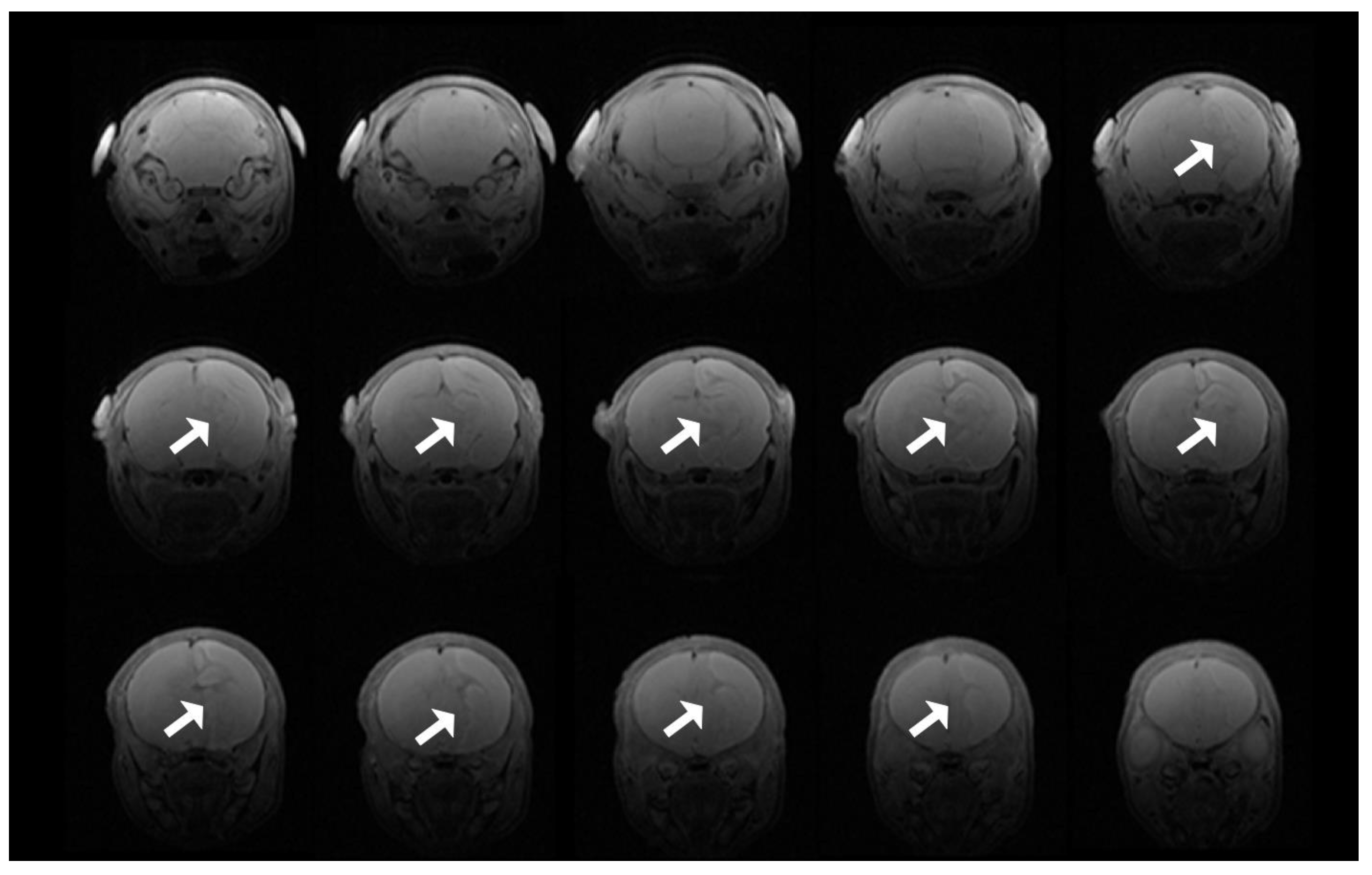
2.5. Magnetic Resonance Imaging and Spectroscopy
2.6. MRS Data Processing
2.7. NAC and Vitamin D Assays
2.8. Statistical Analysis
3. Results
3.1. LPS-HI Morbidity and Mortality
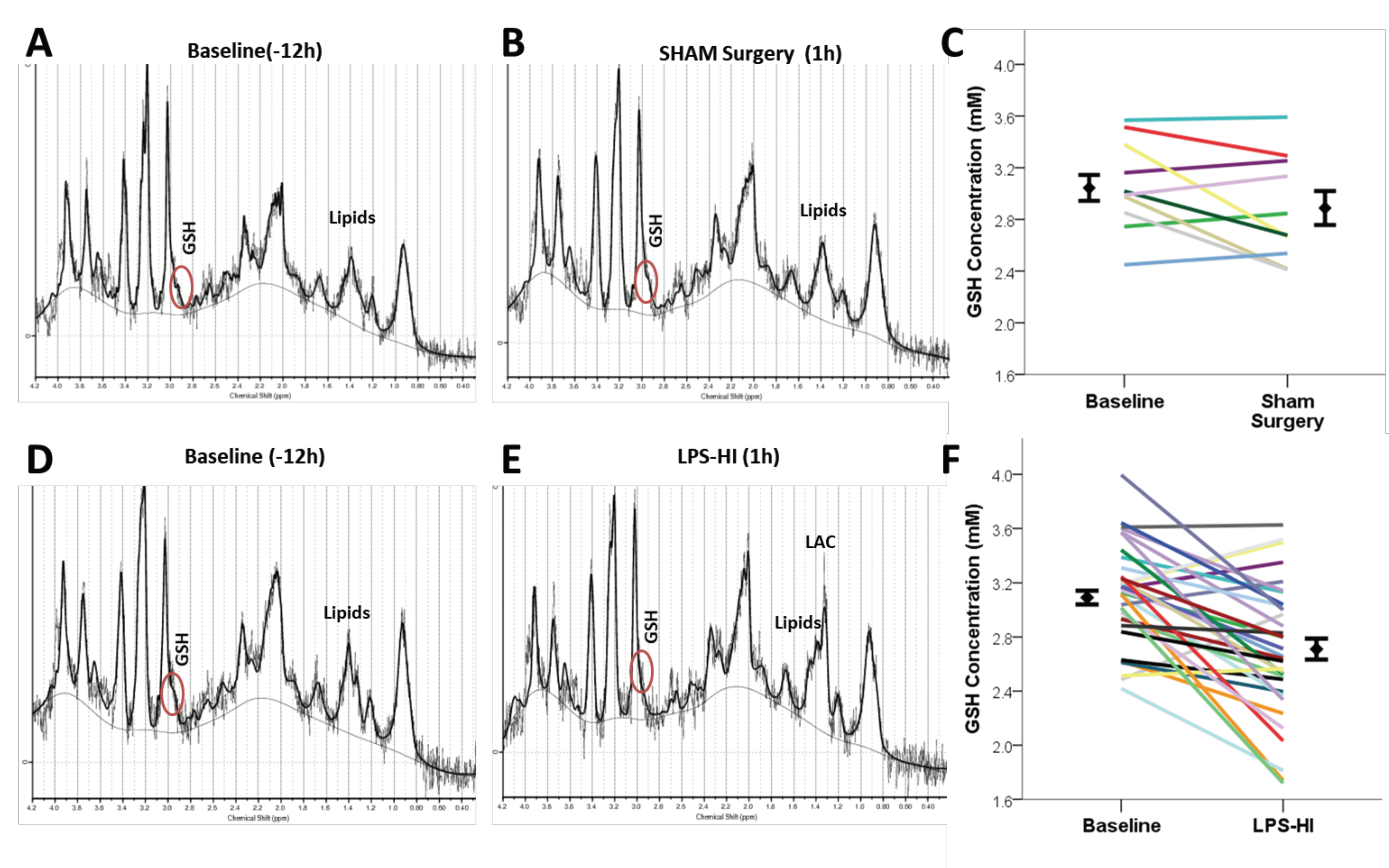
3.2. LPS-HI Acutely Decreases GSH, GLX and tCho
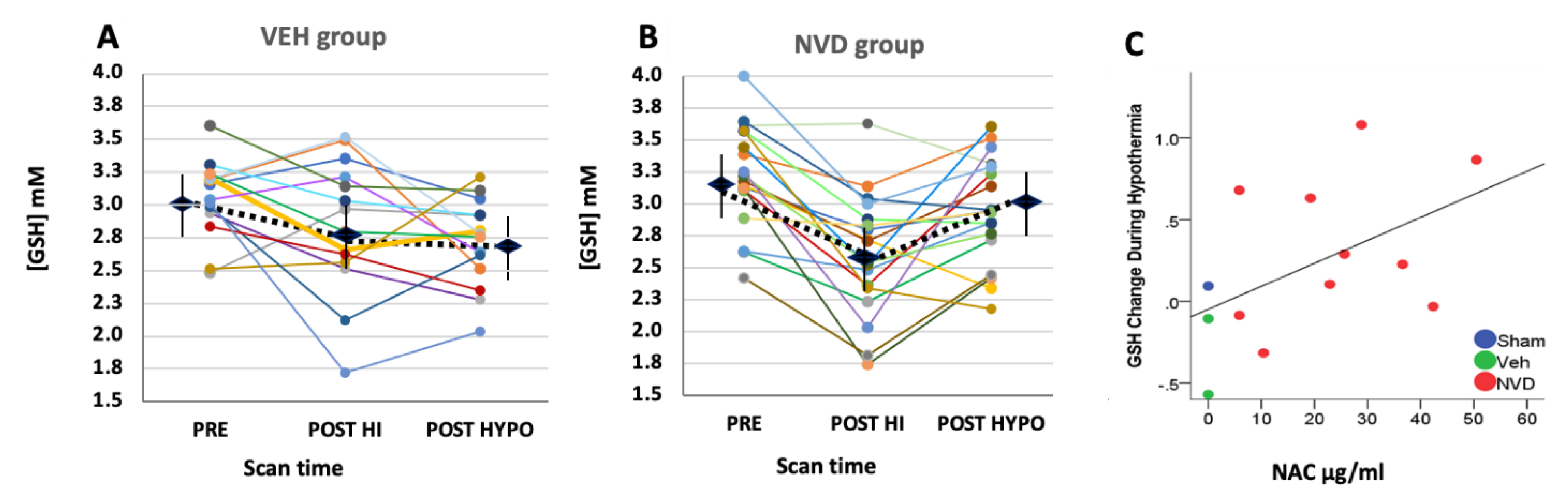
3.3. NVD Improves GSH after LPS-HI Compared with HYPO Alone
3.4. NAC and Active Vitamin D Increase GSH Response Rate in Heterogeneous LPS-HI Injury
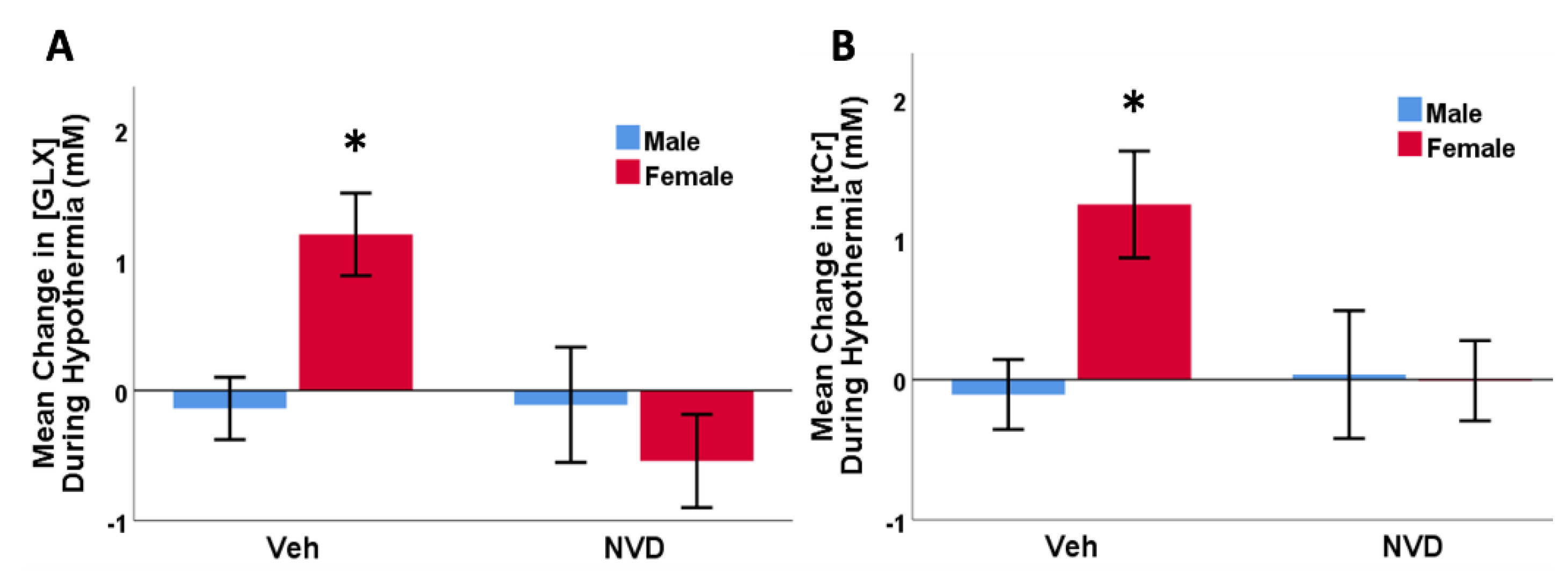
3.5. NAC and Active Vitamin D Suppress Glutamate + Glutamine after LPS-HI Injury
3.6. Lactate Increases after LPS-HI Injury and Persists after HYPO
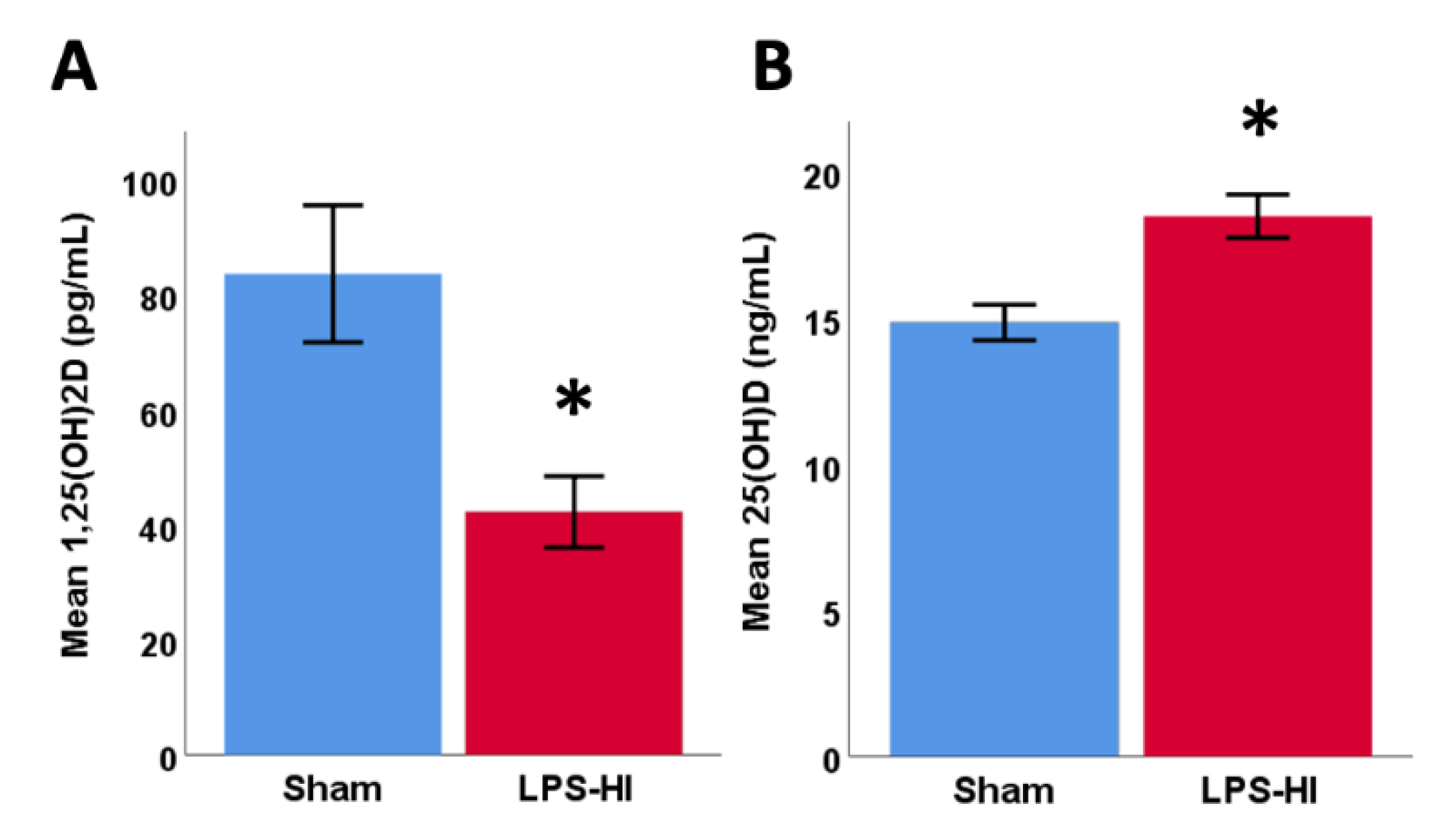
3.7. LPS-HI Injury Decreases Plasma 1,25(OH)2D
3.8. T2 Evidence of Infarcts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cho | total choline (choline + phosphocholine) |
| tCr | total creatine (creatine + phosphocreatine) |
| GLMM | generalized linear mixed model |
| Gln | glutamine |
| Glu | glutamate |
| GLX | glutamate + glutamine |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| HI | hypoxic ischemic |
| HIE | hypoxic ischemic encephalopathy |
| Ins | inositol |
| LAC | lactate |
| LPS-HI | lipopolysaccharide endotoxin-sensitized HI |
| MRS | magnetic resonance spectroscopy |
| NAA | total N-Acetylaspartate (N-Acetylaspartate + N-acetylaspartylglutamate) |
| NAC | N-acetylcysteine |
| NPM | N-(1-pyrenyl)maleimide |
| NVD | NAC and 1,25(OH)2D |
| PND | postnatal day |
| POST HI | immediately after hypoxia |
| POST HYPO | immediately after hypothermia |
| STEAM | simulated echo acquisition mode |
| Tau | taurine |
| TE | echo time |
| VEH | saline |
References
- Barks, J.D.E.; Liu, Y.-Q.; Shangguan, Y.; Li, J.; Pfau, J.; Silverstein, F.S. Impact of indolent inflammation on neonatal hypoxic-ischemic brain injury in mice. Int. J. Dev. Neurosci. 2007, 26, 57–65. [Google Scholar] [CrossRef]
- Eklind, S.; Mallard, C.; Leverin, A.-L.; Gilland, E.; Blomgren, K.; Mattsby-Baltzer, I.; Hagberg, H. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. Eur. J. Neurosci. 2001, 13, 1101–1106. [Google Scholar] [CrossRef]
- Garnier, Y.; Coumans, A.A.C.A.; Jensen, A.; Hasaart, T.H.M.; Berger, R. Infection-related perinatal brain injury: The pathogenic role of impaired fetal cardiovascular control. J. Soc. Gynecol. Investig. 2003, 10, 450–459. [Google Scholar] [CrossRef]
- Martinello, K.A.; Meehan, C.; Avdic-Belltheus, A.; Lingam, I.; Ragab, S.; Hristova, M.; Tann, C.J.; Peebles, D.; Hagberg, H.; Wolfs, T.G.A.M.; et al. Acute LPS sensitization and continuous infusion exacerbates hypoxic brain injury in a piglet model of neonatal encephalopathy. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tann, C.J.; Martinello, K.A.; Sadoo, S.; Lawn, J.E.; Seale, A.C.; Vega-Poblete, M.; Russell, N.J.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Neonatal Encephalopathy With Group B Streptococcal Disease Worldwide: Systematic Review, Investigator Group Datasets, and Meta-analysis. Clin. Infect. Dis. 2017, 65, S173–S189. [Google Scholar] [CrossRef] [PubMed]
