
Pharmacological Targeting of Heme Oxygenase-1 in Osteoarthritis

Abstract
1. Introduction
2. Osteoarthritis Pathogenesis and Its Relationship with Oxidative Stress
2.1. Osteoarthritis Development
2.2. Oxidative Stress and Antioxidant Signaling in Joints
3. The Role of Heme Oxygenase-1 in Arthritis
3.1. Properties of Heme Oxygenase-1
3.2. Anti-Inflammatory Function of Heme Oxygenase-1
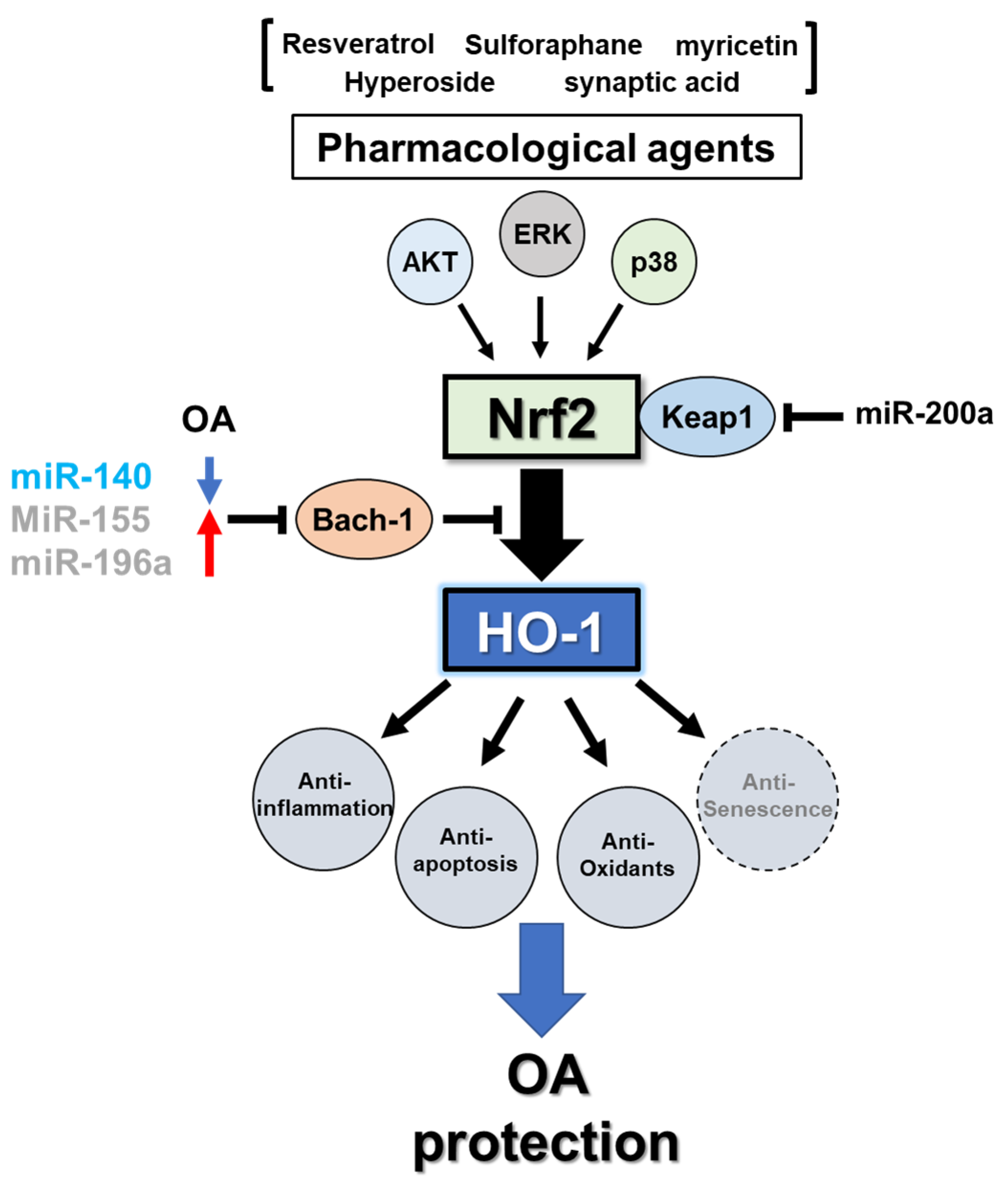
3.3. Regulation of Heme Oxygenase-1
3.3.1. Transcriptional Regulation
3.3.2. MicroRNA-Mediated Post-Transcriptional Regulation
3.4. Heme Oxygenase-1 in Osteoarthritis
3.5. Heme Oxygenase-1 and Osteoarthritis-Associated Cellular Senescence
4. Pharmacological Treatment for OA Protection
4.1. OA Animal Models for Drug Development
4.2. Identifying a Role for HO-1 from a Genetic Modification Mouse Model
4.3. Pharmacological Treatment with HO-1
4.3.1. Intra-Articular Injection-Based Delivery
4.3.2. Intraperitoneal Injection-Based Delivery
4.3.3. Oral Administration
5. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoy, D.G.; Smith, E.; Cross, M.; Sanchez-Riera, L.; Blyth, F.M.; Buchbinder, R.; Woolf, A.D.; Driscoll, T.; Brooks, P.; March, L.M. Reflecting on the global burden of musculoskeletal conditions: Lessons learnt from the Global Burden of Disease 2010 Study and the next steps forward. Ann. Rheum. Dis. 2015, 74, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Del Carlo, M.; Loeser, R.F. Increased Oxidative Stress with Aging Reduces Chondrocyte Survival: Correlation with Intracellular Glutathione Levels. Arthritis Rheum. 2003, 48, 3419–3430. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta Mol. Basis Dis 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Scott, J.L.; Gabrielides, C.; Davidson, R.K.; Swingler, T.E.; Clark, I.M.; Wallis, G.A.; Boot-Handford, R.P.; Kirkwood, T.B.L.; Talyor, R.W.; Young, D.A. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 2010, 69, 1502–1510. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Loeser, R.F.; Carlson, C.S.; Del Carlo, M.; Cole, A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1β and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002, 46, 2349–2357. [Google Scholar] [CrossRef]
- Alcaraz, M.J.; Ferrándiz, M.L. Relevance of Nrf2 and heme oxygenase-1 in articular diseases. Free Radic. Biol. Med 2020, 157, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef]
- Koike, M.; Nojiri, H.; Ozawa, Y.; Watanabe, K.; Muramatsu, Y.; Kaneko, H.; Morikawa, D.; Kobayashi, K.; Saita, Y.; Sasho, T.; et al. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 2015, 5, 11722. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Miyaki, S.; Ishitobi, H.; Hirai, Y.; Nakasa, T.; Igarashi, K.; Lotz, M.K.; Ochi, M. Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Ishitobi, H.; Sanada, Y.; Kato, Y.; Ikuta, Y.; Shibata, S.; Yamasaki, S.; Lotz, M.K.; Matsubara, K.; Miyaki, S.; Adachi, N. Carnosic acid attenuates cartilage degeneration through induction of heme oxygenase-1 in human articular chondrocytes. Eur. J. Pharmacol. 2018, 830, 1–8. [Google Scholar] [CrossRef]
- Sun, J.; Wei, X.; Lu, Y.; Cui, M.; Li, F.; Lu, J.; Liu, Y.; Zhang, X. Glutaredoxin 1 (GRX1) inhibits oxidative stress and apoptosis of chondrocytes by regulating CREB/HO-1 in osteoarthritis. Mol. Immunol. 2017, 90, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.; Choi, A.M.K. Heme oxygenase-1: The “emerging molecule” has arrived. Am. J. Respir. Cell Mol. Biol 2002, 27, 8–16. [Google Scholar] [CrossRef]
- Kuo, S.J.; Yang, W.H.; Liu, S.C.; Tsai, C.H.; Hsu, H.C.; Tang, C.H. Transforming growth factor β1 enhances heme oxygenase 1 expression in human synovial fibroblasts by inhibiting microRNA 519b synthesis. PLoS ONE 2017, 12, e0176052. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, S.; Mizuno, T.; Deie, M.; Igarashi, K.; Hamada, Y.; Ochi, M. Oxidative stress reaction in the meniscus of bach 1 deficient mice: Potential prevention of meniscal degeneration. J. Orthop. Res. 2008, 26, 894–898. [Google Scholar] [CrossRef]
- Ohta, R.; Tanaka, N.; Nakanishi, K.; Kamei, N.; Nakamae, T.; Izumi, B.; Fujioka, Y.; Ochi, M. Heme oxygenase-1 modulates degeneration of the intervertebral disc after puncture in Bach 1 deficient mice. Eur. Spine J. 2012, 21, 1748–1757. [Google Scholar] [CrossRef][Green Version]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- Okada, S.; Muto, A.; Ogawa, E.; Nakanome, A.; Katoh, Y.; Ikawa, S.; Aiba, S.; Igarashi, K.; Okuyama, R. Bach1-dependent and -independent regulation of heme oxygenase-1 in keratinocytes. J. Biol. Chem. 2010, 285, 23581–23589. [Google Scholar] [CrossRef]
- Deshane, J.; Kim, J.; Bolisetty, S.; Hock, T.D.; Hill-Kapturczak, N.; Agarwal, A. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010, 285, 16476–16486. [Google Scholar] [CrossRef]
- Kim, J.; Zarjou, A.; Traylor, A.M.; Bolisetty, S.; Jaimes, E.A.; Hull, T.D.; George, J.F.; Mikhail, F.M.; Agarwal, A. In vivo regulation of the heme oxygenase-1 gene in humanized transgenic mice. Kidney Int. 2012, 82, 278–291. [Google Scholar] [CrossRef]
- Sun, K.; Luo, J.; Jing, X.; Guo, J.; Yao, X.; Hao, X.; Ye, Y.; Liang, S.; Lin, J.; Wang, G.; et al. Astaxanthin protects against osteoarthritis via Nrf2: A guardian of cartilage homeostasis. Aging 2019, 11, 10513–10531. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Feng, W.; Liu, J.; Jiang, L.; Chen, S.; Yuan, T.; Yu, C.; Xie, H.; Geng, D.; Qin, J. 7,8-Dihydroxyflavone activates Nrf2/HO-1 signaling pathways and protects against osteoarthritis. Exp. Ther. Med. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Fu, C.; Yan, Z.; Wu, Y.; Zhan, J.; Lou, Z.; Liao, X.; Pan, J. The protective effect of hesperetin in osteoarthritis: An: In vitro and in vivo study. Food Funct. 2020, 11, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil. Med 2016, 59, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Van Manen, M.D.; Nace, J.; Mont, M.A. Management of primary knee osteoarthritis and indications for total knee arthroplasty for general practitioners. J. Am. Osteopath. Assoc. 2012, 112, 709–715. [Google Scholar] [CrossRef]
