
Pituitary Adenylate Cyclase-Activating Polypeptide: A Potent Therapeutic Agent in Oxidative Stress

 , , ,
, , , 
{kind=link}
Abstract
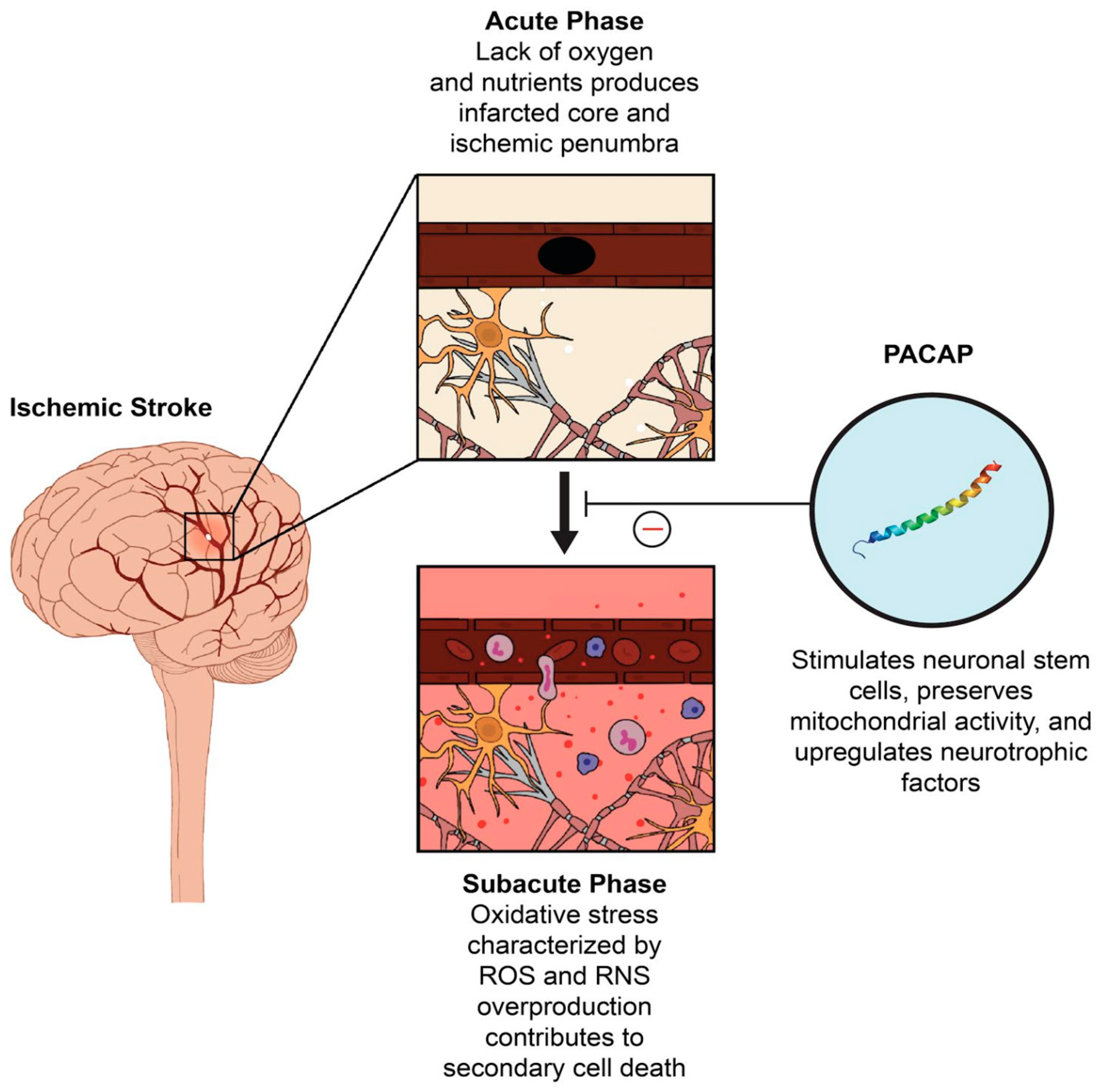
1. Introduction
2. The Role of PACAP in Oxidative Stress
3. PACAP as A Potential Therapeutic Target in Stroke
4. Recent Preclinical Evidence Supporting the Therapeutic Potential of PACAP in Stroke
5. The Therapeutic Role of PACAP in Non-Stroke Nervous System Disorders
5.1. Parkinson’s Disease (PD)
5.2. Alzheimer’s Disease (AD)
5.3. Amyotrophic Lateral Sclerosis (ALS)
5.4. Migraine
5.5. Neuropsychiatric Disorders
6. PACAP’s Limitations: Barriers to Clinical Use
6.1. Potential Side Effects
6.2. Pharmacokinetic Limitations
6.3. Administration Route Problems
7. Clinical Trials Supporting Efficacy of PACAP-related Molecules in Stroke and Neurodegenerative Diseases
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tan, Y.V.; Abad, C.; Lopez, R.; Dong, H.; Liu, S.; Lee, A.; Gomariz, R.; Lecata, J.; Waschek, J. Pituitary adenylyl cyclase-activating polypeptide is an intrinsic regulator of Treg abundance and protects against experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2009, 106, 2012–2017. [Google Scholar] [CrossRef]
- Harch, P.; Andrews, S.; Fogarty, E.; Lucarini, J.; Van, M.K. Case control study: Hyperbaric oxygen treatment of mild traumatic brain injury persistent post-concussion syndrome and post-traumatic stress disorder. Med. Gas. Res. 2017, 7, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.G.; Lin, G.A.; Xie, H.Q.; Pan, H.T.; Huang, B.Q.; Liu, J.D.; Liu, H.; Zhang, N.; Li, L.; et al. The effects of different hyperbaric oxygen manipulations in rats after traumatic brain injury. Neurosci. Lett. 2014, 563, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Miljkovic-Lolic, M.; Silbergleit, R.; Fiskum, G.; Rosenthal, R.E. Neuroprotective effects of hyperbaric oxygen treatment in experimental focal cerebral ischemia are associated with reduced brain leukocyte myeloperoxidase activity. Brain Res. 2003, 971, 90–94. [Google Scholar] [CrossRef]
- Calvert, J.; Zhou, C.; Nanda, A.; Zhang, J. Effect of hyperbaric oxygen on apoptosis in neonatal hypoxia-ischemia rat model. J. Appl. Physiol. 2003, 95. [Google Scholar] [CrossRef]
- Yang, J.L.; Mukda, S.; Chen, S.D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 3–4. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, A.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gomez Izquierdo, L.; Cotan, D.; Navas, P.; Sanchez-Alcazar, J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Chen, Y.; Samal, B.; Hamelink, C.R.; Xiang, C.C.; Chen, Y.; Chen, M.; Vaudry, D.; Brownstein, M.J.; Hallenbeck, J.M.; Eiden, L.E. Neuroprotection by endogenous and exogenous PACAP following stroke. Regul. Pept. 2006, 137, 4–19. [Google Scholar] [CrossRef][Green Version]
- Fang, Y.; Ren, R.; Shi, H.; Huang, L.; Lenahan, C.; Lu, Q.; Tang, L.; Huang, Y.; Tang, J.; Zhang, J.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide: A Promising Neuroprotective Peptide in Stroke. Aging Dis. 2020, 11, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Nogi, H.; Mori, K.; Ohishi, H.; Shigemoto, R.; Yamamoto, K.; Matsuda, T.; Mizuno, N.; Nagata, S.; Baba, A. Distribution of the mRNA for a pituitary adenylate cyclase-activating polypeptide receptor in the rat brain: An in situ hybridization study. J. Comp. Neurol. 1996, 371, 567–577. [Google Scholar] [CrossRef]
- Eftekhari, S.; Salvatore, C.A.; Johansson, S.; Chen, T.B.; Zeng, Z.; Edvinsson, L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res. 2015, 1600, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Miyata, A.; Arimura, A.; Dahl, R.R.; Minamino, N.; Uehara, A.; Jiang, L.; Culler, M.D.; Coy, D.H. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Commun. 1989, 164, 567–574. [Google Scholar] [CrossRef]
- Kimura, C.; Ohkubo, S.; Ogi, K.; Hosoya, M.; Itoh, Y.; Onda, H.; Miyata, A.; Jiang, L.; Dahl, R.; Stibbs, H.; et al. A novel peptide which stimulates adenylate cyclase: Molecular cloning and characterization of the ovine and human cDNAs. Biochem. Biophys. Res. Commun. 1990, 166, 81–89. [Google Scholar] [CrossRef]
- Arimura, A.; Somogyvari-Vigh, A.; Miyata, A.; Mizuno, K.; Coy, D.; Kitada, C. Tissue Distribution of PACAP as Determined by RIA: Highly Abundant in the Rat Brain and Testes. Endocrinology 1991, 129, 2787–2789. [Google Scholar] [CrossRef]
