
Maturation of Mitochondrially Targeted Prx V Involves a Second Cleavage by Mitochondrial Intermediate Peptidase That Is Sensitive to Inhibition by H2O2


Abstract
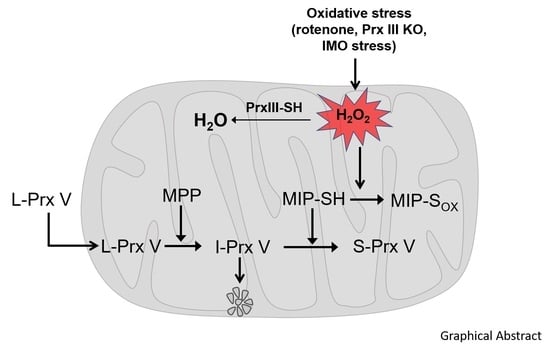
1. Introduction
2. Materials and Methods
2.1. Animals and Antibodies
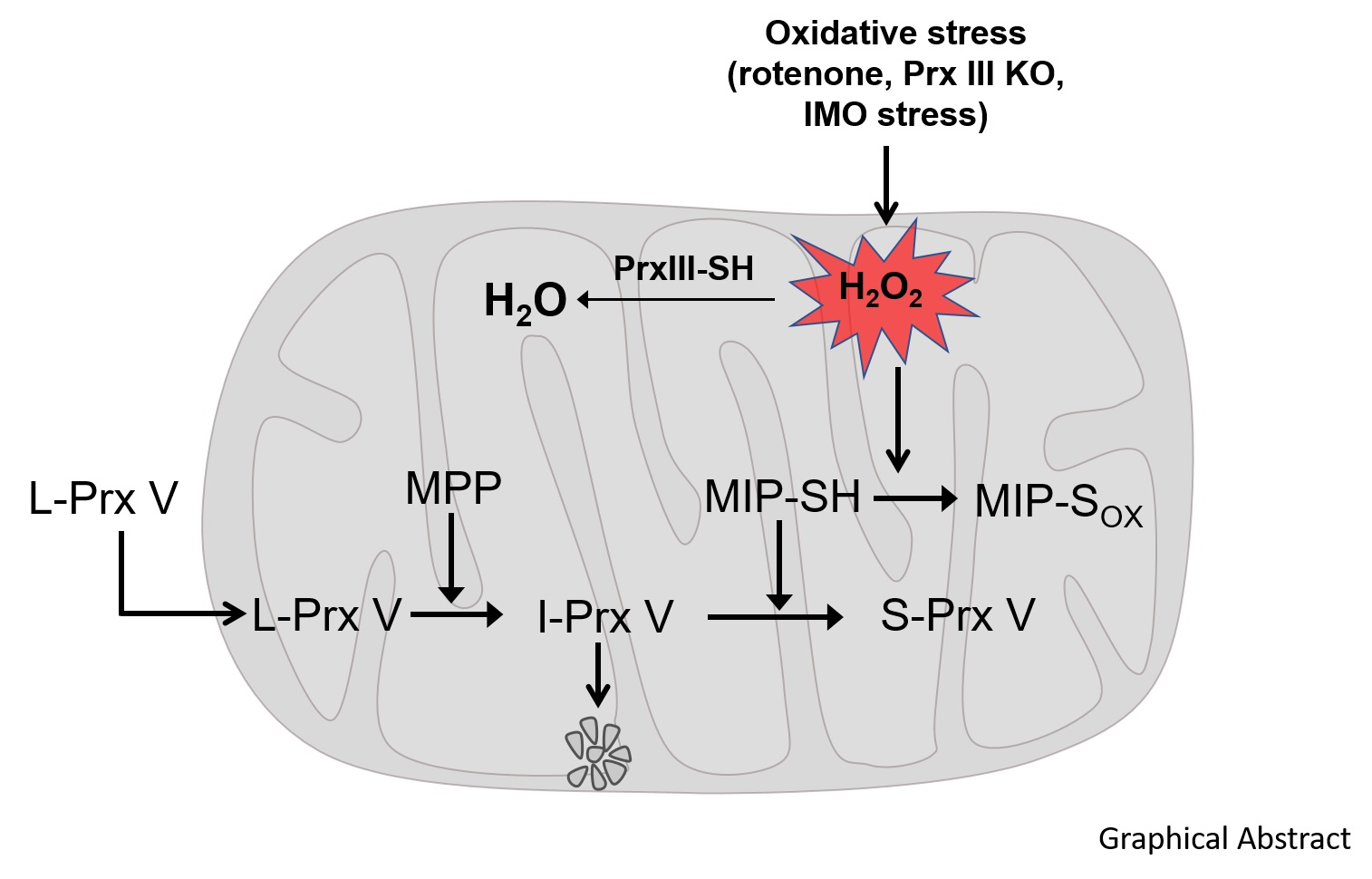
2.2. Subcellular Fractionation
2.3. Immunoblotting
2.4. Purification of Prx V Containing I-Prx V
2.5. RNA Isolation
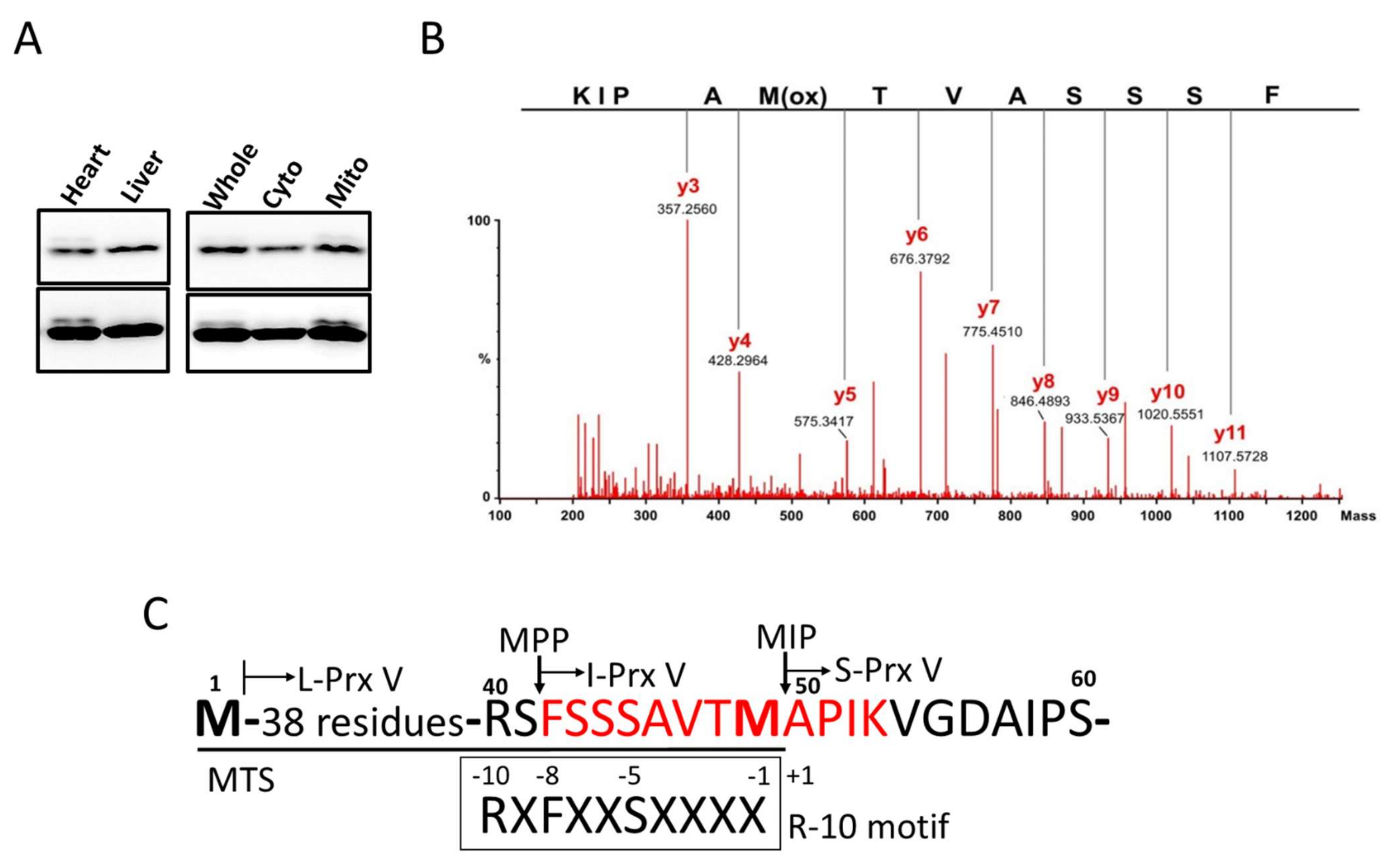
2.6. Mass Spectral Sequencing of I-Prx V Peptide
2.7. Cell Culture and Transfection
2.8. Statistical Analysis
3. Results
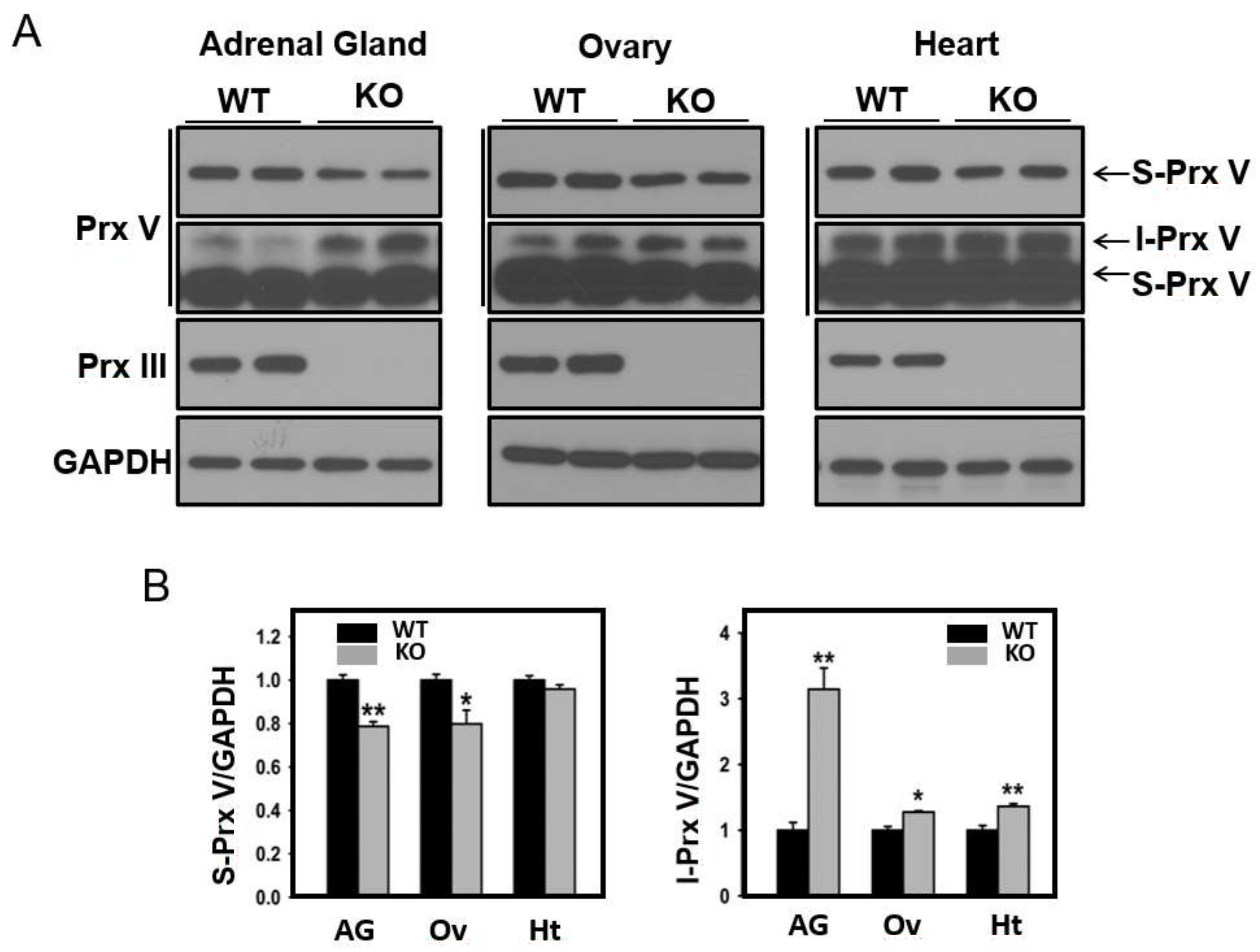
3.1. Determination of the N-Terminal Sequence of I-Prx V Extracted from the Minor Prx V Band
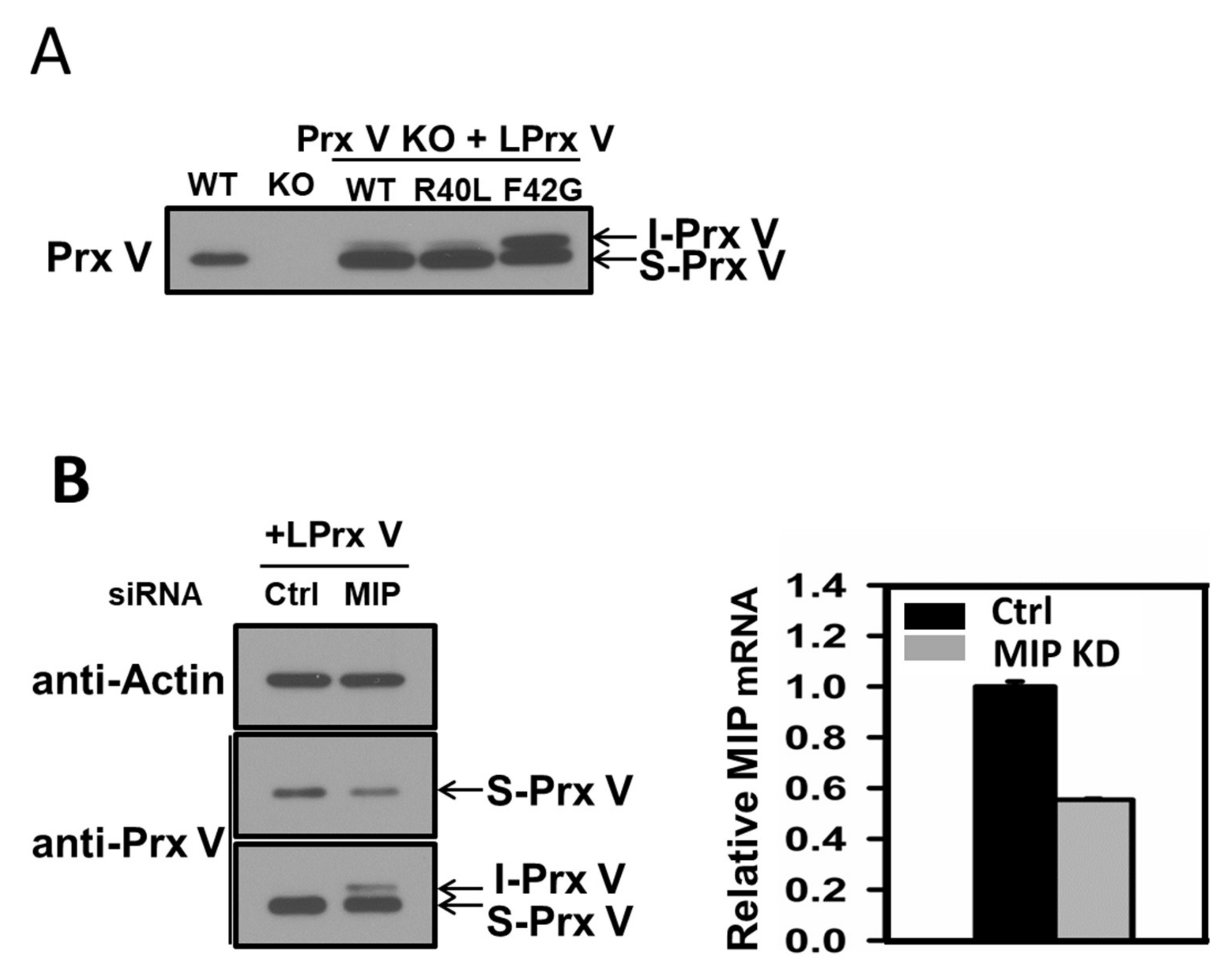
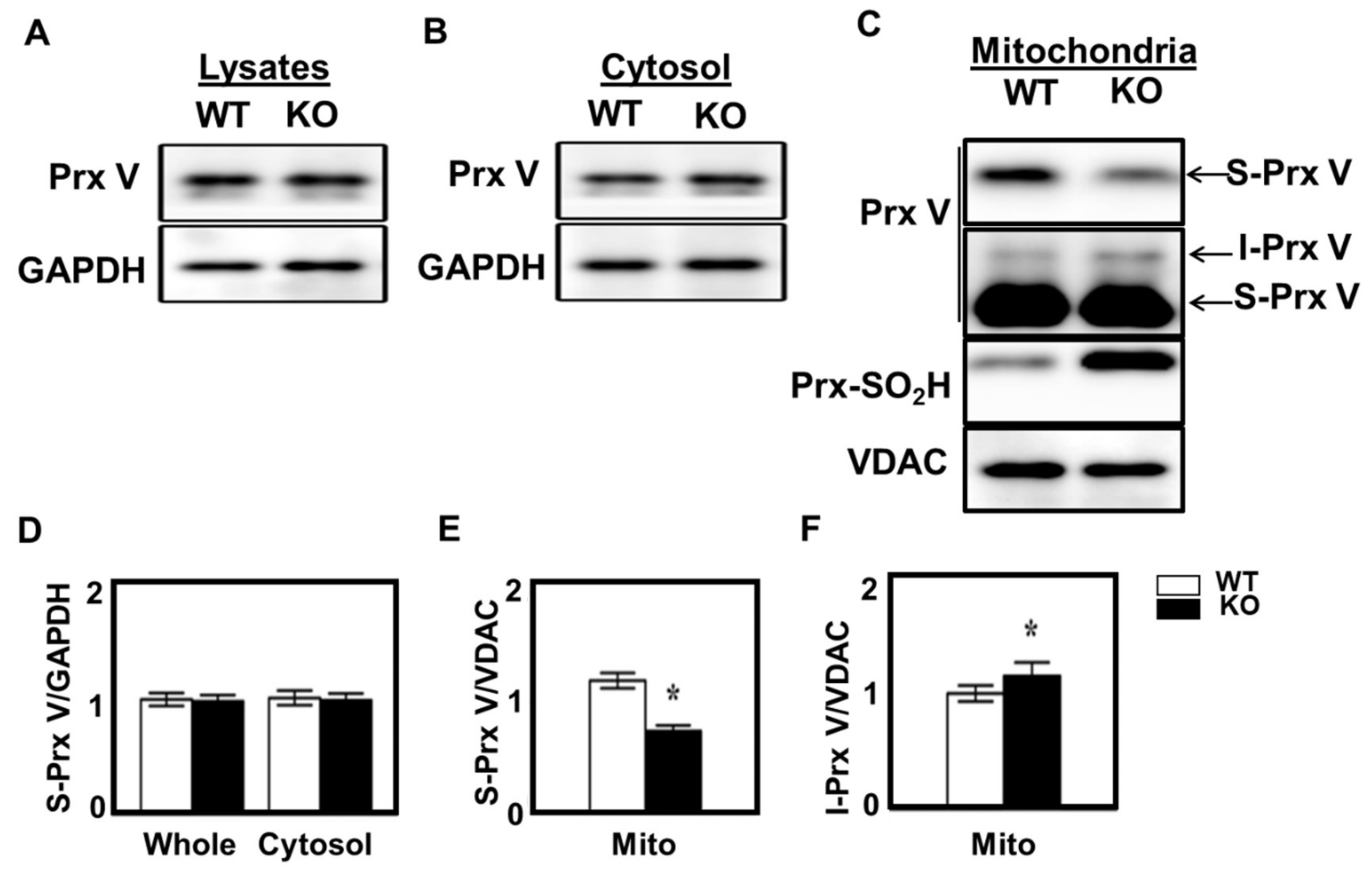
3.2. I-Prx V is Produced during Processing of L-Prx V to S-Prx V
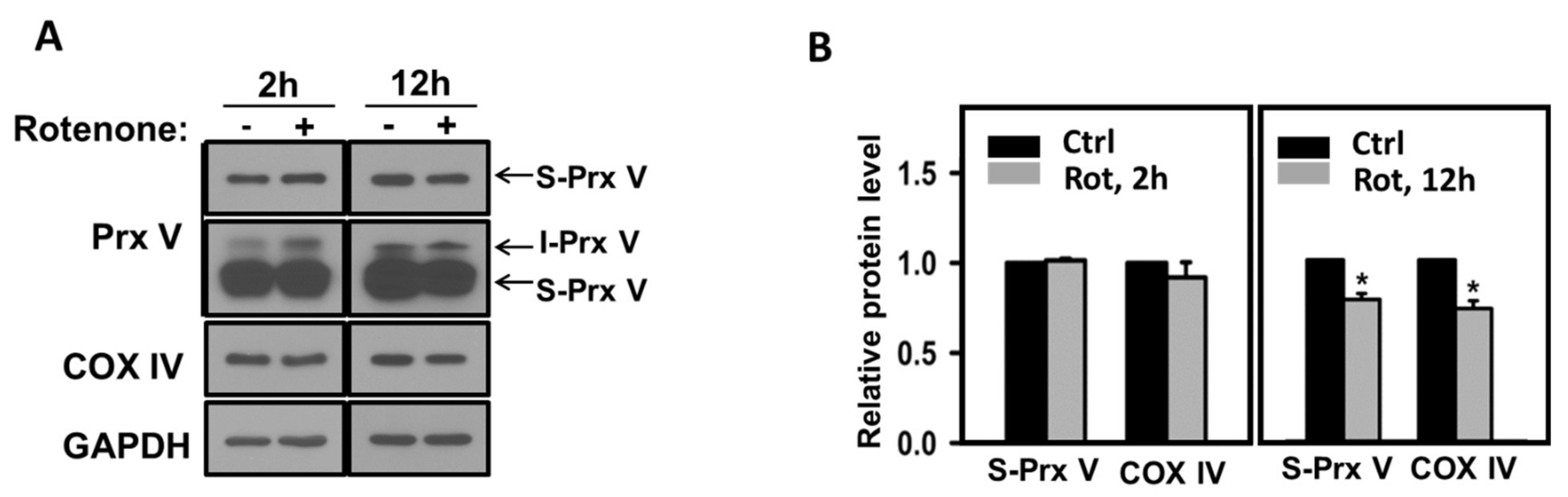
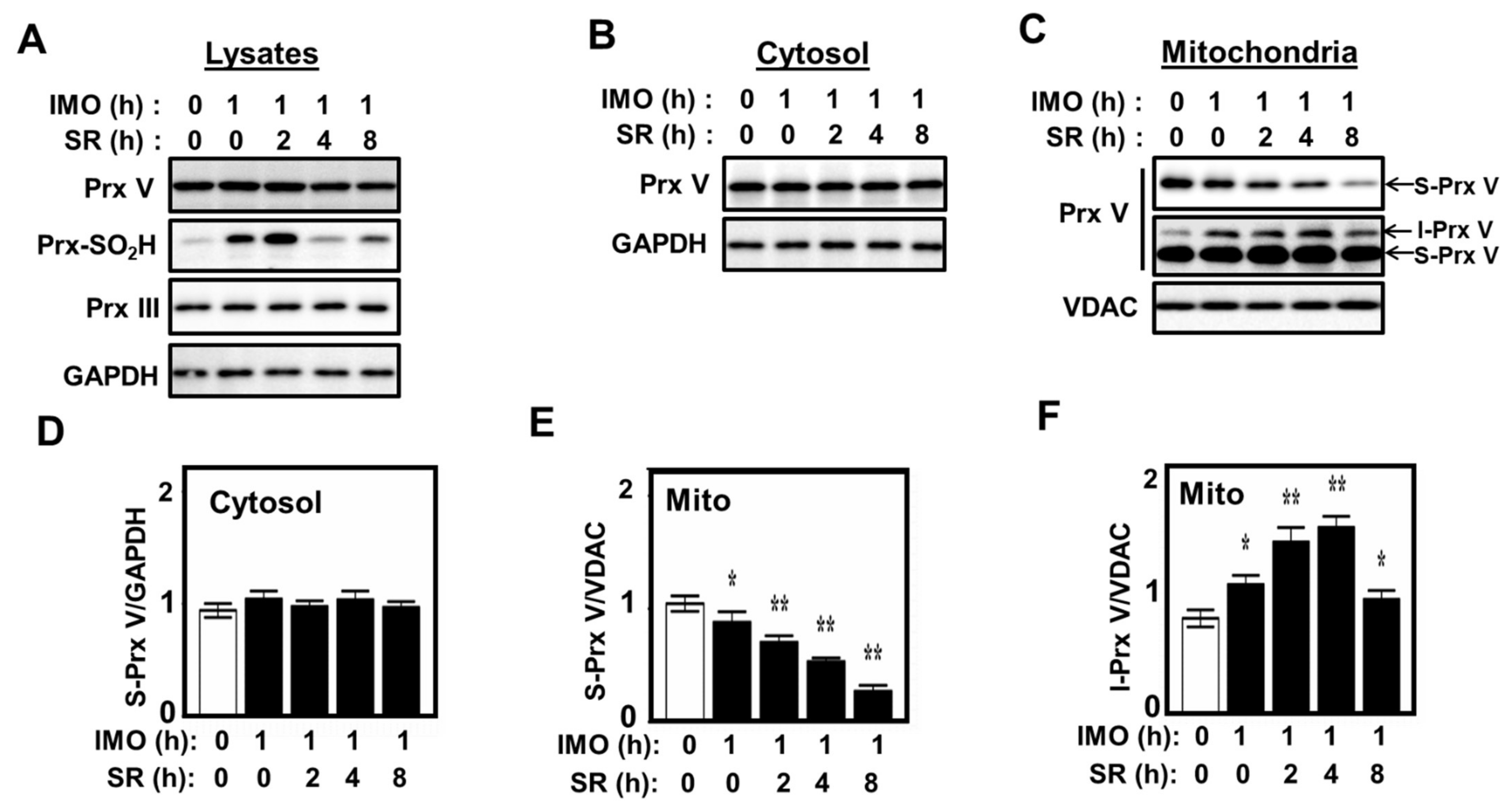
3.3. Effect of Mitochondrial ROS on the Abundance of I-Prx V and S-Prx V
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Prxs | peroxiredoxins |
| L-Prx V | long form Prx V |
| S-Prx V | short form Prx V |
| I-Prx V | intermediate Prx V containing octa peptide to be cleaved by MIP |