- Peat, G.; Thomas, E.; Duncan, R.; Wood, L.; Hay, E.; Croft, P. Clinical classification criteria for knee osteoarthritis: Performance in the general population and primary care. Ann. Rheum. Dis. 2006, 65, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Yamada, N.; Cheung, K.S.C.; Tilley, S.; Clarke, N.M.P.; Oreffo, R.O.C.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef] [PubMed]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.A.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, A.S.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Goldring, M.B. Osteoarthritis and cartilage: The role of cytokines. Curr. Rheumatol. Rep. 2000, 2, 459–465. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Fan, Z.; Söder, S.; Oehler, S.; Fundel, K.; Aigner, T. Activation of interleukin-1 signaling cascades in normal and osteoarthritis articular cartilage. Am. J. Pathol. 2007, 171, 938–946. [Google Scholar] [CrossRef]
- Fan, Z.; Yang, H.; Bau, B.; Söder, S.; Aigner, T. Role of mitogen-activated protein kinases and NFκB on IL-1β-induced effects on collagen type II, MMP-1 and 13 mRNA expression in normal articular human chondrocytes. Rheumatol. Int. 2006, 26, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bryan, J.L.; DeLassus, E.; Chang, L.W.; Liao, W.; Sandell, L.J. CCAAT/enhancer-binding protein β and NF-κB mediate high level expression of chemokine genes CCL3 and CCL4 by human chondrocytes in response to IL-1β. J. Biol. Chem. 2010, 285, 33092–33103. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D. Genetic epidemiology of hip and knee osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 23–32. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol 2000, 279, L1005-28. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Loeser, R.F. The Effects of Aging on the Development of Osteoarthritis. HSS J. 2012, 8, 18–19. [Google Scholar] [CrossRef]
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Weinberg, A.M.; Al-Wasiyah, M.K.; Alqahtani, M.H.; Mobasheri, A. Biomarkers of chondrocyte apoptosis and autophagy in osteoarthritis. Int. J. Mol. Sci. 2015, 16, 20560–20575. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Mori, D.; Kobayashi, H.; Mori, Y.; Nakamoto, H.; Okada, K.; Taniguchi, Y.; Sugita, S.; Yano, F.; Chung, U.I.; et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1–NF-κB pathway. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil 2009, 17, 971–979. [Google Scholar] [CrossRef]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, H.; Wei, J.; Lin, S.; Zong, Z.; Gong, F.; Huang, X.; Sun, J.; Li, P.; Lin, H.; et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res. Ther. 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.M.; Guilak, F.; Weinberg, J.B.; Fermor, B. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthr. Cartil. 2008, 16, 624–630. [Google Scholar] [CrossRef]
- Kim, K.M.K.; Park, S.E.; Lee, M.S.; Kim, K.M.K.; Park, Y.C. Induction of heme oxygenase-1 expression protects articular chondrocytes against cilostazol-induced cellular senescence. Int. J. Mol. Med. 2014, 34, 1335–1340. [Google Scholar] [CrossRef][Green Version]
- García-Arnandis, I.; Guillén, M.I.; Castejón, M.A.; Gomar, F.; Alcaraz, M.J. Haem oxygenase-1 down-regulates high mobility group box 1 and matrix metalloproteinases in osteoarthritic synoviocytes. Rheumatology 2010, 49, 854–861. [Google Scholar] [CrossRef]
- Gavriilidis, C.; Miwa, S.; Von Zglinicki, T.; Taylor, R.W.; Young, D.A. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013, 65, 378–387. [Google Scholar] [CrossRef]
- Wruck, C.J.; Fragoulis, A.; Gurzynski, A.; Brandenburg, L.O.; Kan, Y.W.; Chan, K.; Hassenpflug, J.; Freitag-Wolf, S.; Varoga, D.; Lippross, S.; et al. Role of oxidative stress in rheumatoid arthritis: Insights from the Nrf2-knockout mice. Ann. Rheum. Dis. 2011, 70, 844–850. [Google Scholar] [CrossRef]
- Maicas, N.; Ferrándiz, M.L.; Brines, R.; Ibáñez, L.; Cuadrado, A.; Koenders, M.I.; Van Den Berg, W.B.; Alcaraz, M.J. Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxidants Redox Signal. 2011, 15, 889–901. [Google Scholar] [CrossRef]
- Ferrándiz, M.L.; Nacher-Juan, J.; Alcaraz, M.J. Nrf2 as a therapeutic target for rheumatic diseases. Biochem. Pharmacol. 2018, 152, 338–346. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduño, L.K.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Arai, Y.; Mazda, O.; Kishida, T.; Takahashi, K.A.; Sakao, K.; Saito, M.; Honjo, K.; Imanishi, J.; Kubo, T. N-acetylcysteine prevents nitric oxide-induced chondrocyte apoptosis and cartilage degeneration in an experimental model of osteoarthritis. J. Orthop. Res. 2010, 28, 156–163. [Google Scholar] [CrossRef]
- Aini, H.; Ochi, H.; Iwata, M.; Okawa, A.; Koga, D.; Okazaki, M.; Sano, A.; Asou, Y. Procyanidin B3 prevents articular cartilage degeneration and heterotopic cartilage formation in a mouse surgical osteoarthritis model. PLoS ONE 2012, 7, e37728. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Pan, Z.; Lin, J.; Zhao, Q.; Wang, Y.; Ni, L.; Feng, S.; Tian, N.; Wu, Y.; Sun, L.; et al. S-allyl cysteine reduces osteoarthritis pathology in the tert-butyl hydroperoxide-treated chondrocytes and the destabilization of the medial meniscus model mice via the Nrf2 signaling pathway. Aging 2020, 12, 19254–19272. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Kutty, R.K.; Daniel, R.F.; Ryan, D.E.; Levin, W.; Maines, M.D. Rat liver cytochrome P-450b, P-420b, and P-420c are degraded to biliverdin by heme oxygenase. Arch. Biochem. Biophys. 1988, 260, 638–644. [Google Scholar] [CrossRef]