- Ohtaki, H.; Nakamachi, T.; Dohi, K.; Aizawa, Y.; Takaki, A.; Hodoyama, K.; Yofu, S.; Hashimoto, H.; Shintani, N.; Baba, A.; et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) decreases ischemic neuronal cell death in association with IL-6. Proc. Natl. Acad. Sci. USA 2006, 103, 7488–7493. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Samal, B.; Gerdin, M.J.; Huddleston, D.; Hsu, C.M.; Elkahloun, A.G.; Stroth, N.; Hamelink, C.; Lee, E. Meta-analysis of microarray-derived data from PACAP-deficient adrenal gland in vivo and PACAP-treated chromaffin cells identifies distinct classes of PACAP-regulated genes. Peptides 2007, 28, 1871–1882. [Google Scholar] [CrossRef][Green Version]
- Hawke, Z.; Ivanov, T.R.; Bechtold, D.A.; Dhillon, H.; Lowell, B.B.; Luckman, S.M. PACAP neurons in the hypothalamic ventromedial nucleus are targets of central leptin signaling. J. Neurosci. 2009, 29, 14828–14835. [Google Scholar] [CrossRef]
- Falluel-Morel, A.; Vaudry, D.; Aubert, N.; Galas, L.; Benard, M.; Basille, M.; Fontaine, M.; Fournier, A.; Vaudry, H.; Gonzalez, B. Pituitary adenylate cyclase-activating polypeptide prevents the effects of ceramides on migration, neurite outgrowth, and cytoskeleton remodeling. Proc. Natl. Acad. Sci. USA 2005, 102, 2637–2642. [Google Scholar] [CrossRef]
- Reglodi, D.; Vaczy, A.; Rubio-Beltran, E.; MaassenVanDenBrink, A. Protective effects of PACAP in ischemia. J. Headache Pain 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; de Molliens, M.P.; Schneebeli, S.T.; Brewer, M.; Song, G.; Chatenet, D.; Brass, K.M.; May, V.; Li, J. Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr. Top. Med. Chem. 2019, 19, 1399–1417. [Google Scholar] [CrossRef]
- Rudecki, A.P.; Gray, S.L. PACAP in the Defense of Energy Homeostasis. Trends Endocrinol. Metab. 2016, 27, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Ohtaki, H.; Satoh, A.; Nakamachi, T.; Yofu, S.; Dohi, K.; Mori, H.; Ohara, K.; Miyamoto, K.; Hashimoto, H.; Shintani, N.; et al. Regulation of oxidative stress by pituitary adenylate cyclase-activating polypeptide (PACAP) mediated by PACAP receptor. J. Mol. Neurosci. 2010, 42, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Hamelink, C.; Damadzic, R.; Eskay, R.L.; Gonzalez, B.; Eiden, L.E. Endogenous PACAP acts as a stress response peptide to protect cerebellar neurons from ethanol or oxidative insult. Peptides 2005, 26, 2518–2524. [Google Scholar] [CrossRef][Green Version]
- Kasica, N.; Podlasz, P.; Sundvik, M.; Tamas, A.; Reglodi, D.; Kaleczyc, J. Protective Effects of Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Against Oxidative Stress in Zebrafish Hair Cells. Neurotox Res. 2016, 30, 633–647. [Google Scholar] [CrossRef]
- Vaudry, D.; Pamantung, T.F.; Basille, M.; Rousselle, C.; Fournier, A.; Vaudry, H.; Beauvillain, J.C.; Gonzalez, B.J. PACAP protects cerebellar granule neurons against oxidative stress-induced apoptosis. Eur. J. Neurosci. 2002, 15, 1451–1460. [Google Scholar] [CrossRef]
- Douiri, S.; Bahdoudi, S.; Hamdi, Y.; Cubì, R.; Basille, M.; Fournier, A.; Vaudry, H.; Tonon, M.C.; Amri, M.; Vaudry, D.; et al. Involvement of endogenous antioxidant systems in the protective activity of pituitary adenylate cyclase-activating polypeptide against hydrogen peroxide-induced oxidative damages in cultured rat astrocytes. J. Neurochem. 2016, 137, 913–930. [Google Scholar] [CrossRef]
- Tanaka, J.; Koshimura, K.; Murakami, Y.; Kato, Y. Stimulatory effect of PACAP on neuronal cell survival. Ann. N. Y. Acad. Sci. 1996, 805, 473–475. [Google Scholar] [CrossRef]
- Lioudyno, M.; Skoglosa, Y.; Takei, N.; Lindholm, D. Pituitary adenylate cyclase-activating polypeptide (PACAP) protects dorsal root ganglion neurons from death and induces calcitonin gene-related peptide (CGRP) immunoreactivity in vitro. J. Neurosci. Res. 1998, 51, 243–256. [Google Scholar] [CrossRef]
- Morio, H.; Tatsuno, I.; Hirai, A.; Tamura, Y.; Saito, Y. Pituitary adenylate cyclase-activating polypeptide protects rat-cultured cortical neurons from glutamate-induced cytotoxicity. Brain Res. 1996, 741, 82–88. [Google Scholar] [CrossRef]