- Osredkar, D.; Thoresen, M.; Maes, E.; Flatebø, T.; Elstad, M.; Sabir, H. Hypothermia is not neuroprotective after infection-sensitized neonatal hypoxic–ischemic brain injury. Resuscitation. 2014, 85, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef]
- Chevin, M.; Guiraut, C.; Maurice-Gelinas, C.; DesLauriers, J.; Grignon, S.; Sébire, G. Neuroprotective effects of hypothermia in inflammatory-sensitized hypoxic-ischemic encephalopathy. Int. J. Dev. Neurosci. 2016, 55, 1–8. [Google Scholar] [CrossRef]
- Falck, M.; Osredkar, D.; Maes, E.; Flatebø, T.; Wood, T.R.; Sabir, H.; Thoresen, M. Hypothermic Neuronal Rescue from Infection-Sensitised Hypoxic-Ischaemic Brain Injury Is Pathogen Dependent. Dev. Neurosci. 2017, 39, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Wintermark, P.; Boyd, T.; Gregas, M.C.; Labrecque, M.; Hansen, A. Placental pathology in asphyxiated newborns meeting the criteria for therapeutic hypothermia. Am. J. Obstet. Gynecol. 2010, 203, 579.e1–579.e9. [Google Scholar] [CrossRef]
- Martinello, K.; Meehan, C.; Avdic-Belltheus, A.; Mutshiya, T.; Yang, Q.; Price, D.; Sokolska, M.; Bainbridge, A.; Hristova, M.; Tann, C.J.; et al. Hypothermia is not neuroprotective in a piglet model of LPS sensitized neonatal encephalopathy. J. Paediatr. Child Health 2019, 55, 33. [Google Scholar] [CrossRef]
- Lingam, I.; Meehan, C.; Avdic-Belltheus, A.; Martinello, K.; Hristova, M.; Kaynezhad, P.; Bauer, C.; Tachtsidis, I.; Golay, X.; Robertson, N.J. Short-term effects of early initiation of magnesium infusion combined with cooling after hypoxia–ischemia in term piglets. Pediatr. Res. 2019, 86, 699–708. [Google Scholar] [CrossRef]
- Robertson, N.J.; Martinello, K.; Lingam, I.; Avdic-Belltheus, A.; Meehan, C.; Alonso-Alconada, D.; Ragab, S.; Bainbridge, A.; Sokolska, M.; Tachrount, M.; et al. Melatonin as an adjunct to therapeutic hypothermia in a piglet model of neonatal encephalopathy: A translational study. Neurobiol. Dis. 2019, 121, 240–251. [Google Scholar] [CrossRef]
- Azzopardi, D.; Robertson, N.J.; Bainbridge, A.; Cady, E.; Charles-Edwards, G.; Deierl, A.; Fagiolo, G.; Franks, N.P.; Griffiths, J.; Hajnal, J.; et al. Moderate hypothermia within 6 h of birth plus inhaled xenon versus moderate hypothermia alone after birth asphyxia (TOBY-Xe): A proof-of-concept, open-label, randomised controlled trial. Lancet Neurol. 2016, 15, 145–153. [Google Scholar] [CrossRef]