- Elbirt, K.K.; Bonkovsky, H.L. Heme oxygenase: Recent advances in understanding its regulation and role. Proc. Assoc. Am. Physicians 1999, 111, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Dennery, P.A. Signaling function of heme oxygenase proteins. Antioxid. Redox Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef]
- Vitali, S.H.; Fernandez-Gonzalez, A.; Nadkarni, J.; Kwong, A.; Rose, C.; Alex Mitsialis, S.; Kourembanas, S. Heme oxygenase-1 dampens the macrophage sterile inflammasome response and regulates its components in the hypoxic lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L125–L134. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M.K. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res 2016, 167, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Willis, D.; Moore, A.R.; Frederick, R.; Willoughby, D.A. Heme oxygenase: A novel target for the modulation of the inflammatory response. Nat. Med. 1996, 2, 87–90. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef]
- Kawashima, A.; Oda, Y.; Yachie, A.; Koizumi, S.; Nakanishi, I. Heme oxygenase-1 deficiency: The first autopsy case. Hum. Pathol. 2002, 33, 125–130. [Google Scholar] [CrossRef]
- Burnett, A.L.; Johns, D.G.; Kriegsfeld, L.J.; Klein, S.L.; Calbin, D.C.; Demas, G.E.; Schramm, L.P.; Tonegawa, S.; Nelson, R.J.; Snyder, S.H.; et al. Ejaculatory abnormalities in mice with targeted disruption of the gene for heme oxygenase-2. Nat. Med. 1998, 4, 84–87. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Uchiyama, K.; Yoshikawa, T. Heme oxygenase-1: A novel therapeutic target for gastrointestinal diseases. J. Clin. Biochem. Nutr. 2011, 48, 126–133. [Google Scholar] [CrossRef]
- Sikorski, E.M.; Hock, T.; Hill-Kapturczak, N.; Agarwal, A. The story so far: Molecular regulation of the heme oxygenase-1 gene in renal injury. Am. J. Physiol. Ren. Physiol. 2004, 286. [Google Scholar] [CrossRef]
- Ann Applegate, L.; Luscher, P.; Tyrrell, R.M. Induction of Heme Oxygenase: A General Response to Oxidant Stress in Cultured Mammalian Cells. Cancer Res. 1991, 51, 974–978. [Google Scholar]
- Zhou, J.; Terluk, M.R.; Basso, L.; Mishra, U.R.; Orchard, P.J.; Cloyd, J.C.; Schröder, H.; Kartha, R.V. N-acetylcysteine provides cytoprotection in murine oligodendrocytes through heme oxygenase-1 activity. Biomedicines 2020, 8, 240. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Zhang, X.; Bedard, E.L.; Potter, R.; Zhong, R.; Alam, J.; Choi, A.M.K.; Lee, P.J. Mitogen-activated protein kinases regulate HO-1 gene transcription after ischemia-reperfusion lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-Activated Protein Kinases: A Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar]
- Naidu, S.; Vijayan, V.; Santoso, S.; Kietzmann, T.; Immenschuh, S. Inhibition and Genetic Deficiency of p38 MAPK Up-Regulates Heme Oxygenase-1 Gene Expression via Nrf2. J. Immunol. 2009, 182, 7048–7057. [Google Scholar] [CrossRef]
- Salom, M.G.; Bonacasa, B. Humanized HO-1 BAC transgenic mice: A new model for the study of HO-1 gene regulation. Kidney Int. 2012, 82, 253–255. [Google Scholar] [CrossRef][Green Version]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Buscher, C.; Weis, A.; Wohrle, M.; Bretzel, R.G.; Cohen, A.M.; Federlin, K. Islet transplantation in experimental diabetes of the rat. XII. Effect on diabetic retinopathy. Morphological findings and morphometrical evaluation. Horm. Metab. Res. 1989, 21, 227–231. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Chattopadhyay, A. Nrf2–ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, S.; Du, Z.; Ke, A.; Liang, Q.; Li, X. KLF2 Protects against Osteoarthritis by Repressing Oxidative Response through Activation of Nrf2/ARE Signaling in Vitro and in Vivo. Oxid. Med. Cell. Longev. 2019, 2019, 8564681. [Google Scholar] [CrossRef]
- Das, M.; Laha, D.; Kanji, S.; Joseph, M.; Aggarwal, R.; Iwenofu, O.H.; Pompili, V.J.; Jain, M.K.; Das, H. Induction of Krüppel-like factor 2 reduces K/BxN serum-induced arthritis. J. Cell. Mol. Med. 2019, 23, 1386–1395. [Google Scholar] [CrossRef]
- Wu, W.D.; Geng, P.B.; Zhu, J.; Li, J.W.; Zhang, L.; Chen, W.L.; Zhang, D.F.; Lu, Y.; Xu, X.H. KLF2 regulates eNOS uncoupling via Nrf2/HO-1 in endothelial cells under hypoxia and reoxygenation. Chem. Biol. Interact. 2019, 305, 105–111. [Google Scholar] [CrossRef]
- Fledderus, J.O.; Boon, R.A.; Volger, O.L.; Hurttila, H.; Ylä-Herttuala, S.; Pannekoek, H.; Levonen, A.L.; Horrevoets, A.J.G. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C. Posttranscriptional Regulation of MicroRNA Biogenesis in Animals. Mol. Cell. 2010, 38, 323–332. [Google Scholar] [CrossRef]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S.; et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Asahara, H. Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Munk, R.; Panda, A.C.; Grammatikakis, I.; Gorospe, M.; Abdelmohsen, K. Senescence-Associated MicroRNAs. Int. Rev. Cell Mol. Biol. 2017, 334, 177–205. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, K.S.; Lee, D.K.; Kim, J.; Kwak, S.N.; Ha, K.S.; Choe, J.; Won, M.H.; Cho, B.R.; Jeoung, D.; et al. Hypoxia-responsive MicroRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxidants Redox Signal. 2014, 21, 2469–2482. [Google Scholar] [CrossRef]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef]
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef]
- Beckman, J.D.; Chen, C.; Nguyen, J.; Thayanithy, V.; Subramanian, S.; Steer, C.J.; Vercellotti, G.M. Regulation of heme oxygenase-1 protein expression by miR-377 in Combination with miR-217. J. Biol. Chem. 2011, 286, 3194–3202. [Google Scholar] [CrossRef]
- Pulkkinen, K.H.; Ylä-Herttuala, S.; Levonen, A.L. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic. Biol. Med. 2011, 51, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, S.; Alvarez-Garcia, O.; Muramatsu, Y.; Flamigni, F.; Lotz, M.K. MicroRNA-155 suppresses autophagy in chondrocytes by modulating expression of autophagy proteins. Osteoarthr. Cartil. 2016, 24, 1082–1091. [Google Scholar] [CrossRef]