- Kong, L.Y.; Maderdrut, J.L.; Jeohn, G.H.; Hong, J.S. Reduction of lipopolysaccharide-induced neurotoxicity in mixed cortical neuron/glia cultures by femtomolar concentrations of pituitary adenylate cyclase-activating polypeptide. Neuroscience 1999, 91, 493–500. [Google Scholar] [CrossRef]
- Reglodi, D.; Somogyvari-Vigh, A.; Vigh, S.; Kozicz, T.; Arimura, A. Delayed systemic administration of PACAP38 is neuroprotective in transient middle cerebral artery occlusion in the rat. Stroke 2000, 31, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Arimura, A. Perspectives on pituitary adenylate cyclase activating polypeptide (PACAP) in the neuroendocrine, endocrine, and nervous systems. Jpn. J. Physiol. 1998, 48, 301–331. [Google Scholar] [CrossRef] [PubMed]
- Tatsuno, I.; Morio, H.; Tanaka, T.; Uchida, D.; Hirai, A.; Tamura, Y.; Saito, Y. Pituitary adenylate cyclase-activating polypeptide (PACAP) is a regulator of astrocytes: PACAP stimulates proliferation and production of interleukin 6 (IL-6), but not nerve growth factor (NGF), in cultured rat astrocyte. Ann. N. Y. Acad. Sci. 1996, 805, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Gottschall, P.E.; Tatsuno, I.; Arimura, A. Regulation of interleukin-6 (IL-6) secretion in primary cultured rat astrocytes: Synergism of interleukin-1 (IL-1) and pituitary adenylate cyclase activating polypeptide (PACAP). Brain Res. 1994, 637, 197–203. [Google Scholar] [CrossRef]
- Fulda, S.; Lorenzo, G.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Tuazon, J.P.; Ji, X.; Borlongan, C.V. Pituitary Adenylate Cyclase Activating Polypeptide Elicits Neuroprotection Against Acute Ischemic Neuronal Cell Death Associated with NMDA Receptors. Cell Physiol. Biochem. 2018, 51, 1982–1995. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, C.F.; Morales, M.; Chiang, Y.H.; Hoffer, J. Protective effects of glial cell line-derived neurotrophic factor in ischemic brain injury. Ann. N. Y. Acad. Sci. 2002, 962, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, C.Z.; Anastasio, N.C.; Johnson, K.M. Brain-derived Neurotrophic Factor Prevents Phencyclidine-induced Apoptosis in Developing Brain by Parallel Activation of both the ERK and PI-3K/Akt Pathways. Neuropharmacology 2010, 58, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 2001, 3. [Google Scholar] [CrossRef]
- Kaneko, Y.; Pappas, C.; Taijri, N.; Borlongan, C.V. Oxytocin modulates GABA A R subunits to confer neuroprotection in stroke in vitro. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Nakamachi, T.; Watanabe, J.; Sugiyama, K.; Ohtaki, H.; Murai, N.; Sasaki, S.; Xu, Z.; Hashimoto, H.; Seki, T.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Is Involved in Adult Mouse Hippocampal Neurogenesis After Stroke. J. Mol. Neurosci. 2016, 59, 270–279. [Google Scholar] [CrossRef]
- Dejda, A.; Seaborn, T.; Bourgault, S.; Touzani, O.; Fournier, A.; Vaudry, H.; Vaudry, D. PACAP and a novel stable analog protect rat brain from ischemia: Insight into the mechanisms of action. Peptides 2011, 32, 1207–1216. [Google Scholar] [CrossRef]
- Brifault, C.; Gras, M.; Liot, D.; May, V.; Vaudry, D.; Wurtz, O. Delayed pituitary adenylate cyclase-activating polypeptide delivery after brain stroke improves functional recovery by inducing m2 microglia/macrophage polarization. Stroke 2015, 46, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Lazarovici, P.; Cohen, G.; Arien-Zakay, H.; Chen, J.; Zhang, C.; Chopp, M.; Jiang, H. Multimodal neuroprotection induced by PACAP38 in oxygen-glucose deprivation and middle cerebral artery occlusion stroke models. J. Mol. Neurosci. 2012, 48, 526–540. [Google Scholar] [CrossRef]
- Zaccaro, M.C.; Ivanisevic, L.; Perez, P.; Meakin, S.O.; Saragovi, H.U. p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J. Biol. Chem. 2001, 276, 31023–31029. [Google Scholar] [CrossRef]
- Schomerus, C.; Maronde, E.; Laedtke, E.; Korf, H.W. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) induce phosphorylation of the transcription factor CREB in subpopulations of rat pinealocytes: Immunocytochemical and immunochemical evidence. Cell Tissue Res. 1996, 286, 305–313. [Google Scholar]
- Vaudry, D.; Gonzalezm, B.J.; Basille, M.; Anouar, Y.; Fournier, A.; Vaudry, H. Pituitary adenylate cyclase-activating polypeptide stimulates both c-fos gene expression and cell survival in rat cerebellar granule neurons through activation of the protein kinase A pathway. Neuroscience 1998, 84, 801–812. [Google Scholar] [CrossRef]