| Srx | sulfiredoxin |
| MTS | mitochondrial targeting sequence |
| MPP | mitochondrial processing peptidase |
| MIP | mitochondrial intermediate peptidase |
| ROS | reactive oxygen species |
| COX IV | cytochrome c oxidase subunit 4 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| VDAC | voltage-dependent anion channels |
| ACTH | adrenocorticotropic hormone |
| Oct1 | octapeptidyl aminopeptidase |
References
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Woo, H.A.; Kang, S.W.; Kim, H.K.; Yang, K.S.; Chae, H.Z.; Rhee, S.G. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J. Biol. Chem. 2003, 278, 47361–47364. [Google Scholar] [CrossRef]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Woo, H.A.; Lee, S.M.; Park, S.; Rhee, S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004, 279, 50994–51001. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Clippe, A.; Bogard, C.; Arsalane, K.; Wattiez, R.; Hermans, C.; Duconseille, E.; Falmagne, P.; Bernard, A. Cloning and characterization of AOEB166, a novel mammalian antioxidant enzyme of the peroxiredoxin family. J. Biol. Chem. 1999, 274, 30451–30458. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Avraham, S.; Jiang, S.; London, R.; Van Veldhoven, P.P.; Subramani, S.; Rogers, R.A.; Avraham, H. Characterization of human and murine PMP20 peroxisomal proteins that exhibit antioxidant activity in vitro. J. Biol. Chem. 1999, 274, 29897–29904. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Nhu, N.T.; Berck, J.; Clippe, A.; Duconseille, E.; Cherif, H.; Boone, C.; Van der Eecken, V.; Bernard, A.; Banmeyer, I.; Knoops, B. Human peroxiredoxin 5 gene organization, initial characterization of its promoter and identification of alternative forms of mRNA. Biochim. Biophys. Acta 2007, 1769, 472–483. [Google Scholar] [CrossRef]
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta 2002, 1592, 63–77. [Google Scholar] [CrossRef]
- Mossmann, D.; Meisinger, C.; Vogtle, F.N. Processing of mitochondrial presequences. Biochim. Biophys. Acta 2012, 1819, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Isaya, G.; Kalousek, F.; Fenton, W.A.; Rosenberg, L.E. Cleavage of precursors by the mitochondrial processing peptidase requires a compatible mature protein or an intermediate octapeptide. J. Cell Biol. 1991, 113, 65–76. [Google Scholar] [CrossRef]