- Li, H.; Liu, P.; Gong, Y.; Liu, J.; Ruan, F. Expression and function of miR-155 in rat synovial fibroblast model of rheumatoid arthritis. Exp. Ther. Med. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Su, L.C.; Huang, A.F.; Jia, H.; Liu, Y.; Xu, W.D. Role of microRNA-155 in rheumatoid arthritis. Int. J. Rheum. Dis. 2017, 20, 1631–1637. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Alivernini, S.; Ballantine, L.E.; Asquith, D.L.; Millar, N.L.; Gilchrist, D.S.; Reilly, J.; Ierna, M.; Fraser, A.R.; Stolarski, B.; et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 11193–11198. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic Control of Autophagy. Cell. 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- Vinatier, C.; Domínguez, E.; Guicheux, J.; Caramés, B. Role of the inflammation-autophagy-senescence integrative network in Osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Watari, T.; Naito, K.; Sakamoto, K.; Kurosawa, H.; Nagaoka, I.; Kaneko, K. Evaluation of the effect of oxidative stress on articular cartilage in spontaneously osteoarthritic STR/OrtCrlj mice by measuring the biomarkers for oxidative stress and type II collagen degradation/synthesis. Exp. Ther. Med. 2011, 2, 245–250. [Google Scholar] [CrossRef]
- Khan, N.M.; Ahmad, I.; Haqqi, T.M. Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK signaling axis. Free Radic. Biol. Med. 2018, 116, 159–171. [Google Scholar] [CrossRef]
- Cai, D.; Huff, T.W.; Liu, J.; Yuan, T.; Wei, Z.; Qin, J. Alleviation of cartilage destruction by sinapic acid in experimental osteoarthritis. BioMed Res. Int. 2019, 2019, 5689613. [Google Scholar] [CrossRef]
- Abusarah, J.; Benabdoune, H.; Shi, Q.; Lussier, B.; Martel-Pelletier, J.; Malo, M.; Fernandes, J.C.; de Souza, F.P.; Fahmi, H.; Benderdour, M. Elucidating the Role of Protandim and 6-Gingerol in Protection Against Osteoarthritis. J. Cell. Biochem. 2017, 118, 1003–1013. [Google Scholar] [CrossRef]
- Fernández, P.; Guillén, M.I.; Gomar, F.; Alcaraz, M.J. Expression of heme oxygenase-1 and regulation by cytokines in human osteoarthritic chondrocytes. Biochem. Pharmacol. 2003, 66, 2049–2052. [Google Scholar] [CrossRef]
- Guillén, M.I.; Megías, J.; Gomar, F.; Alcaraz, M.J. Haem oxygenase-1 regulates catabolic and anabolic processes in osteoarthritic chondrocytes. J. Pathol. 2008, 214, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F.; Nguyen, M.V.C.; Grange, L.; Morel, F.; Lardy, B. Heme Oxygenase-1 Regulates Matrix Metalloproteinase MMP-1 Secretion and Chondrocyte Cell Death via Nox4 NADPH Oxidase Activity in Chondrocytes. PLoS ONE 2013, 8, e0066478. [Google Scholar] [CrossRef]
- Wei, Y.; Jia, J.; Jin, X.; Tong, W.; Tian, H. Resveratrol ameliorates inflammatory damage and protects against osteoarthritis in a rat model of osteoarthritis. Mol. Med. Rep. 2018, 17, 1493–1498. [Google Scholar] [CrossRef]
- Clérigues, V.; Murphy, C.L.; Guillén, M.I.; Alcaraz, M.J. Haem oxygenase-1 induction reverses the actions of interleukin-1β on hypoxia-inducible transcription factors and human chondrocyte metabolism in hypoxia. Clin. Sci. 2013, 125, 99–108. [Google Scholar] [CrossRef]
- Pan, X.; Chen, T.; Zhang, Z.; Chen, X.; Chen, C.; Chen, L.; Wang, X.; Ying, X. Activation of Nrf2/HO-1 signal with Myricetin for attenuating ECM degradation in human chondrocytes and ameliorating the murine osteoarthritis. Int. Immunopharmacol. 2019, 75. [Google Scholar] [CrossRef]
- Hinoi, E.; Takarada, T.; Fujimori, S.; Wang, L.; Iemata, M.; Uno, K.; Yoneda, Y. Nuclear factor E2 p45-related factor 2 negatively regulates chondrogenesis. Bone 2007, 40, 337–344. [Google Scholar] [CrossRef]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic. Biol. Med. 2015, 88, 168–178. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2021, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. 2011, 29, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Brown, T.D.; Heiner, A.D.; Buckwalter, J.A. Chondrocyte senescence, joint loading and osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S96–S103. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, F.; Tong, Z.; Konstantopoulos, K. Response of chondrocytes to shear stress: Antagonistic effects of the binding partners Toll-like receptor 4 and caveolin-1. FASEB J. 2011, 25, 3401–3415. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, P.; Lee, N.H.; Goldring, M.B.; Konstantopoulos, K. Prolonged Application of High Fluid Shear to Chondrocytes Recapitulates Gene Expression Profiles Associated with Osteoarthritis. PLoS ONE 2010, 5, e0015174. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.K.; Sohn, D.H.; Kim, K.M.K.; Park, Y.C. Inhibition of protein kinase CK2 facilitates cellular senescence by inhibiting the expression of HO-1 in articular chondrocytes. Int. J. Mol. Med. 2019, 43, 1033–1040. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, H.; Porvasnik, S.L.; Terada, N.; Agarwal, A.; Cheng, Y.; Visner, G.A. Heme oxygenase-1 mediates the protective effects of rapamycin in monocrotaline-induced pulmonary hypertension. Lab. Investig. 2006, 86, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Song, D.H.; Kim, S.H.; Jung, Y.; Kim, S.J. Development and characterization of various osteoarthritis models for tissue engineering. PLoS ONE 2018, 13, e0194288. [Google Scholar] [CrossRef]
- Kuyinu, E.L.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Animal models of osteoarthritis: Classification, update, and measurement of outcomes. J. Orthop. Surg. Res. 2016, 11. [Google Scholar] [CrossRef]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.A.; Glasson, S.S.; Trubetskoy, O.V.; Haimes, H.B. Spontaneous Osteoarthritis in Dunkin Hartley Guinea Pigs: Histologic, Radiologic, and Biochemical Changes. Lab. Anim. Sci. 1997, 47, 598–601. [Google Scholar] [PubMed]