- Tanaka, J.; Koshimura, K.; Murakami, Y.; Sohmiya, M.; Yanaihara, N.; Kato, Y. Neuronal protection from apoptosis by pituitary adenylate cyclase-activating polypeptide. Regul. Pept. 1997, 72, 1–8. [Google Scholar] [CrossRef]
- Jin, K.; Mao, X.O.; Simon, R.P.; Greenberg, D.A. Cyclic AMP response element binding protein (CREB) and CREB binding protein (CBP) in global cerebral ischemia. J. Mol. Neurosci. 2001, 16, 49–56. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Stumm, R.; Kolodziej, A.; Prinz, V.; Endres, M.; Wu, D.F.; Hollt, V. Pituitary adenylate cyclase-activating polypeptide is up-regulated in cortical pyramidal cells after focal ischemia and protects neurons from mild hypoxic/ischemic damage. J. Neurochem. 2007, 103, 1666–1681. [Google Scholar] [CrossRef]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Muller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473–480. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Balel, C.; Wang, M.; Jia, N.; Zhang, W.; et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 2010, 140, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.P.; Swan, J.H.; Griffiths, T.; Meldrum, B.S. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 1984, 226, 850–852. [Google Scholar] [CrossRef]
- Kortesi, T.; Tuka, B.; Tajti, J.; Bagoly, T.; Fulop, F.; Helyes, Z.; Vecsei, L. Kynurenic Acid Inhibits the Electrical Stimulation Induced Elevated Pituitary Adenylate Cyclase-Activating Polypeptide Expression in the TNC. Front. Neurol. 2018, 8, 745. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Okazawa, M.; Nakanishi, S. Gene regulation via excitation and BDNF is mediated by induction and phosphorylation of the Etv1 transcription factor in cerebellar granule cells. Proc. Natl. Acad. Sci. USA 2012, 109, 8734–8739. [Google Scholar] [CrossRef]
- Hansen, H.H.; Briem, T.; Dzietko, M.; Sifringer, M.; Voss, A.; Rzeski, W.; Zdzisinska, B.; Thor, F.; Heumann, R.; Stepulak, A.; et al. Mechanisms leading to disseminated apoptosis following NMDA receptor blockade in the developing rat brain. Neurobiol. Dis. 2004, 16, 440–453. [Google Scholar] [CrossRef]
- Botanas, C.J.; de la Peña, J.B.; Dela Peña, I.J.; Reinholdgher, T.; Yoon, R.; Kim, H.J.; Lee, Y.S.; Jang, C.G.; Cheong, J.H. Methoxetamine, a ketamine derivative, produced conditioned place preference and was self-administered by rats: Evidence of its abuse potential. Pharmacol. Biochem. Behav. 2015, 133, 31–36. [Google Scholar] [CrossRef]
- de la Peña, J.B.; dela Peña, I.J.; Lee, H.L.; dela Peña, I.; Shin, Y.C.; Sohn, R.A.; Cheong, H.J. Pre-exposure to ethanol, but not to caffeine and nicotine, induced place preference and self-administration of the NMDA receptor antagonist-benzodiazepine combination, Zoletil(R). Pharmacol. Biochem. Behav. 2013, 110, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.K.; Boyadjieva, N.I.; Chen, C.P.; Ortiguela, M.; Reuhl, K.; Clement, E.M.; Kuhn, P.; Marano, J. Cyclic adenosine monophosphate differentiated beta-endorphin neurons promote immune function and prevent prostate cancer growth. Proc. Natl. Acad. Sci. USA 2008, 105, 9105–9110. [Google Scholar] [CrossRef]
- Waschek, J.A.; Cohen, J.R.; Chi, G.C.; Proszynski, T.J.; Niewiadomski, P. PACAP Promotes Matrix-Driven Adhesion of Cultured Adult Murine Neural Progenitors. ASN Neuro 2017, 9, 1759091417708720. [Google Scholar] [CrossRef] [PubMed]
- Ressler, K.J.; Mercer, K.B.; Bradley, B.; Jovanovic, T.; Mahan, A.; Kerley, K.; Norrholm, S.D.; Kilaru, V.; Smith, A.K.; Myers, A.J.; et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 2011, 470, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Polanco, M.J.; Parodi, S.; Piol, D.; Stack, C.; Chivet, M.; Contestabile, A.; Miranda, H.C.; Lievens, P.M.; Espinoza, S.; Jochum, T.; et al. Adenylyl cyclase activating polypeptide reduces phosphorylation and toxicity of the polyglutamine-expanded androgen receptor in spinobulbar muscular atrophy. Sci. Transl. Med. 2016, 8, 370ra181. [Google Scholar] [CrossRef] [PubMed]
- Lamine, A.; Poujol de Molliens, M.; Letourneau, M.; Hebert, T.E.; Vaudry, D.; Fournier, A.; Chatenet, D. The amidated PACAP1-23 fragment is a potent reduced-size neuroprotective agent. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129410. [Google Scholar] [CrossRef]
