
Carbonyl Stress in Red Blood Cells and Hemoglobin


Abstract
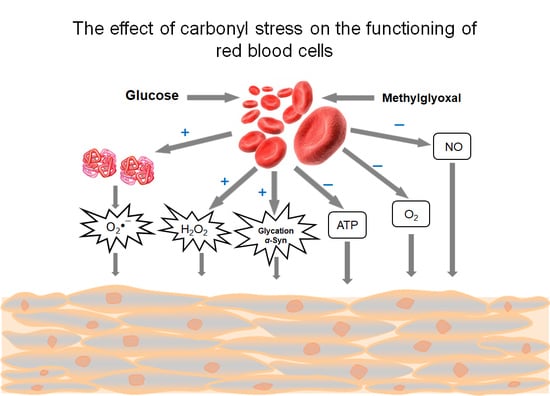
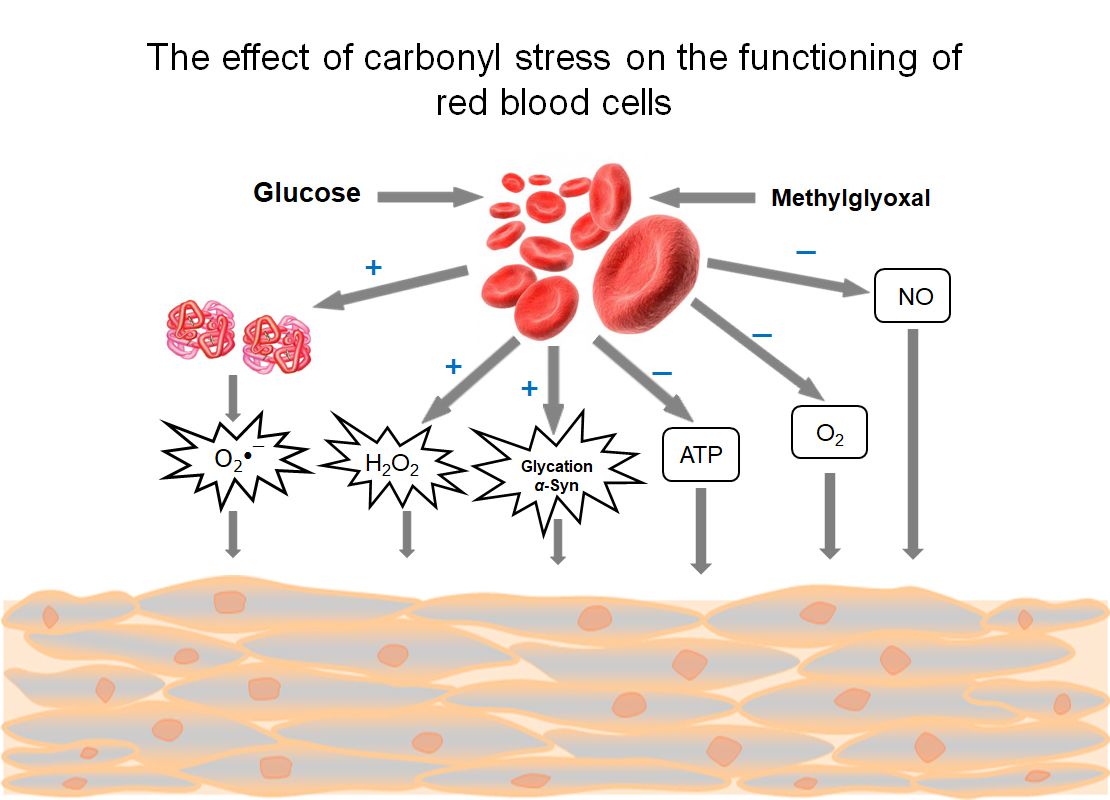{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genesis of Carbonyl Stress in Red Blood Cells
3. Inhibition and Inactivation of the Glycolytic Enzymes
3.1. Glyceraldehyde-3-Phosphate Dehydrogenase
3.2. Triosophosphate Isomerase
3.3. Glyoxalase System
4. Effects of Methylglyoxal on Red Blood Cells
4.1. Methylglyoxal—Highly Reactive Dicarbonyl Metabolite
4.2. Pathological Effects Caused by MG Action on RBC
5. Carbonyl Stress and Hemoglobin
5.1. Glycated Hemoglobin
5.2. Hemoglobin in the Development of Carbonyl Stress Consequences
5.3. HbA1 and MG—the Hyperglycemic Biomarkers
6. Relationship of Carbonyl Stress with Oxidative and Nitrosative Stresses
7. Pharmacological Interventions and Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Selye, H. The Stress of Life; McGraw-Hill: New York, NY, USA, 1956. [Google Scholar]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988, 254, 751–755. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Hooper, N.I.; Jennings, P.E.; Florkowski, C.M.; Jones, A.F.; Lunec, J.; Barnett, A.H. The human red blood cell glyoxalase system in diabetes mellitus. Diabetes Res. Clin. Pract. 1989, 7, 115–120. [Google Scholar] [CrossRef]
- Baynes, J.W. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P.H. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. Lond. 1994, 87, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Biswas, U.K.; Kumar, A. Study on the changes of Carbonic Anhydrase activity in insulin resistance and the effect of methylglyoxal. J. Pak. Med. Assoc. 2012, 62, 417–421. [Google Scholar] [PubMed]
- Berlanga, J.; Cibrian, D.; Guillén, I.; Freyre, F.; Alba, J.S.; Lopez-Saura, P.; Merino, N.; Aldama, A.; Quintela, A.M.; Triana, M.E.; et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 2005, 109, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wu, L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Dicarbonyl intermediates in the Maillard reaction. Ann. N. Y. Acad. Sci. 2005, 1043, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.M.; Wu, L. Free radical generation by methylglyoxal in tissues. Drug Metabol. Drug Interact. 2008, 23, 151–173. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. The tandem of free radicals and methylglyoxal. Chem. Biol. Interact. 2008, 171, 251–271. [Google Scholar] [CrossRef]
- Constantin, A.; Constantinescu, E.; Dumitrescu, M.; Calin, A.; Popov, D. Effects of ageing on carbonyl stress and antioxidant defense in RBCs of obese Type 2 diabetic patients. J. Cell. Mol. Med. 2005, 9, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Taniquchi, N.; Kinoshita, N.; Arai, K.; Iizuka, S.; Usui, M.; Naito, T. Inactivation of erythrocyte Cu-Zn-superoxide dismutase through nonenzymatic glycosylation. Prog. Clin. Biol. Res. 1989, 304, 277–290. [Google Scholar] [PubMed]
- Fujii, J.; Myint, T.; Okado, A.; Kaneto, H.; Taniguchi, N. Oxidative stress caused by glycation of Cu,Zn-superoxide dismutase and its effects on intracellular components. Nephrol. Dial. Transplant. 1996, 11, 34–40. [Google Scholar] [CrossRef]
- Mohr, S.; Stamler, J.S.; Brüne, B. Mechanism of covalent modification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosating agents. FEBS Lett. 1994, 348, 223–227. [Google Scholar] [CrossRef]