- Lampropoulou-Adamidou, K.; Lelovas, P.; Karadimas, E.V.; Liakou, C.; Triantafillopoulos, I.K.; Dontas, I.; Papaioannou, N.A. Useful animal models for the research of osteoarthritis. Eur. J. Orthop. Surg. Traumatol. 2014, 24, 263–271. [Google Scholar] [CrossRef]
- Yan, J.Y.; Zhang, Y.Z.; Tian, F.M.; Wang, W.Y.; Cheng, Y.; Xu, H.F.; Song, H.P.; Zhang, L. Age dependent changes in cartilage matrix, subchondral bone mass, and estradiol levels in blood serum, in naturally occurring osteoarthritis in guinea pigs. Int. J. Mol. Sci. 2014, 15, 13578–13595. [Google Scholar] [CrossRef]
- Mason, R.M.; Chambers, M.G.; Flannelly, J.; Gaffen, J.D.; Dudhia, J.; Bayliss, M.T. The STR/ort mouse and its use as a model of osteoarthritis. Osteoarthr. Cartil. 2001, 9, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Staines, K.A.; Poulet, B.; Wentworth, D.N.; Pitsillides, A.A. The STR/ort mouse model of spontaneous osteoarthritis–an update. Osteoarthr. Cartil. 2017, 25, 802–808. [Google Scholar] [CrossRef]
- Bapat, S.; Hubbard, D.; Munjal, A.; Hunter, M.; Fulzele, S. Pros and cons of mouse models for studying osteoarthritis. Clin. Transl. Med. 2018, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Vitters, E.L.; Van de Putte, L.B.A.; Van den Berg, W.B. Development of osteoarthritic lesions in mice by “metabolic” and “mechanical” alterations in the knee joints. Am. J. Pathol. 1989, 135, 1001–1014. [Google Scholar]
- Hayami, T.; Pickarski, M.; Zhuo, Y.; Wesolowski, G.A.; Rodan, G.A.; Duong, L.T. Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone 2006, 38, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Guingamp, C.; Gegout-Pottie, P.; Philippe, L.; Terlain, B.; Netter, P.; Gillet, P. Mono-iodoacetate-induced experimental osteoarthritis: A dose-response study of loss of mobility, morphology, and biochemistry. Arthritis Rheum. 1997, 40, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.E.; Evans, M.G.; Bove, S.; Morenko, B.; Kilgore, K. Mono-Iodoacetate-Induced Histologic Changes in Subchondral Bone and Articular Cartilage of Rat Femorotibial Joints: AN Animal Model of Osteoarthritis. Toxicol. Pathol. 2003, 31, 619–624. [Google Scholar] [CrossRef]
- Vaamonde-Garcia, C.; Courties, A.; Pigenet, A.; Laiguillon, M.C.; Sautet, A.; Houard, X.; Kerdine-Römer, S.; Meijide, R.; Berenbaum, F.; Sellam, J. The nuclear factor-erythroid 2-related factor/heme oxygenase-1 axis is critical for the inflammatory features of type 2 diabetes–associated osteoarthritis. J. Biol. Chem. 2017, 292, 14505–14515. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Matsushita, T.; Takayama, K.; Matsumoto, T.; Nishida, K.; Kuroda, R.; Kurosaka, M. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 2014, 73, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed]
- Brines, R.; Maicas, N.; Ferrándiz, M.L.; Loboda, A.; Jozkowicz, A.; Dulak, J.; Alcaraz, M.J. Heme Oxygenase-1 Regulates the Progression of K/BxN Serum Transfer Arthritis. PLoS ONE 2012, 7, e0052435. [Google Scholar] [CrossRef]
- Brines, R.; Catalán, L.; Alcaraz, M.J.; Ferrándiz, M.L. Myeloid heme oxygenase-1 regulates the acute inflammatory response to zymosan in the mouse air pouch. Oxid. Med. Cell. Longev. 2018, 2018, 5053091. [Google Scholar] [CrossRef]
- Hama, M.; Kirino, Y.; Takeno, M.; Takase, K.; Miyazaki, T.; Yoshimi, R.; Ueda, A.; Itoh-Nakadai, A.; Muto, A.; Igarashi, K.; et al. Bach1 regulates osteoclastogenesis in a mouse model via both heme oxygenase 1-dependent and heme oxygenase 1-independent pathways. Arthritis Rheum. 2012, 64, 1518–1528. [Google Scholar] [CrossRef]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M.K. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Takeno, M.; Iwasaki, M.; Kobayashi, H.; Kirino, Y.; Ueda, A.; Nagahama, K.; Aoki, I.; Ishigatsubo, Y. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin. Exp. Immunol. 2004, 138, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Hashiba, T.; Tsuburai, T.; Ikehara, K.; Matsuse, T.; Ishigatsubo, Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum. Gene Ther. 2001, 12, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Jin, S.S.; Lin, Z.; Chen, R.; Pan, T.; Kang, X.; Huang, H.; Lin, C.; Pan, J. Sauchinone inhibits IL-1β induced catabolism and hypertrophy in mouse chondrocytes to attenuate osteoarthritis via Nrf2/HO-1 and NF-κB pathways. Int. Immunopharmacol. 2018, 62, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Qiao, J.; Guan, D.; Chen, T. Anti-Inflammatory Effects of Licochalcone A on IL-1β-Stimulated Human Osteoarthritis Chondrocytes. Inflammation 2017, 40, 1894–1902. [Google Scholar] [CrossRef]
- Yan, Z.; Qi, W.; Zhan, J.; Lin, Z.; Lin, J.; Xue, X.; Pan, X.; Zhou, Y. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J. Cell. Mol. Med. 2020, 24, 13046–13057. [Google Scholar] [CrossRef]
- Li, X.; Lin, J.; Ding, X.; Xuan, J.; Hu, Z.; Wu, D.; Zhu, X.; Feng, Z.; Ni, W.; Wu, A. The protective effect of sinapic acid in osteoarthritis: In vitro and in vivo studies. J. Cell. Mol. Med. 2019, 23, 1940–1950. [Google Scholar] [CrossRef]
- Zheng, G.; Zhan, Y.; Tang, Q.; Chen, T.; Zheng, F.; Wang, H.; Wang, J.; Wu, D.; Li, X.; Zhou, Y.Y.; et al. Monascin inhibits IL-1β induced catabolism in mouse chondrocytes and ameliorates murine osteoarthritis. Food Funct. 2018, 9, 1454–1464. [Google Scholar] [CrossRef]
- Khan, N.M.; Haseeb, A.; Ansari, M.Y.; Devarapalli, P.; Haynie, S.; Haqqi, T.M. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radic. Biol. Med. 2017, 106, 288–301. [Google Scholar] [CrossRef]
- Xue, X.H.; Xue, J.X.; Hu, W.; Shi, F.L.; Yang, Y. Nomilin targets the Keap1-Nrf2 signalling and ameliorates the development of osteoarthritis. J. Cell. Mol. Med. 2020, 24, 8579–8588. [Google Scholar] [CrossRef]
- Tang, Q.; Feng, Z.; Tong, M.; Xu, J.; Zheng, G.; Shen, L.; Shang, P.; Zhang, Y.; Liu, H. Piceatannol inhibits the IL-1β-induced inflammatory response in human osteoarthritic chondrocytes and ameliorates osteoarthritis in mice by activating Nrf2. Food Funct. 2017, 8, 3926–3937. [Google Scholar] [CrossRef]