- Fang, A.Y.; Gonzalez, F.F.; Sheldon, R.A.; Ferriero, D.M. Effects of combination therapy using hypothermia and erythropoietin in a rat model of neonatal hypoxia–ischemia. Pediatr. Res. 2012, 73, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Chevin, M.; Guiraut, C.; Sébire, G. Effect of hypothermia on interleukin-1 receptor antagonist pharmacodynamics in inflammatory-sensitized hypoxic-ischemic encephalopathy of term newborns. J. Neuroinflamm. 2018, 15, 214. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Svedin, P.; Nie, C.; Lapatto, R.; Zhu, C.; Gustavsson, M.; Sandberg, M.; Karlsson, J.-O.; Romero, R.; Hagberg, H.; et al. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann. Neurol. 2007, 61, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Osredkar, D.; Sabir, H.; Falck, M.; Wood, T.K.; Maes, E.; Flatebø, T.; Puchades, M.; Thoresen, M. Hypothermia Does Not Reverse Cellular Responses Caused by Lipopolysaccharide in Neonatal Hypoxic-Ischaemic Brain Injury. Dev. Neurosci. 2015, 37, 390–397. [Google Scholar] [CrossRef]
- Packer, L.; Weber, S.U.; Rimbach, G. Molecular Aspects of α-Tocotrienol Antioxidant Action and Cell Signalling. J. Nutr. 2001, 131, 369S–373S. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Bolaños, J.P. Glutathione and γ-glutamylcysteine in the antioxidant and survival functions of mitochondria. Biochem. Soc. Trans. 2013, 41, 106–110. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrell, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ 2002, 9, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Empey, P.E.; Ji, J.; Chao, H.; Kochanek, P.M.; Bayır, H.; Clark, R.S. Probenecid and N-Acetylcysteine Prevent Loss of Intracellular Glutathione and Inhibit Neuronal Death after Mechanical Stretch Injury In Vitro. J. Neurotrauma 2016, 33, 1913–1917. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Sekhon, B.; Jatana, M.; Giri, S.; Gilg, A.G.; Sekhon, C.; Singh, I.; Singh, A.K. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J. Neurosci. Res. 2004, 76, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Maher, P.; Schubert, D. A Role for 12-lipoxygenase in Nerve Cell Death Caused by Glutathione Depletion. Neuron 1997, 19, 453–463. [Google Scholar] [CrossRef]
- Kirkland, R.A.; Franklin, J.L. Evidence for Redox Regulation of Cytochrome c Release during Programmed Neuronal Death: Antioxidant Effects of Protein Synthesis and Caspase Inhibition. J. Neurosci. 2001, 21, 1949–1963. [Google Scholar] [CrossRef]
- Lowe, D.W.; Fraser, J.L.; Rollins, L.G.; Bentzley, J.; Nie, X.; Martin, R.; Singh, I.; Jenkins, D. Vitamin D improves functional outcomes in neonatal hypoxic ischemic male rats treated with N -acetylcysteine and hypothermia. Neuropharmacology 2017, 123, 186–200. [Google Scholar] [CrossRef]
- Jain, S.K.; Micinski, D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem. Biophys. Res. Commun. 2013, 437, 7–11. [Google Scholar] [CrossRef]