- Nonaka, N.; Banks, W.A.; Shioda, S. Pituitary adenylate cyclase-activating polypeptide: Protective effects in stroke and dementia. Peptides 2020, 130, 170332. [Google Scholar] [CrossRef]
- Zhang, Y.; Malmberg, A.B.; Yaksh, T.L.; Sjölund, B.; Sundler, F.; Håkanson, R. Capsaicin-evoked release of pituitary adenylate cyclase activating peptide (PACAP) and calcitonin gene-related peptide (CGRP) from rat spinal cord in vivo. Regul. Pept. 1997, 69, 83–87. [Google Scholar] [CrossRef]
- Nakamachi, T.; Ohtaki, H.; Yofu, S.; Dohi, K.; Watanabe, J.; Hayashi, D.; Matsuno, R.; Nonaka, N.; Itabashi, K.; Shioda, S. Pituitary adenylate cyclase-activating polypeptide (PACAP) type 1 receptor (PAC1R) co-localizes with activity-dependent neuroprotective protein (ADNP) in the mouse brains. Regul. Pept. 2008, 145, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Hata, R.; Hossmann, K.-A. Delayed up-regulation of Zac1 and PACAP type I receptor after transient focal cerebral ischemia in mice. Brain Res. Mol. Brain Res. 1998, 61, 207–210. [Google Scholar] [CrossRef]
- Dejda, A.; Sokołowska, P.; Nowak, J.Z. Neuroprotective potential of three neuropeptides PACAP, VIP and PHI. Pharmacol. Rep. 2005, 57, 307–320. [Google Scholar]
- Mao, S.S.; Hua, R.; Zhao, X.P.; Qin, X.; Sun, Z.Q.; Zhang, Y.; Wu, Y.Q.; Jia, M.X.; Cao, J.L.; Zhang, Y.M. Exogenous administration of PACAP alleviates traumatic brain injury in rats through a mechanism involving the TLR4/MyD88/NF-κB pathway. J. Neurotrauma 2012, 29, 1941–1959. [Google Scholar] [CrossRef] [PubMed]
- Staines, D.R.; Brenu, E.W.; Marshall-Gradisnik, S. Postulated vasoactive neuropeptide immunopathology affecting the blood-brain/blood-spinal barrier in certain neuropsychiatric fatigue-related conditions: A role for phosphodiesterase inhibitors in treatment? Neuropsychiatr. Dis. Treat. 2009, 5, 81–89. [Google Scholar] [PubMed]
- Fang, Y.; Shi, H.; Ren, R.; Huang, L.; Okada, T.; Lenahan, C.; Gamdzyk, M.; Travis, Z.D.; Lu, Q.; Tang, L.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide Attenuates Brain Edema by Protecting Blood-Brain Barrier and Glymphatic System After Subarachnoid Hemorrhage in Rats. Neurotherapeutics 2020. [Google Scholar] [CrossRef]
- Karatas, H.; Yemisci, M.; Eren-Kocak, E.; Dalkara, T. Brain Peptides for the Treatment of Neuropsychiatric Disorders. Curr. Pharm. Des. 2018, 24, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Ladjimi, M.H.; Barbouche, R.; Ben Barka, Z.; Vaudry, D.; Lefranc, B.; Leprince, J.; Troadec, J.D.; Ben Rhouma, K.; Sakly, M.; Tebourbi, O.; et al. Comparison of the effects of PACAP-38 and its analog, acetyl-[Ala, Ala] PACAP-38-propylamide, on spatial memory, post-learning BDNF expression and oxidative stress in rat. Behav. Brain Res. 2019, 359, 247–257. [Google Scholar] [CrossRef]
- Huang, Y.J.; Huang, T.H.; Yadav, V.K.; Sumitra, M.R.; Tzeng, D.T.; Wei, P.L.; Shih, J.W.; Wu, A.T. Preclinical investigation of ovatodiolide as a potential inhibitor of colon cancer stem cells via downregulating sphere-derived exosomal beta-catenin/STAT3/miR-1246 cargoes. Am. J. Cancer Res. 2020, 10, 2337–2354. [Google Scholar]
- Abid, H.; Cartier, D.; Hamieh, A.; Francois-Bellan, A.M.; Bucharles, C.; Pothion, H.; Manecka, D.L.; Leprince, J.; Adriouch, S.; Boyer, O.; et al. AMPK Activation of PGC-1alpha/NRF-1-Dependent SELENOT Gene Transcription Promotes PACAP-Induced Neuroendocrine Cell Differentiation Through Tolerance to Oxidative Stress. Mol. Neurobiol. 2019, 56, 4086–4101. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Musumeci, G.; Reglodi, D.; D’Agata, V. Effects of Pacap on Schwann Cells: Focus on Nerve Injury. Int. J. Mol. Sci. 2020, 21, 8233. [Google Scholar] [CrossRef] [PubMed]
- Shioda, S.; Nakamachi, T. PACAP as a neuroprotective factor in ischemic neuronal injuries. Peptides 2015, 72, 202–207. [Google Scholar] [CrossRef]
- Delgado, M.; Abad, C.; Martinez, C.; Juarranz, M.G.; Leceta, J.; Ganea, D.; Gomariz, R.P. PACAP in immunity and inflammation. Ann. N. Y. Acad. Sci. 2003, 992, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Nakamachi, T.; Farkas, J.; Watanabe, J.; Ohtaki, H.; Dohi, K.; Arata, S.; Shioda, S. Role of PACAP in neural stem/progenitor cell and astrocyte—From neural development to neural repair. Curr. Pharm. Des. 2011, 17, 973–984. [Google Scholar] [CrossRef]