- Branda, S.S.; Isaya, G. Prediction and identification of new natural substrates of the yeast mitochondrial intermediate peptidase. J. Biol. Chem. 1995, 270, 27366–27373. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, J.P.; Hodges, P.E.; Rosenberg, L.E. Survey of amino-terminal proteolytic cleavage sites in mitochondrial precursor proteins: Leader peptides cleaved by two matrix proteases share a three-amino acid motif. Proc. Natl. Acad. Sci. USA 1989, 86, 4056–4060. [Google Scholar] [CrossRef] [PubMed]
- Vogtle, F.N.; Prinz, C.; Kellermann, J.; Lottspeich, F.; Pfanner, N.; Meisinger, C. Mitochondrial protein turnover: Role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol. Biol. Cell 2011, 22, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.; Palma, F.R.; Barros, M.H.; Tsuchida, E.T.; Turano, H.G.; Alegria, T.G.P.; Demasi, M.; Netto, L.E.S. Proteolytic cleavage by the inner membrane peptidase (IMP) complex or Oct1 peptidase controls the localization of the yeast peroxiredoxin Prx1 to distinct mitochondrial compartments. J. Biol. Chem. 2017, 292, 17011–17024. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.H.; Trumpower, B.L. Intermediate length Rieske iron-sulfur protein is present and functionally active in the cytochrome bc1 complex of Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 9253–9257. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein. Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef]