- Orosz, F.; Wágner, G.; Liliom, K.; Kovács, J.; Baróti, K.; Horányi, M.; Farkas, T.; Hollán, S.; Ovádi, J. Enhanced association of mutant triosephosphate isomerase to red cell membranes and to brain microtubules. Proc. Natl. Acad. Sci. USA 2000, 97, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Vander Jagt, D.L.; Hunsaker, L.A.; Campos, N.M.; Baack, B.R. D-lactate production in erythrocytes infected with Plasmodium falciparum. Mol. Biochem. Parasitol. 1990, 42, 277–284. [Google Scholar] [CrossRef]
- Dumont, L.; Richardson, M.B.; van der Peet, P.; Marapana, D.S.; Triglia, T.; Dixon, M.W.A.; Cowman, A.F.; Williams, S.J.; Tilley, L.; McConville, M.J.; et al. The metabolic repair enzyme phosphoglycolate phosphatase regulates central carbon 2 metabolism and fosmidomycin sensitivity in Plasmodium falciparum. mBio 2019, 10, 02060-19. [Google Scholar] [CrossRef] [PubMed]
- Urscher, M.; Alisch, R.; Deponte, M. The glyoxalase system of malaria parasites--implications for cell biology and general glyoxalase research. Semin. Cell. Dev. Biol. 2011, 22, 262–270. [Google Scholar] [CrossRef]
- Baskaran, S.; Rajan, D.P.; Balasubramanian, K.A. Formation of methylglyoxal by bacteria isolated from human faeces. J. Med. Microbiol. 1989, 28, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.-G.; Lee, J.-W.; Han, J.-S.; Lee, B.; Jeong, J.-H.; Park, S.-H.; Kim, J.-H.; Jang, S.; Park, M.; Kim, S.-Y.; et al. Bacteria-derived metabolite, methylglyoxal, modulates the longevity of C. elegans through TORC2/SGK-1/DAF-16 signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 17142–17150. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, G.; Maresca, M.; Bonsignore, A. The effect of methylglyoxal on the glycolytic enzymes. FEBS Lett. 1980, 117, 17–18. [Google Scholar] [CrossRef]
- Lee, H.J.; Howell, S.K.; Sanford, R.J.; Beisswenger, P. Methylglyoxal can modify GAPDH activity and structure. Ann. N. Y. Acad. Sci. 2005, 1043, 135–145. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Dan’shina, P.V.; Schmalhausen, E.V.; Arutiunov, D.Y.; Pleten’, A.P.; Muronetz, V.I. Acceleration of glycolysis in the presence of the non-phosphorylating and the oxidized phosphorylating glyceraldehyde-3-phosphate dehydrogenases. Biochem. Mosc. 2003, 68, 593–600. [Google Scholar] [CrossRef]
- Souza, J.M.; Radi, R. Glyceraldehyde-3-phosphate dehydrogenase inactivation by peroxynitrite. Arch. Biochem. Biophys. 1998, 360, 187–194. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. J. Alzheimers Dis. 2010, 20, 369–393. [Google Scholar] [CrossRef]
- Molina y Vedia, L.; McDonald, B.; Reep, B.; Brüne, B.; Di Silvio, M.; Billiar, T.R.; Lapetina, E.G. Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J. Biol. Chem. 1992, 267, 24929–24932. [Google Scholar] [CrossRef]
- Galli, F.; Rovidati, S.; Ghibelli, L.; Canestrari, F. S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase decreases the enzyme affinity to the erythrocyte membrane. Nitric Oxide 1998, 2, 17–27. [Google Scholar] [CrossRef]
- Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E.G.; Brune, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1999, 274, 9427–9430. [Google Scholar] [CrossRef]
- Yim, H.S.; Kang, S.O.; Hah, Y.C.; Chock, P.B.; Yim, M.B. Free radicals generated during the glycation reaction of amino acids by methylglyoxal. A model study of protein-cross-linked free radicals. J. Biol. Chem. 1995, 270, 28228–28233. [Google Scholar] [CrossRef]