- Woodley, P.K.; Min, Q.; Li, Y.; Mulvey, N.F.; Parkinson, D.B.; Dun, X.P. Distinct VIP and PACAP Functions in the Distal Nerve Stump During Peripheral Nerve Regeneration. Front. Neurosci. 2019, 13, 1326. [Google Scholar] [CrossRef] [PubMed]
- Maurel, P.; Salzer, J.L. Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J. Neurosci. 2000, 20, 4635–4645. [Google Scholar] [CrossRef]
- Harrisingh, M.C.; Perez-Nadales, E.; Parkinson, D.B.; Malcolm, D.S.; Mudge, A.W.; Lloyd, A.C. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflamm. 2011, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Poujol de Molliens, M.; Jamadagni, P.; Letourneau, M.; Devost, D.; Hebert, T.E.; Patten, S.A.; Fournier, A.; Chatenet, D. Design and biological assessment of membrane-tethering neuroprotective peptides derived from the pituitary adenylate cyclase-activating polypeptide type 1 receptor. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129398. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Li, J.; Lin, Z.; Ouyang, Z.; Huang, X.; Reglodi, D.; Vaudry, D. TAT-tagging of VIP exerts positive allosteric modulation of the PAC1 receptor and enhances VIP neuroprotective effect in the MPTP mouse model of Parkinson’s disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129626. [Google Scholar] [CrossRef]
- Hajji, K.; Mteyrek, A.; Sun, J.; Cassar, M.; Mezghani, S.; Leprince, J.; Vaudry, D.; Masmoudi-Kouki, O.; Birman, S. Neuroprotective effects of PACAP against paraquat-induced oxidative stress in the Drosophila central nervous system. Hum. Mol. Genet. 2019, 28. [Google Scholar] [CrossRef]
- Ye, D.; Shi, Y.; Xu, Y.; Huang, J. PACAP Attenuates Optic Nerve Crush-Induced Retinal Ganglion Cell Apoptosis Via Activation of the CREB-Bcl-2 Pathway. J. Mol. Neurosci. 2019, 68, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Du, Y.F.; Chen, L. Neuropeptides Exert Neuroprotective Effects in Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 11, 493. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Maugeri, G.; Saccone, S.; Federico, C.; Cavallaro, S.; Reglodi, D.; D’Agata, V. PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2943. [Google Scholar] [CrossRef]
- Bonaventura, G.; Iemmolo, R.; D’Amico, A.G.; La Cognata, V.; Costanzo, E.; Zappia, M.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. PACAP and PAC1R are differentially expressed in motor cortex of amyotrophic lateral sclerosis patients and support survival of iPSC-derived motor neurons. J. Cell Physiol. 2018, 233, 3343–3351. [Google Scholar] [CrossRef]
- Nonaka, N.; Banks, W.A.; Mizushima, H.; Shioda, S.; Morley, J.E. Regional differences in PACAP transport across the blood-brain barrier in mice: A possible influence of strain, amyloid beta protein, and age. Peptides 2002, 23, 2197–2202. [Google Scholar] [CrossRef]
- Xu, X.; Liu, L.; Zhao, L.; Li, B.; Jing, X.; Qu, Z.; Zhu, Y.; Zhang, Y.; Li, Z.; Fisher, M.; et al. Effect of Electroacupuncture on Hyperalgesia and Vasoactive Neurotransmitters in a Rat Model of Conscious Recurrent Migraine. Evid. Based Complement. Alternat. Med. 2019, 2019, 9512875. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, X.; Wu, H.; Zhang, M.; Hu, G.; Dong, Z.; Yu, S. Dynamic changes in CGRP, PACAP, and PACAP receptors in the trigeminovascular system of a novel repetitive electrical stimulation rat model: Relevant to migraine. Mol. Pain 2019, 15, 1744806918820452. [Google Scholar] [CrossRef]
- Hanci, F.; Kilinc, Y.B.; Kilinc, E.; Turay, S.; Dilek, M.; Kabakus, N. Plasma levels of vasoactive neuropeptides in pediatric patients with migraine during attack and attack-free periods. Cephalalgia 2020, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Lutfy, K.; Shankar, G. Emerging evidence for the role of pituitary adenylate cyclase-activating peptide in neuropsychiatric disorders. Prog. Mol. Biol. Transl. Sci. 2019, 167, 143–157. [Google Scholar]
- Hammack, S.E.; Cheung, J.; Rhodes, K.M.; Schutz, K.C.; Falls, W.A.; Braas, K.M.; May, V. Chronic stress increases pituitary adenylate cyclase-activating peptide (PACAP) and brain-derived neurotrophic factor (BDNF) mRNA expression in the bed nucleus of the stria terminalis (BNST): Roles for PACAP in anxiety-like behavior. Psychoneuroendocrinology 2009, 34, 833–843. [Google Scholar] [CrossRef]