- Sriram, S.M.; Kim, B.Y.; Kwon, Y.T. The N-end rule pathway: Emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol. 2011, 12, 735–747. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Wortelkamp, S.; Zahedi, R.P.; Becker, D.; Leidhold, C.; Gevaert, K.; Kellermann, J.; Voos, W.; Sickmann, A.; Pfanner, N.; et al. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 2009, 139, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, E.G.; Yi, H.J.; Kim, N.H.; Rhee, S.G.; Woo, H.A. Ablation of Peroxiredoxin V Exacerbates Ischemia/Reperfusion-Induced Kidney Injury in Mice. Antioxidants 2020, 9, 769. [Google Scholar] [CrossRef] [PubMed]
- Kil, I.S.; Lee, S.K.; Ryu, K.W.; Woo, H.A.; Hu, M.C.; Bae, S.H.; Rhee, S.G. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef]
- Kim, S.; Na, S.; Sim, J.W.; Park, H.; Jeong, J.; Kim, H.; Seo, Y.; Seo, J.; Lee, K.J.; Paek, E. MODi: A powerful and convenient web server for identifying multiple post-translational peptide modifications from tandem mass spectra. Nucleic Acids Res. 2006, 34, W258–W263. [Google Scholar] [CrossRef] [PubMed]