- Liu, F.C.; Wang, C.C.; Lu, J.W.; Lee, C.H.; Chen, S.C.; Ho, Y.J.; Peng, Y.J. Chondroprotective effects of genistein against osteoarthritis induced joint inflammation. Nutrients 2019, 11, 1180. [Google Scholar] [CrossRef]
- Zhou, S.; Shi, J.; Wen, H.; Xie, W.; Han, X.; Li, H. A chondroprotective effect of moracin on IL-1ß-induced primary rat chondrocytes and an osteoarthritis rat model through Nrf2/HO-1 and NF-κB axes. Food Funct. 2020, 11, 7935–7945. [Google Scholar] [CrossRef]
- Li, Y.N.; Fan, M.L.; Liu, H.Q.; Ma, B.; Dai, W.L.; Yu, B.Y.; Liu, J.H. Dihydroartemisinin derivative DC32 inhibits inflammatory response in osteoarthritic synovium through regulating Nrf2/NF-κB pathway. Int. Immunopharmacol. 2019, 74. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.K.; Jupp, O.; De Ferrars, R.; Kay, C.D.; Culley, K.L.; Norton, R.; Driscoll, C.; Vincent, T.L.; Donell, S.T.; Bao, Y.; et al. Sulforaphane represses matrix-degrading proteases and protects cartilage from destruction in vitro and in vivo. Arthritis Rheum. 2013, 65, 3130–3140. [Google Scholar] [CrossRef]
- Fujita, N.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Takayama, K.; Kurosaka, M.; Kuroda, R. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 2011, 29, 511–515. [Google Scholar] [CrossRef]
- Luo, Z.; Zheng, B.; Jiang, B.; Xue, X.; Xue, E.; Zhou, Y. Peiminine inhibits the IL-1β induced inflammatory response in mouse articular chondrocytes and ameliorates murine osteoarthritis. Food Funct. 2019, 10, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, L.; Zhang, Y.; Cui, L.; Shen, G. Intra-articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF-2α. J. Orthop. Res. 2015, 33, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, W.; Wu, P.; Deng, Z.; Zeng, C.; Li, H.; Yang, T.; Lei, G. The expression of SIRT1 in articular cartilage of patients with knee osteoarthritis and its correlation with disease severity. J. Orthop. Surg. Res. 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Luo, P.; Li, X.; Liu, P.; Li, Y.; Xu, J. Nrf2/ARE is a key pathway for curcumin-mediated protection of TMJ chondrocytes from oxidative stress and inflammation. Cell Stress Chaperones 2020, 25, 395–406. [Google Scholar] [CrossRef]
- Yang, G.; Fan, M.; Zhu, J.; Ling, C.; Wu, L.; Zhang, X.; Zhang, M.; Li, J.; Yao, Q.; Gu, Z.; et al. A multifunctional anti-inflammatory drug that can specifically target activated macrophages, massively deplete intracellular H2O2, and produce large amounts CO for a highly efficient treatment of osteoarthritis. Biomaterials 2020, 255. [Google Scholar] [CrossRef]
- Kyostio-Moore, S.; Bangari, D.S.; Ewing, P.; Nambiar, B.; Berthelette, P.; Sookdeo, C.; Hutto, E.; Moran, N.; Sullivan, J.; Matthews, G.L.; et al. Local gene delivery of heme oxygenase-1 by adeno-associated virus into osteoarthritic mouse joints exhibiting synovial oxidative stress. Osteoarthr. Cartil. 2013, 21, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C.; Altman, R.D.; April, K.T.; Benkhalti, M.; Guyatt, G.; McGowan, J.; Towheed, T.; Welch, V.; Wells, G.; Tugwell, P.; et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res. 2012, 64, 465–474. [Google Scholar] [CrossRef]
- Son, Y.O.; Kim, H.E.; Choi, W.S.; Chun, C.H.; Chun, J.S. RNA-binding protein ZFP36L1 regulates osteoarthritis by modulating members of the heat shock protein 70 family. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Son, Y.O.; Park, S.; Kwak, J.S.; Won, Y.; Choi, W.S.; Rhee, J.; Chun, C.H.; Ryu, J.H.; Kim, D.K.; Choi, H.S.; et al. Estrogen-related receptor γ causes osteoarthritis by upregulating extracellular matrix-degrading enzymes. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Ulrich-Vinther, M. Gene therapy methods in bone and joint disorders. Evaluation of the adeno-associated virus vector in experimental models of articular cartilage disorders, periprosthetic osteolysis and bone healing. Acta Orthop. Suppl. 2007, 78, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.A.; Lee, H.H.; Haleem, A.M.; Martins, C.; Yuan, Z.; Qiao, C.; Xiao, X.; Chu, C.R. Single intra-articular injection of adeno-associated virus results in stable and controllable in vivo transgene expression in normal rat knees. Osteoarthr. Cartil. 2011, 19, 1058–1065. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Gene Delivery to Joints by Intra-Articular Injection. Hum. Gene Ther. 2018, 29, 2–14. [Google Scholar] [CrossRef]
- Lee, C.W.; Chi, M.C.; Hsu, L.F.; Yang, C.M.; Hsu, T.H.; Chuang, C.C.; Lin, W.N.; Chu, P.M.; Lee, I.T. Carbon monoxide releasing molecule-2 protects against particulate matter-induced lung inflammation by inhibiting TLR2 and 4/ROS/NLRP3 inflammasome activation. Mol. Immunol. 2019, 112, 163–174. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M.K. Carbon monoxide has anti-inflammatory effects involving the mitogen- activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Tabe, H.; Shimoi, T.; Boudes, M.; Abe, S.; Coulibaly, F.; Kitagawa, S.; Mori, H.; Ueno, T. Photoactivatable CO release from engineered protein crystals to modulate NF-κB activation. Chem. Commun. 2016, 52, 4545–4548. [Google Scholar] [CrossRef] [PubMed]
- Marker, C.L.; Pomonis, J.D. The monosodium iodoacetate model of osteoarthritis pain in the rat. Methods Mol. Biol. 2012, 851, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar] [PubMed]
- Pan, S.; Li, S.; Hu, Y.; Zhang, H.; Liu, Y.; Jiang, H.; Fang, M.; Li, Z.; Xu, K.; Zhang, H.; et al. Resveratrol post-treatment protects against neonatal brain injury after hypoxia-ischemia. Oncotarget 2016, 7, 79247–79261. [Google Scholar] [CrossRef]
- Hsu, D.Z.; Chu, P.Y.; Jou, I.M. Daily sesame oil supplement attenuates joint pain by inhibiting muscular oxidative stress in osteoarthritis rat model. J. Nutr. Biochem. 2016, 29, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739. [Google Scholar] [CrossRef]