- Roman, C.W.; Lezak, K.R.; Hartsock, M.J.; Falls, W.A.; Braas, K.M.; Howard, A.B.; Hammack, S.E.; May, V. PAC1 receptor antagonism in the bed nucleus of the stria terminalis (BNST) attenuates the endocrine and behavioral consequences of chronic stress. Psychoneuroendocrinology 2014, 47, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Dias, B.G.; Ressler, K.J. PACAP and the PAC1 receptor in post-traumatic stress disorder. Neuropsychopharmacology 2013, 38, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Missig, G.; Roman, C.W.; Vizzard, M.A.; Braas, K.M.; Hammack, S.E.; May, V. Parabrachial nucleus (PBn) pituitary adenylate cyclase activating polypeptide (PACAP) signaling in the amygdala: Implication for the sensory and behavioral effects of pain. Neuropharmacology 2014, 86, 38–48. [Google Scholar] [CrossRef]
- Missig, G.; Mei, L.; Vizzard, M.A.; Braas, K.M.; Waschek, J.A.; Ressler, K.J.; Hammack, S.E.; May, V. Parabrachial pituitary adenylate cyclase-activating polypeptide activation of amygdala endosomal extracellular signal-regulated kinase signaling regulates the emotional component of pain. Biol. Psychiatry 2017, 81, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Lamine, A.; Létourneau, M.; Doan, N.D.; Maucotel, J.; Couvineau, A.; Vaudry, H.; Chatenet, D.; Vaudry, D.; Fournier, A. Characterizations of a synthetic pituitary adenylate cyclase-activating polypeptide analog displaying potent neuroprotective activity and reduced in vivo cardiovascular side effects in a Parkinson’s disease model. Neuropharmacology 2016, 108, 440–450. [Google Scholar] [CrossRef]
- Sarszegi, Z.; Szabo, D.; Gaszner, B.; Konyi, A.; Reglodi, D.; Nemeth, J.; Lelesz, B.; Polgar, B.; Jungling, A.; Tamas, A. Examination of Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) as a Potential Biomarker in Heart Failure Patients. J. Mol. Neurosci. 2019, 68, 368–376. [Google Scholar] [CrossRef]
- Farnham, M.M.; Inglott, M.A.; Pilowsky, P.M. Intrathecal PACAP-38 causes increases in sympathetic nerve activity and heart rate but not blood pressure in the spontaneously hypertensive rat. Am. J. Physiol Heart Circ. Physiol. 2011, 200, H214–H222. [Google Scholar] [CrossRef]
- Ghanizada, H.; Al-Karagholi, M.A.; Arngrim, N.; Ghanizada, M.; Larsson, H.B.W.; Amin, F.M.; Ashina, M. Effect of pituitary adenylate cyclase-activating polypeptide-27 on cerebral hemodynamics in healthy volunteers: A 3T MRI study. Peptides 2019, 121, 170134. [Google Scholar] [CrossRef]
- Syed, A.U.; Koide, M.; Braas, K.M.; May, V.; Wellman, G.C. Pituitary adenylate cyclase-activating polypeptide (PACAP) potently dilates middle meningeal arteries: Implications for migraine. J. Mol. Neurosci. 2012, 48, 574–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seiglie, M.P.; Smith, K.L.; Blasio, A.; Cottone, P.; Sabino, V. Pituitary adenylate cyclase-activating polypeptide induces a depressive-like phenotype in rats. Psychopharmacology 2015, 232, 3821–3831. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Chatenet, D.; Wurtz, O.; Doan, N.D.; Leprince, J.; Vaudry, H.; Fournier, A.; Vaudry, D. Strategies to convert PACAP from a hypophysiotropic neurohormone into a neuroprotective drug. Curr Pharm. Des. 2011, 17, 1002–1024. [Google Scholar] [CrossRef]
- Bourgault, S.; Vaudry, D.; Botia, B.; Couvineau, A.; Laburthe, M.; Vaudry, H.; Fournier, A. Novel stable PACAP analogs with potent activity towards the PAC1 receptor. Peptides 2008, 29, 919–932. [Google Scholar] [CrossRef]
- Igarashi, H.; Ito, T.; Mantey, S.A.; Pradhan, T.K.; Hou, W.; Coy, D.H.; Jensen, R.T. Development of simplified vasoactive intestinal peptide analogs with receptor selectivity and stability for human vasoactive intestinal peptide/pituitary adenylate cyclase-activating polypeptide receptors. J. Pharmacol. Exp. Ther. 2005, 315, 370–381. [Google Scholar] [CrossRef]
- Reglodi, D.; Atlasz, T.; Jungling, A.; Szabo, E.; Kovari, P.; Manavalan, S.; Tamas, A. Alternative Routes of Administration of the Neuroprotective Pituitary Adenylate Cyclase Activating Polypeptide. Curr. Pharm. Des. 2018, 24, 3892–3904. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug delivery to the brain. J. Cereb. Blood Flow Metab. 1997, 17, 713–731. [Google Scholar] [CrossRef]