- Garcion, E.; Sindji, L.; Leblondel, G.; Brachet, P.; Darcy, F. 1,25-dihydroxyvitamin D3 regulates the synthesis of gamma-glutamyl transpeptidase and glutathione levels in rat primary astrocytes. J. Neurochem. 1999, 73, 859–866. [Google Scholar] [CrossRef]
- Soher, B.J.; Semanchuk, P.; Todd, D.; Steinberg, J.; Young, K. Vespa: Integrated applications for RF pulse design, spectral simulation and MRS data analysis. Proc. Int. Soc. Magn. Reson. Med. 2011, 19, 1410. [Google Scholar]
- Provencher, S.W. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med. 1993, 30, 672–679. [Google Scholar] [CrossRef]
- Kreis, R.; Hofmann, L.; Kuhlmann, B.; Boesch, C.; Bossi, E.; Hüppi, P. Brain metabolite composition during early human brain development as measured by quantitative in vivo1H magnetic resonance spectroscopy. Magn. Reson. Med. 2002, 48, 949–958. [Google Scholar] [CrossRef]
- Wiest, D.B.; Chang, E.; Fanning, D.; Garner, S.; Cox, T.; Jenkins, D.D. Antenatal Pharmacokinetics and Placental Transfer of N-Acetylcysteine in Chorioamnionitis for Fetal Neuroprotection. J. Pediatr. 2014, 165, 672–677.e2. [Google Scholar] [CrossRef]
- Lowe, D.W.; Hollis, B.W.; Wagner, C.L.; Bass, T.; A Kaufman, D.; Horgan, M.J.; Givelichian, L.M.; Sankaran, K.; Yager, J.Y.; Katikaneni, L.D.; et al. Vitamin D insufficiency in neonatal hypoxic–ischemic encephalopathy. Pediatr. Res. 2017, 82, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Hollis, B.W.; Singh, A.K.; Singh, I. Interference with RhoA-ROCK signaling mechanism in autoreactive CD4+ T cells enhances the bioavailability of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2012, 181, 993–1006. [Google Scholar] [CrossRef][Green Version]
- Bouts, M.J.R.J.; Westmoreland, S.V.; De Crespigny, A.J.; Liu, Y.; Vangel, M.; Dijkhuizen, R.M.; Wu, O.; D’Arceuil, H.E. Magnetic resonance imaging-based cerebral tissue classification reveals distinct spatiotemporal patterns of changes after stroke in non-human primates. BMC Neurosci. 2015, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Moss, H.G.; Brown, T.R.; Wiest, D.B.; Jenkins, D.D. N-Acetylcysteine rapidly replenishes central nervous system glutathione measured via magnetic resonance spectroscopy in human neonates with hypoxic-ischemic encephalopathy. Br. J. Pharmacol. 2018, 38, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lowe, D.W.; Rollins, L.G.; Bentzley, J.; Fraser, J.L.; Martin, R.; Singh, I.; Jenkins, D. Sex-specific effects of N-acetylcysteine in neonatal rats treated with hypothermia after severe hypoxia-ischemia. Neurosci. Res. 2016, 108, 24–33. [Google Scholar] [CrossRef]