- Isaya, G.; Kalousek, F.; Rosenberg, L.E. Amino-terminal octapeptides function as recognition signals for the mitochondrial intermediate peptidase. J. Biol. Chem. 1992, 267, 7904–7910. [Google Scholar] [CrossRef]
- Kalousek, F.; Isaya, G.; Rosenberg, L.E. Rat liver mitochondrial intermediate peptidase (MIP): Purification and initial characterization. EMBO J. 1992, 11, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, A.; Dong, K.W.; Glucksman, M.J.; Orlowski, M.; Roberts, J.L. Molecular cloning and primary structure of rat testes metalloendopeptidase EC 3.4.24.15. Biochemistry 1990, 29, 10323–10329. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kang, D. The Role of Peroxiredoxins in the Transduction of H2O2 Signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Pecina, P.; Demont, J.; Vojtiskova, A.; Simonnet, H.; Houstek, J.; Godinot, C. Inhibition of cytochrome c oxidase subunit 4 precursor processing by the hypoxia mimic cobalt chloride. Biochem. Biophys. Res. Commun. 2006, 344, 1086–1093. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, J.; Han, S.J.; Park, I.; Huu, T.N.; Kim, J.S.; Woo, H.A.; Lee, S.R. The critical role of redox regulation of PTEN and peroxiredoxin III in alcoholic fatty liver. Free Radic. Biol. Med. 2020, 162, 141–148. [Google Scholar] [CrossRef]