- Pericles, C.; Banks, W.A.; Begley, D.; Scarpa, M.; Dickson, P. Intrathecal delivery of protein therapeutics to the brain: A critical reassessment. Pharmacol. Ther. 2014, 144, 114–122. [Google Scholar] [CrossRef]
- Meredith, M.E.; Salameh, T.S.; Banks, W.A. Intranasal Delivery of Proteins and Peptides in the Treatment of Neurodegenerative Diseases. AAPS J. 2015, 17, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 2012, 9, 566–582. [Google Scholar] [CrossRef]
- Merkus, F.W.; Verhoef, J.C.; Marttin, E.; Romeijn, S.G.; van der Kuy, P.H.; Hermens, W.A.; Schipper, N.G. Cyclodextrins in nasal drug delivery. Adv. Drug Deliv. Rev. 1999, 36, 41–57. [Google Scholar] [CrossRef]
- Nonaka, N.; Farr, S.A.; Nakamachi, T.; Morley, J.E.; Nakamura, M.; Shioda, S.; Banks, W.A. Intranasal administration of PACAP: Uptake by brain and regional brain targeting with cyclodextrins. Peptides 2012, 36, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.M.; Hougaard, A.; Magon, S.; Asghar, M.S.; Ahmad, N.N.; Rostrup, E.; Sprenger, T.; Ashina, M. Change in brain network connectivity during PACAP38-induced migraine attacks: A resting-state functional MRI study. Neurology 2016, 86, 180–187. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29—Identifier NCT02829502, The Effect of GLP-1 Receptor Agonist on Cerebral Blood Flow Velocity in Stroke (EGRABIS1). 2018. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02829502 (accessed on 26 January 2021).
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29—Identifier NCT01792193, Gut-Derived Neuropeptides in Cerebrospinal Fluid of Patients with Parkinson’s Disease and Healthy Controls. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01792193 (accessed on 17 March 2016).
- Zhou, J.N.; Hofman, M.A.; Swaab, D.F. VIP neurons in the human SCN in relation to sex, age, and Alzheimer’s disease. Neurobiol. Aging 1995, 16, 571–576. [Google Scholar] [CrossRef]
- Filatov, E.; Short, L.I.; Forster, M.A.M.; Harris, S.S.; Schien, E.N.; Hughes, M.C.; Cline, D.L.; Appleby, C.J.; Gray, S.L. Contribution of thermogenic mechanisms by male and female mice lacking Pituitary Adenylate Cyclase-Activating Polypeptide in response to cold acclimation. Am. J. Physiol. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Toth, D.; Szabo, E.; Tamas, A.; Juhasz, T.; Horvath, G.; Fabian, E.; Opper, B.; Szabo, D.; Maugeri, G.; D’Amico, A.G.; et al. Protective Effects of PACAP in Peripheral Organs. Front. Endocrinol. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Lauretta, G.; Ravalli, S.; Szychlinska, M.A.; Castorina, A.; Maugeri, G.; D’Amico, A.G.; D’Agata, V.; Musumeci, G. Current knowledge of pituitary adenylate cyclase activating polypeptide (PACAP) in articular cartilage. Histol. Histopathol. 2020, 35, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cao, L.; Yi, P.Q.; Xu, C.; Su, J.; Chen, P.Z.; Li, M.; Chen, J.Y. Pituitary adenylate cyclase-activating polypeptide ameliorates radiation-induced cardiac injury. Am. J. Transl. Res. 2019, 11, 6585–6599. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadanandan, N.; Cozene, B.; Park, Y.J.; Farooq, J.; Kingsbury, C.; Wang, Z.-J.; Moscatello, A.; Saft, M.; Cho, J.; Gonzales-Portillo, B.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide: A Potent Therapeutic Agent in Oxidative Stress. Antioxidants 2021, 10, 354. https://doi.org/10.3390/antiox10030354
Sadanandan N, Cozene B, Park YJ, Farooq J, Kingsbury C, Wang Z-J, Moscatello A, Saft M, Cho J, Gonzales-Portillo B, et al. Pituitary Adenylate Cyclase-Activating Polypeptide: A Potent Therapeutic Agent in Oxidative Stress. Antioxidants. 2021; 10(3):354. https://doi.org/10.3390/antiox10030354
Chicago/Turabian StyleSadanandan, Nadia, Blaise Cozene, You Jeong Park, Jeffrey Farooq, Chase Kingsbury, Zhen-Jie Wang, Alexa Moscatello, Madeline Saft, Justin Cho, Bella Gonzales-Portillo, and et al. 2021. "Pituitary Adenylate Cyclase-Activating Polypeptide: A Potent Therapeutic Agent in Oxidative Stress" Antioxidants 10, no. 3: 354. https://doi.org/10.3390/antiox10030354
APA StyleSadanandan, N., Cozene, B., Park, Y. J., Farooq, J., Kingsbury, C., Wang, Z.-J., Moscatello, A., Saft, M., Cho, J., Gonzales-Portillo, B., & Borlongan, C. V. (2021). Pituitary Adenylate Cyclase-Activating Polypeptide: A Potent Therapeutic Agent in Oxidative Stress. Antioxidants, 10(3), 354. https://doi.org/10.3390/antiox10030354