- Du, L.; Hickey, R.W.; Bayir, H.; Watkins, S.C.; Tyurin, V.A.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Ren, J.; Gibson, G.; et al. Starving Neurons Show Sex Difference in Autophagy. J. Biol. Chem. 2009, 284, 2383–2396. [Google Scholar] [CrossRef]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels – A Novel Biomarker for Mild Cognitive Impairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Zhang, Y.; Thomasson, D.M.; Latour, L.L.; Baker, E.H.; Shen, J.; Warach, S. Measurement of glutathione in normal volunteers and stroke patients at 3T using J-difference spectroscopy with minimized subtraction errors. J. Magn. Reson. Imaging 2009, 30, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Ratiney, H.; Hammond-Rosenbluth, K.E.; Pelletier, D.; Nelson, S.J. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn. Reson. Imaging 2010, 28, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Dalton, T.P.; Chen, Y.; Schneider, S.N.; Nebert, D.W.; Shertzer, H.G. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free. Radic. Biol. Med. 2004, 37, 1511–1526. [Google Scholar] [CrossRef] [PubMed]
- Ibi, M.; Sawada, H.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Depletion of Intracellular Glutathione Increases Susceptibility to Nitric Oxide in Mesencephalic Dopaminergic Neurons. J. Neurochem. 2002, 73, 1696–1703. [Google Scholar] [CrossRef]
- Franco, R.; A Cidlowski, J. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Franco, R.; Panayiotidis, M.I.; Cidlowski, J.A. Glutathione Depletion Is Necessary for Apoptosis in Lymphoid Cells Independent of Reactive Oxygen Species Formation. J. Biol. Chem. 2007, 282, 30452–30465. [Google Scholar] [CrossRef]
- Hilton, G.; Nuñez, J.L.; McCarthy, M. Sex differences in response to kainic acid and estradiol in the hippocampus of newborn rats. Neuroscience 2003, 116, 383–391. [Google Scholar] [CrossRef]
- Fu, J.; Xue, R.; Gu, J.; Xiao, Y.; Zhong, H.; Pan, X.; Ran, R. Neuroprotective effect of calcitriol on ischemic/reperfusion injury through the NR3A/CREB pathways in the rat hippocampus. Mol. Med. Rep. 2013, 8, 1708–1714. [Google Scholar] [CrossRef]
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New clues about vitamin D functions in the nervous system. Trends Endocrinol. Metab. 2002, 13, 100–105. [Google Scholar] [CrossRef]
- Groves, N.J.; McGrath, J.J.; Burne, T.H. Vitamin D as a Neurosteroid Affecting the Developing and Adult Brain. Annu. Rev. Nutr. 2014, 34, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Yue, H.; Wang, L.; Ding, C.; Wu, L.; Wu, Y.; Gao, F.; Qin, G. Vitamin D supplement ameliorates hippocampal metabolism in diabetic rats. Biochem. Biophys. Res. Commun. 2017, 490, 239–246. [Google Scholar] [CrossRef]
- Ibi, M.; Sawada, H.; Nakanishi, M.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Protective effects of 1α,25-(OH)2D3 against the neurotoxicity of glutamate and reactive oxygen species in mesencephalic culture. Neuropharmacology 2001, 40, 761–771. [Google Scholar] [CrossRef]