- Hanukoglu, I. Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab. Rev. 2006, 38, 171–196. [Google Scholar] [CrossRef] [PubMed]
- Rosol, T.J.; Yarrington, J.T.; Latendresse, J.; Capen, C.C. Adrenal gland: Structure, function, and mechanisms of toxicity. Toxicol. Pathol. 2001, 29, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef] [PubMed]
- Eldomery, M.K.; Akdemir, Z.C.; Vögtle, F.-N.; Charng, W.-L.; Mulica, P.; Rosenfeld, J.A.; Gambin, T.; Gu, S.; Burrage, L.C.; Al Shamsi, A.; et al. MIPEP recessive variants cause a syndrome of left ventricular non-compaction, hypotonia, and infantile death. Genome Med. 2016, 8, 106. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| GAPDH | AGAACATCATCCCTGCATCC | GGTCCTCAGTGTAGCCCAAG |
| LPrx V | AGAAGCAGGTTGGGAGTGTG | CTTTCTTGCCCTTGAACAGC |
| SPrx V | GGCATTTACACCTGGCTGTT | CGACGATTCCCAAAGAGAGA |
| MIP | TTTCAGCGAGCAGACAAACC | TCCCAGTGACGTGTTGGTAA |
| Target Gene | Sense | Anti-Sense |
|---|---|---|
| Scrambled | AUGAACGUGAAUUGCUCAATT | UUGAGCAAUUCACGUUCAUTT |
| LPrx V | GCUAUAUACUCGUCGGUGGTT | CCACCGACGAGUAUAUAGCTT |
| SPrx V | GGAAGGAGACAGACUUAUUTT | AAUAAGUCUGUCUCCUUCCTT |