- Deng, Z.; Li, Y.; Liu, H.; Xiao, S.; Li, L.; Tian, J.; Cheng, C.; Zhang, G.; Zhang, F. The role of sirtuin 1 and its activator, resveratrol in osteoarthritis. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Lei, M.; Wang, J.G.; Xiao, D.M.; Fan, M.; Wang, D.P.; Xiong, J.Y.; Chen, Y.; Ding, Y.; Liu, S.L. Resveratrol inhibits interleukin 1β-mediated inducible nitric oxide synthase expression in articular chondrocytes by activating SIRT1 and thereby suppressing nuclear factor-κB activity. Eur. J. Pharmacol. 2012, 674, 73–79. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, Z.; Yan, Z.; Wang, Z.; Fu, X.; Yu, K. Sinomenine contributes to the inhibition of the inflammatory response and the improvement of osteoarthritis in mouse-cartilage cells by acting on the Nrf2/HO-1 and NF-κB signaling pathways. Int. Immunopharmacol. 2019, 75. [Google Scholar] [CrossRef]
- Luo, Z.; Hu, Z.; Bian, Y.; Su, W.; Li, X.; Li, S.; Wu, J.; Shi, L.; Song, Y.; Zheng, G.; et al. Scutellarin Attenuates the IL-1β-Induced Inflammation in Mouse Chondrocytes and Prevents Osteoarthritic Progression. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Sun, K.; Luo, J.; Jing, X.; Xiang, W.; Guo, J.; Yao, X.; Liang, S.; Guo, F.; Xu, T. Hyperoside ameliorates the progression of osteoarthritis: An in vitro and in vivo study. Phytomedicine 2021, 80, 153387. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Panicker, S.; Borgia, J.; Fhied, C.; Mikecz, K.; Oegema, T.R. Oral glucosamine modulates the response of the liver and lymphocytes of the mesenteric lymph nodes in a papain-induced model of joint damage and repair. Osteoarthr. Cartil. 2009, 17, 1014–1021. [Google Scholar] [CrossRef]
- Wang, Y.J.; Shen, M.; Wang, S.; Wen, X.; Han, X.R.; Zhang, Z.F.; Li, H.; Wang, F.; Wu, D.M.; Lu, J.; et al. Inhibition of the TGF-β1/Smad signaling pathway protects against cartilage injury and osteoarthritis in a rat model. Life Sci. 2017, 189, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.L.; Chen, Y.W.; Der Hsiao, L.; Chen, Y.L.; Yang, C.M. Heme oxygenase 1 attenuates interleukin-1β-induced cytosolic phospholipase A2 expression via a decrease in NADPH oxidase/reactive oxygen species/activator protein 1 activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012, 64, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Glehr, M.; Fritsch-Breisach, M.; Lohberger, B.; Walzer, S.M.; Moazedi-Fuerst, F.; Rinner, B.; Gruber, G.; Graninger, W.; Leithner, A.; Windhager, R. Influence of resveratrol on rheumatoid fibroblast-like synoviocytes analysed with gene chip transcription. Phytomedicine 2013, 20, 310–318. [Google Scholar] [CrossRef]
- Jin, S.E.; Ha, H.; Shin, H.K.; Seo, C.S. Anti-allergic and anti-inflammatory effects of Kuwanon G and Morusin on MC/9 mast cells and HaCaT keratinocytes. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Gu, R.; Huang, Z.; Liu, H.; Qing, Q.; Zhuan, Z.; Yang, L.; Su, Z.; Huang, W. Moracin attenuates LPS-induced inflammation in nucleus pulposus cells via Nrf2/HO-1 and NF-κB/TGF-β pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Kershaw, J.; Kim, K.H. The Therapeutic Potential of Piceatannol, a Natural Stilbene, in Metabolic Diseases: A Review. J. Med. Food 2017, 20, 427–438. [Google Scholar] [CrossRef]
- Jin, C.Y.; Moon, D.O.; Lee, K.J.; Kim, M.O.; Lee, J.D.; Choi, Y.H.; Park, Y.M.; Kim, G.Y. Piceatannol attenuates lipopolysaccharide-induced NF-κB activation and NF-κB-related proinflammatory mediators in BV2 microglia. Pharmacol. Res. 2006, 54, 461–467. [Google Scholar] [CrossRef]
- Jeong, S.O.; Son, Y.; Lee, J.H.; Cheong, Y.K.; Park, S.H.; Chung, H.T.; Pae, H.O. Resveratrol analog piceatannol restores the palmitic acid-induced impairment of insulin signaling and production of endothelial nitric oxide via activation of anti-inflammatory and antioxidative heme oxygenase-1 in human endothelial cells. Mol. Med. Rep. 2015, 12, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Malaise, O.; Tachikart, Y.; Constantinides, M.; Mumme, M.; Ferreira-Lopez, R.; Noack, S.; Krettek, C.; Noël, D.; Wang, J.; Jorgensen, C.; et al. Mesenchymal stem cell senescence alleviates their intrinsic and senosuppressive paracrine properties contributing to osteoarthritis development. Aging 2019, 11, 9128–9146. [Google Scholar] [CrossRef] [PubMed]
- Nagira, K.; Ikuta, Y.; Shinohara, M.; Sanada, Y.; Omoto, T.; Kanaya, H.; Nakasa, T.; Ishikawa, M.; Adachi, N.; Miyaki, S.; et al. Histological scoring system for subchondral bone changes in murine models of joint aging and osteoarthritis. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Murakami, S.; Suzuki, T.; Harigae, H.; Romeo, P.-H.; Yamamoto, M.; Motohashi, H. NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.S.; Kappas, A. Chemoprevention of neonatal jaundice: Potency of tin-protoporphyrin in an animal model. Science 1982, 217, 1250–1252. [Google Scholar] [CrossRef] [PubMed]
- Kappas, A. A Method for Interdicting the Development of Severe Jaundice in Newborns by Inhibiting the Production of Bilirubin. Pediatrics 2004, 113, 119–123. [Google Scholar] [CrossRef]


{kind=link}
| Risk Factor | Primary or Secondary | Mechanism |
|---|---|---|
| Aging | Primary | cellular senescence, mitochondrial dysfunction |
| Genetics | Primary | GDF5, DVWA etc, See (review [43]) |
| Obesity | Secondary | mechanical stress, inflammatory mediators |
| Trauma | Secondary | joint instability, pro-inflammation |
| Overuse | Secondary | wear and tear, pro-inflammation |
| Varus and valgus alignment | Secondary | chronic overload, wear and tear |
| C57/B6 Mouse | Method | Time for OA Development | Mechanism | Secondary or Primary | Ref |
|---|---|---|---|---|---|
| C57/B6 mouse | DMM surgery | 8 weeks | Joint instability and inflammation | Secondary OA | [143] |
| C57/B6 mouse | MIA injection | 4~6 weeks | Inflammation and synovitis | Secondary OA | [150] |