- Balagopalakrishna, C.; Manoharan, P.T.; Abugo, O.O.; Rifkind, J.M. Production of superoxide from hemoglobin-bound oxygen under hypoxic conditions. Biochemistry 1996, 35, 6393–6398. [Google Scholar] [CrossRef]
- Gomez-Mejiba, S.E.; Zhai, Z.; Muñoz, M.D.; Vedova, C.D.; Ranguelova, K.; Ashby, M.T.; Ramirez, D.C. Radicalization of glyceraldehyde-3-phosphate dehydrogenase by HOCl in living cells. Enz. Eng. 2015, 4, 134. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Melnikova, A.K.; Barinova, K.V.; Schmalhausen, E.V. Inhibitors of glyceraldehyde 3-phosphate dehydrogenase and unexpected effects of its reduced activity. Biochem. Mosc. 2019, 84, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, G.; Maresca, M.; Buzzi, E. Inhibition of the glycolytic pathway by methylglyoxal in human platelets. Cell Biochem. Funct. 1989, 7, 65–70. [Google Scholar] [CrossRef]
- Agrawal, R.; Smart, T.; Nobre-Cardoso, J.; Richards, C.; Bhatnagar, R.; Tufail, A.; Shima, D.; Jones, P.H.; Pavesio, C. Assessment of red blood cell deformability in type 2 diabetes mellitus and diabetic retinopathy by dual optical tweezers stretching technique. Sci. Rep. 2016, 6, 15873. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stit mulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, S.; Hemshekhar, M.; Naveenkumar, S.K.; Kumar, K.S.S.; Sundaram, M.S.; Girish, K.S.; Rangappa, K.S. Novel oxolane derivative DMTD mitigates high glucose-induced erythrocyte apoptosis by regulating oxidative stress. Toxicol. Appl. Pharmacol. 2017, 334, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Kosmachevskaya, O.V.; Shumaev, K.B.; Nasybullina, E.I.; Gubkina, S.A.; Topunov, A.F. Interaction of S-nitrosoglutathione with methemoglobin under conditions of modeling carbonyl stress. Hemoglobin 2013, 37, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation end products: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef]
- Lee, C.; Yim, M.B.; Chock, P.B.; Yim, H.S.; Kang, S.O. Oxidation-reduction properties of methylglyoxal-modified protein in relation to free radical generation. J. Biol. Chem. 1998, 273, 25272–25278. [Google Scholar] [CrossRef]
- Kalapos, M.P. Methylglyoxal in living organisms: Chemistry, biochemistry, toxicology and biological implications. Toxicol. Lett. 1999, 110, 145–175. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolo, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl stress at the crossroads of healthy and unhealthy aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Jahan, I.; Ng, R. Suppression of the accumulation of triosephosphates and increased formation of methylglyoxal in human red blood cells during hyperglycaemia by thiamine in vitro. J. Biochem. 2001, 129, 543–549. [Google Scholar] [CrossRef]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metab. 2018, 27, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Howell, S.K.; Smith, K.; Szwergold, B.S. Glyceraldehyde-3-phosphate dehydrogenase activity as an independent modifier of methylglyoxal levels in diabetes. Biochim. Biophys. Acta 2003, 1637, 98–106. [Google Scholar] [CrossRef]
- Orosz, F.; Oláh, J.; Ovádi, J. Triosephosphate isomerase deficiency: New insights into an enigmatic disease. Biochim. Biophys. Acta 2009, 1792, 1168–1174. [Google Scholar] [CrossRef]