| MIP | GCCGGGAUCCGGGCCCGAATT | UUCGGGCCCGGAUCCCGGCTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, J.; Park, J.; Woo, H.A.; Rhee, S.G. Maturation of Mitochondrially Targeted Prx V Involves a Second Cleavage by Mitochondrial Intermediate Peptidase That Is Sensitive to Inhibition by H2O2. Antioxidants 2021, 10, 346. https://doi.org/10.3390/antiox10030346
Sim J, Park J, Woo HA, Rhee SG. Maturation of Mitochondrially Targeted Prx V Involves a Second Cleavage by Mitochondrial Intermediate Peptidase That Is Sensitive to Inhibition by H2O2. Antioxidants. 2021; 10(3):346. https://doi.org/10.3390/antiox10030346
Chicago/Turabian StyleSim, Juhyun, Jiyoung Park, Hyun Ae Woo, and Sue Goo Rhee. 2021. "Maturation of Mitochondrially Targeted Prx V Involves a Second Cleavage by Mitochondrial Intermediate Peptidase That Is Sensitive to Inhibition by H2O2" Antioxidants 10, no. 3: 346. https://doi.org/10.3390/antiox10030346
APA StyleSim, J., Park, J., Woo, H. A., & Rhee, S. G. (2021). Maturation of Mitochondrially Targeted Prx V Involves a Second Cleavage by Mitochondrial Intermediate Peptidase That Is Sensitive to Inhibition by H2O2. Antioxidants, 10(3), 346. https://doi.org/10.3390/antiox10030346