| C57/B6 mouse | Papain injection | 4~6 weeks | Inflammation and synovitis | Secondary OA | [150] |
| STR/ORT mouse | Aging | 28 weeks~ | Spontaneous (prone to obesity) | Primary OA | [147,148] |
| SD rat | ACLT surgery | 8 weeks | Joint instability and inflammation | Secondary OA | [151] |
| SD rat | MIA injection | 4~6 weeks | Inflammation and synovitis | Secondary OA | [152,153] |
| Target Gene | Modification | Methods | Target Joint | OA Progression | Mechanisms | Ref |
|---|---|---|---|---|---|---|
| Nrf2 | KO | MIA injecrtion or DMM | Knee joint | Promote | Reduction in HO-1, NQO1 | [11] |
| Bach-1 | KO | Aging or DMM | Knee joint | Prevent | mild induction of HO-1 | [13] |
| Nrf2 | KO | AIA | Knee joint | Promote | Reduction in HO-1, NQO1 | [56,57] |
| Bach-1 | KO | Aging | Meniscus | Prevent | Mild induction of HO-1 | [19] |
| Bach-1 | KO | Aging or puncture | Intervertebral disc | Prevent | Mild induction of HO-1 | [20] |
| Nrf2 | KO | IL-1β stimuli in a high glucose condition | Mouse primary chondrocyte | Higher sensitivity | Reduction in HO-1 | [154] |
| SOD2 | cKO (Col2a1 Cre) | Aging or DMM | Knee joint | Promote | Mitochondrial dysfunction | [12] |
| SIRT1 | cKO (Col2a1 Cre) | Aging or DMM | Knee joint | Promote | Activation of NF-κB signaling | [155] |
| Pharmacological Treatment in Secondary OA Model | ||||||
|---|---|---|---|---|---|---|
| Drug | Experimental Model | Dose and Methods | Nrf2/HO-1 Expression in Cartilage | Detection | Ref. | |
| Oral gavage | Piceatannol | DMM mouse | 10 mg/kg/day, p.o for 8weeks | Nrf2 (immunohisto) in cartilage | MMP13, Col2 (immunohisto) in cartilage | [171] |
| Sauchinone | DMM mouse | 10 mg/kg/day, p.o for 8weeks | - | - | [164] | |
| Myricetin | DMM mouse | 20 mg/kg/2day, p.o for 8 weeks | Nrf2 (immunohisto) in cartilage | p-Akt (immunohisto) in cartilage | [128] | |
| Sinapic acid | DMM (with fat pad resection) mouse | 10 mg/kg/2day, p.o for 8 weeks | HO-1 (qPCR, WB), Nrf2 (WB) in cartilage | MMP13, ADAMTS5 (qPCR) in cartilage | [121] | |
| Licochalcone A (Lico A) | DMM mouse | 10 mg/kg/day, p.o for 8 weeks | Nrf2 (immunohisto) in cartilage | IL-1β, IL18 (ELISA) | [165,166] | |
| Genistein | ACLT Rat | Standard feeding with oral genistein (40 mg/kg) | - | - | [172] | |
| Moracin | ACLT Rat | 30 mg/kg/2day, p.o for 8 weeks | Nrf2 (immunohisto) in cartilage | Col2 (immunohisto) in cartilage | [173] | |
| DC32 [(9α,12α-dihydroartemisinyl) bis(2-chlorocinnmate) | Papain-induced OA mouse | 6.25 mg, 12.5 mg, 25 mg/kg/day, p.o for 4 weeks | - | Col2a1, MMP13 (qPCR), TNF-α (q-PCR, WB) in cartilage | [174] | |
| Sulforaphane | DMM mouse | feeding with AIN-93G containing 0.18 or 0.6 mg/kg | - | Col2, Col10 (immunohisto) in cartilage | [175] | |
| S-allyl cysteine | DMM mouse | 100 mg/kg/day, p.o. for 8 weeks | Nrf2 (immunohisto) in cartilage | p16 (immunohisto) in cartilage | [63] | |
| hesperetin | DMM mouse | 10 mg/kg/day, p.o for 8 weeks | Nrf2 (immunohisto) in cartilage | - | [27] | |
| Sinapic acid | DMM mouse | 20 mg/kg/day, p.o for 14 days | Nrf2 (immunohisto) in cartilage | MMP13, Col2a1 (immunohisto) in cartilage | [167] | |
| Monascin | DMM mouse | 10 mg kg/day, p.o for 8 weeks | Nrf2 (immunohisto) in cartilage | - | [168] | |
| Nomilin | DMM mouse | 20 mg/kg/day, p.o for 8 weeks | Nrf2 (immunohisto) in cartilage | - | [170] | |
| Intrapenetorial injection | Sinomenine | DMM mouse | 10 mg/kg/day, i.p for 2 months after 1 month surgery | - | - | [176] |
| Peiminine | DMM mouse | 5 mg/kg/day, i.p for 8 weeks. | Nrf2 (immunohisto) in cartilage | - | [177] | |
| 7,8-dihydroxyflavone (7,8-DHF) | DMM mouse | 5 mg/kg/week, for 8 weeks | Nrf2, HO-1 (qPCR, WB) in cartilage | MMP1, 3, 13, IL-1β, TNF-α (qPCR) in cartilage | [26] | |
| Hyperoside | DMM mouse | 20 mg/kg/2days, for 4 or 8 weeks | Nrf2 (immunohisto) in cartilage | - | [178] | |
| Scutellarin | DMM mouse | 50 mg/kg/day, i.p for 8 weeks | - | PGE2, Cox2 (qPCR) in cartilage | [179] | |
| Intra-articular injection | Resveratorol | MIA-induced arthritis Rat | 50mg/kg/3days, for 8 weeks | Nrf2/HO-1 (WB) in joint | Cas3/9 (ELISA) in joint | [126] |
| Adenovirus-KLF2 | MIA-nduced OA Rat | 1 × 109 PHUs/10 μL for 3 weeks | Nrf2 (immunohisto) in cartilage | Tunel staining, MMP13 (immunohisto) in cartilage | [94] | |
| Curcumine | Freund’s adjuvant injection TMJ OA Rat | 40 μM/week, for 1 or 4 weeks | Nrf2 (immunohisto) in TMJ | MMP13, 9, IL-1β, iNOS (immunohisto) in TMJ cartilage | [180] | |
| FA-HA modified CORMs | MIA-nduced OA Rat | 1 mg, 1.5 mg, 2.5 mg/mL/4days, for 23 days | - | TNF-α, IL-1β, IL-6 (ELISA) in joint | [181] | |
| Astaxanthin | DMM mouse | 20 mg/kg/2 week, for 8 weeks | Nrf2 (immunohisto) in cartilage | - | [25] | |
| Pharmacological treatment in primary OA model | ||||||
| Drug | Experimental model | Dose and Methods | Nrf2/HO-1 expression in cartilage | Detection | Ref | |
| Intra-articular injection | rAAV/HO-1 (adenovirus) | STR/ORT OA model mouse (13–15 weeks ~25–27 week) | 5 × 1010 drp rAAV (one shot) | HO1 in synovium (immunohisto) | β-gal staining | [182] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanada, Y.; Tan, S.J.O.; Adachi, N.; Miyaki, S. Pharmacological Targeting of Heme Oxygenase-1 in Osteoarthritis. Antioxidants 2021, 10, 419. https://doi.org/10.3390/antiox10030419
Sanada Y, Tan SJO, Adachi N, Miyaki S. Pharmacological Targeting of Heme Oxygenase-1 in Osteoarthritis. Antioxidants. 2021; 10(3):419. https://doi.org/10.3390/antiox10030419
Chicago/Turabian StyleSanada, Yohei, Sho Joseph Ozaki Tan, Nobuo Adachi, and Shigeru Miyaki. 2021. "Pharmacological Targeting of Heme Oxygenase-1 in Osteoarthritis" Antioxidants 10, no. 3: 419. https://doi.org/10.3390/antiox10030419
APA StyleSanada, Y., Tan, S. J. O., Adachi, N., & Miyaki, S. (2021). Pharmacological Targeting of Heme Oxygenase-1 in Osteoarthritis. Antioxidants, 10(3), 419. https://doi.org/10.3390/antiox10030419