- Kajta, M.; Makarewicz, D.; Zieminska, E.; Jantas, D.; Domin, H.; Lasoń, W.; Kutner, A.; Łazarewicz, J.W. Neuroprotection by co-treatment and post-treating with calcitriol following the ischemic and excitotoxic insult in vivo and in vitro. Neurochem. Int. 2009, 55, 265–274. [Google Scholar] [CrossRef]
- Balden, R.; Selvamani, A.; Sohrabji, F. Vitamin D Deficiency Exacerbates Experimental Stroke Injury and Dysregulates Ischemia-Induced Inflammation in Adult Rats. Endocrinology 2012, 153, 2420–2435. [Google Scholar] [CrossRef] [PubMed]
- Brewer, L.D.; Thibault, V.; Chen, K.-C.; Langub, M.C.; Landfield, P.W.; Porter, N.M. Vitamin D Hormone Confers Neuroprotection in Parallel with Downregulation of L-Type Calcium Channel Expression in Hippocampal Neurons. J. Neurosci. 2001, 21, 98–108. [Google Scholar] [CrossRef]
- Daumas, A.; Daubail, B.; Legris, N.; Jacquin-Piques, A.; Sensenbrenner, B.; Denimal, D.; Lemaire-Ewing, S.; Duvillard, L.; Giroud, M.; Béjot, Y. Association between Admission Serum 25-Hydroxyvitamin D Levels and Functional Outcome of Thrombolyzed Stroke Patients. J. Stroke Cerebrovasc. Dis. 2016, 25, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the Vitamin D receptor and 1α-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef]
- De Abreu, D.A.F.; Eyles, D.; Féron, F. Vitamin D, a neuro-immunomodulator: Implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology 2009, 34, S265–S277. [Google Scholar] [CrossRef]
- Paintlia, M.K.; Singh, I.; Singh, A.K. Effect of vitamin D3 intake on the onset of disease in a murine model of human Krabbe disease. J. Neurosci. Res. 2014, 93, 28–42. [Google Scholar] [CrossRef]
- Park, K.-Y.; Chung, P.-W.; Kim, Y.B.; Moon, H.-S.; Suh, B.-C.; Won, Y.S.; Kim, J.-M.; Youn, Y.C.; Kwon, O.-S. Serum Vitamin D Status as a Predictor of Prognosis in Patients with Acute Ischemic Stroke. Cerebrovasc. Dis. 2015, 40, 73–80. [Google Scholar] [CrossRef]
- Turetsky, A.; Goddeau, R.P.; Henninger, N. Low Serum Vitamin D Is Independently Associated with Larger Lesion Volumes after Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2015, 24, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Mandell, E.; Seedorf, G.J.; Ryan, S.; Gien, J.; Cramer, S.D.; Abman, S.H. Antenatal endotoxin disrupts lung vitamin D receptor and 25-hydroxyvitamin D 1α-hydroxylase expression in the developing rat. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1018–L1026. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.; Sarıaydın, M.; Aslan, Y.; Şebnem, K.; Dereci, S.; Kart, C.; Yaman, S.Ö.; Kural, B. Status of vitamin D, antioxidant enzymes, and antioxidant substances in neonates with neonatal hypoxic-ischemic encephalopathy. J. Matern. Neonatal Med. 2015, 29, 1–5. [Google Scholar] [CrossRef]
- Konety, B.R.; Somogyi, G.; Atan, A.; Muindi, J.; Chancellor, M.B.; Getzenberg, R.H. Evaluation of intraprostatic metabolism of 1,25-dihydroxyvitamin D(3) (calcitriol) using a microdialysis technique. Urology 2002, 59, 947–952. [Google Scholar] [CrossRef]
- LaMonte, G.; Tang, X.; Chen, J.L.-Y.; Wu, J.; Ding, C.-K.C.; Keenan, M.M.; Sangokoya, C.; Kung, H.-N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.B.; Empey, P.E.; Bayır, H.; Rosario, B.L.; Poloyac, S.M.; Kochanek, P.M.; Nolin, T.D.; Au, A.K.; Horvat, C.M.; Wisniewski, S.R.; et al. Phase I randomized clinical trial of N-acetylcysteine in combination with an adjuvant probenecid for treatment of severe traumatic brain injury in children. PLoS ONE 2017, 12, e0180280. [Google Scholar] [CrossRef]
- Nemere, I.; Wilson, C.; Jensen, W.; Steinbeck, M.; Rohe, B.; Farach-Carson, M.C. Mechanism of 24,25-dihydroxyvitamin D3-mediated inhibition of rapid, 1,25-dihydroxyvitamin D3-induced responses: Role of reactive oxygen species. J. Cell. Biochem. 2006, 99, 1572–1581. [Google Scholar] [CrossRef]
- Igarashi, H.; Suzuki, Y.; Huber, V.J.; Ida, M.; Nakada, T. N-acetylaspartate Decrease in Acute Stage of Ischemic Stroke: A Perspective from Experimental and Clinical Studies. Magn. Reson. Med. Sci. 2015, 14, 13–24. [Google Scholar] [CrossRef]
- Xu, S.; Waddell, J.; Zhu, W.; Shi, D.; Marshall, A.D.; McKenna, M.C.; Gullapalli, R.P. In vivo longitudinal proton magnetic resonance spectroscopy on neonatal hypoxic-ischemic rat brain injury: Neuroprotective effects of acetyl-L-carnitine. Magn. Reson. Med. 2015, 74, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Xarrié, E.; Davila, M.; Candiota, A.P.; Delgado-Mederos, R.; Ortega-Martorell, S.; Julià-Sapé, M.; Arús, C.; Martí-Fàbregas, J. Brain metabolic pattern analysis using a magnetic resonance spectra classification software in experimental stroke. BMC Neurosci. 2017, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Mlynárik, V.; Kohler, I.; Gambarota, G.; Vaslin, A.; Clarke, P.G.; Gruetter, R. Quantitative proton spectroscopic imaging of the neurochemical profile in rat brain with microliter resolution at ultra-short echo times. Magn. Reson. Med. 2007, 59, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zijden, J.P.; Bouts, M.J.R.J.; Wu, O.; Roeling, T.A.P.; Bleys, R.L.; Van Der Toorn, A.; Dijkhuizen, R.M. Manganese-Enhanced MRI of Brain Plasticity in Relation to Functional Recovery after Experimental Stroke. Br. J. Pharmacol. 2007, 28, 832–840. [Google Scholar] [CrossRef]
- Shemesh, N.; Rosenberg, J.T.; Dumez, J.-N.; Muniz, J.A.; Grant, S.C.; Frydman, L. Metabolic properties in stroked rats revealed by relaxation-enhanced magnetic resonance spectroscopy at ultrahigh fields. Nat. Commun. 2014, 5, 4958. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSH | GLX | tCr | NAA | tCho | Ins | Tau | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SHAM | LPS-HI | SHAM | LPS-HI | SHAM | LPS-HI | SHAM | LPS-HI | SHAM | LPS-HI | SHAM | LPS-HI | SHAM | LPS-HI | |
| PRE PND 6 | 3.06 ± 0.11 | 3.06 ± 0.19 * | 9.08 ± 0.19 | 9.00 ± 0.39 * | 8.64 ± 0.17 | 8.58 ± 0.22 | 4.98 ± 0.19 | 4.75 ± 0.20 | 3.02 ± 0.07 | 2.89 ± 0.17 | 3.20 ± 0.42 | 3.13 ± 0.98 | 17.22 ± 0.48 | 16.70 ± 1.08 |
| PND 7/ POST HI | 2.89 ± 0.11 | 2.65 ± 0.19 * | 9.57 ± 0.20 | 7.59 ± 0.40 * | 8.44 ± 0.18 | 8.41 ± 023 | 4.80 ± 0.09 | 5.02 ± 0.10 | 3.01 ± 0.07 | 2.71 ± 0.18 | 2.95 ± 0.57 | 3.15 ± 0.90 | 16.25 ± 0.54 | 16.18 ± 1.6 |
| p value | ns | <0.0001 | ns | <0.0001 | ns | ns | ns | ns | ns | <0.001 | ns | ns | ns | ns |
| GSH | GLX | tCr | NAA | tCho | Ins | Tau | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VEH | NVD | VEH | NVD | VEH | NVD | VEH | NVD | VEH | NVD | VEH | NVD | VEH | NVD | |
| PRE PND 6 | 2.97 ± 0.22 *^ | 3.16 ± 0.32 *^ | 8.94 ± 0.26 *^ | 9.02 ± 0.74 *^ | 8.65 ± 0.27 | 8.52 ± 0.35 | 4.96 ± 0.12 | 5.14 ± 0.13 | 2.82 ± 0.24 | 2.97 ± 0.25 *^ | 3.32 ± 0.91 | 3.16 ± 0.91 | 16.70 ± 1.08 | 16.18 ± 1.60 |
| POST HI | 2.72 ± 0.22 ^ | 2.58 ± 0.32 * | 7.41 ± 0.28 * | 7.77 ± 0.74 * | 8.37± 0.28 * | 8.44 ± 0.35 | 4.74 ± 0.19 | 4.94 ± 0.16 | 2.66 ± 0.24 | 2.75 ± 0.25 * | 2.86 ± 0.92 | 2.85 ± 0.91 | 15.85 ± 1.10 | 15.74 ± 1.62 |
| POST HYPO | 2.65 ± 0.22 * | 2.90 ± 0.32 *^ | 8.16 ± 0.29 *^ | 7.54 ± 0.74 ^ | 8.96 ± 0.28 * | 8.67 ± 0.35 | 4.94 ± 0.16 | 5.14 ± 0.13 | 2.71 ± 0.24 | 2.72 ± 0.25 ^ | 3.42 ± 0.92 | 2.83 ± 0.91 | 16.45 ± 1.10 | 15.59 ± 1.62 |
| p value | <0.05 | <0.01 | <0.005 | <0.0001 | ≤ 0.01 | ns | ns | ns | ns | <0.005 | ns | ns | ns | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adams, L.E.; Moss, H.G.; Lowe, D.W.; Brown, T.; Wiest, D.B.; Hollis, B.W.; Singh, I.; Jenkins, D.D. NAC and Vitamin D Restore CNS Glutathione in Endotoxin-Sensitized Neonatal Hypoxic-Ischemic Rats. Antioxidants 2021, 10, 489. https://doi.org/10.3390/antiox10030489
Adams LE, Moss HG, Lowe DW, Brown T, Wiest DB, Hollis BW, Singh I, Jenkins DD. NAC and Vitamin D Restore CNS Glutathione in Endotoxin-Sensitized Neonatal Hypoxic-Ischemic Rats. Antioxidants. 2021; 10(3):489. https://doi.org/10.3390/antiox10030489
Chicago/Turabian StyleAdams, Lauren E., Hunter G. Moss, Danielle W. Lowe, Truman Brown, Donald B. Wiest, Bruce W. Hollis, Inderjit Singh, and Dorothea D. Jenkins. 2021. "NAC and Vitamin D Restore CNS Glutathione in Endotoxin-Sensitized Neonatal Hypoxic-Ischemic Rats" Antioxidants 10, no. 3: 489. https://doi.org/10.3390/antiox10030489
APA StyleAdams, L. E., Moss, H. G., Lowe, D. W., Brown, T., Wiest, D. B., Hollis, B. W., Singh, I., & Jenkins, D. D. (2021). NAC and Vitamin D Restore CNS Glutathione in Endotoxin-Sensitized Neonatal Hypoxic-Ischemic Rats. Antioxidants, 10(3), 489. https://doi.org/10.3390/antiox10030489