- De la Mora-de la Mora, I.; Torres-Larios, A.; Enríquez-Flores, S.; Méndez, S.T.; Castillo-Villanueva, A.; Gómez-Manzo, S.; López-Velázquez, G.; Marcial-Quino, J.; Torres-Arroyo, A.; García-Torres, I.; et al. Structural effects of protein aging: Terminal marking by deamidation in human triosephosphate isomerase. PLoS ONE 2015, 10, 0123379. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.Q.; Yüksel, K.U.; Gracy, R.W. Relationship between the catalytic center and the primary degradation site of triosephosphate isomerase: Effects of active site modification and deamidation. Arch. Biochem. Biophys. 1992, 293, 382–389. [Google Scholar] [CrossRef]
- Enríquez-Flores, S.; Flores-López, L.A.; García-Torres, I.; de la Mora-de la Mora, I.; Cabrera, N.; Gutiérrez-Castrellón, P.; Martínez-Pérez, Y.; López-Velázquez, G. Deamidated human triosephosphate isomerase is a promising druggable target. Biomolecules 2020, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Hipkiss, A.R. Energy metabolism and ageing regulation: Metabolically driven deamidation of triosephosphate isomerase may contribute to proteostatic dysfunction. Ageing Res. Rev. 2011, 10, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Hipkiss, A.R. The human erythrocyte can become both a metabolic “Achilles’ Heel” and a “Trojan Horse”: Likely consequences of persistent excessive glycolysis. Integr. Food Nutr. Metab. 2019, 6, 1–2. [Google Scholar] [CrossRef]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Guix, F.X.; Clarimón, J.; Miscione, G.P.; Boada, M.; et al. Methylglyoxal produced by amyloid-β peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 273–288. [Google Scholar] [CrossRef]
- Brandhorst, T.T.; Kean, I.R.L.; Lawry, S.M.; Wiesner, D.L.; Klein, B.S. Phenylpyrrole fungicides act on triosephosphate isomerase to induce methylglyoxal stress and alter hybrid histidine kinase activity. Sci. Rep. 2019, 9, 5047. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Talesa, V.N. Glyoxalases in urological malignancies. Int. J. Mol. Sci. 2018, 19, 415. [Google Scholar] [CrossRef]
- Honek, J.F. Glyoxalase biochemistry. Biomol. Concepts 2015, 6, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.D.; Berners-Price, S.J.; Bulliman, B.T.; Kuchel, P.W. Kinetic analysis of the human erythrocyte glyoxalase system using 1H NMR and a computer model. Eur. J. Biochem. 1990, 193, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Leone, A.; Raciti, G.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-glyoxalase 1 balance: The root of vascular damage. Int. J. Mol. Sci. 2017, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- -Morgenstern, J.; Campos, M.C.; Nawroth, P.; Fleming, T. The glyoxalase system—New insights into an ancient metabolism. Antioxidants 2020, 9, 939. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.E.; Lo, T.W.; Thornalley, P.J. A simplified method for the purification of human red blood cell glyoxalase. I. Characteristics, immunoblotting, and inhibitor studies. J. Protein. Chem. 1993, 12, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.E.; Lo, T.W.; Thornalley, P.J. Purification and characterisation of glyoxalase II from human red blood cells. Eur. J. Biochem. 1993, 213, 1261–1267. [Google Scholar] [CrossRef]
- Birkenmeier, G.; Stegemann, C.; Hoffmann, R.; Günther, R.; Huse, K.; Birkemeyer, C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS ONE 2010, 5, 10399. [Google Scholar] [CrossRef]
- Mitsumoto, A.; Kim, K.R.; Oshima, G.; Kunimoto, M.; Okawa, K.; Iwamatsu, A.; Nakagawa, Y. Glyoxalase I is a novel nitric-oxide-responsive protein. Biochem. J. 1999, 344, 837–844. [Google Scholar] [CrossRef]
- Mitsumoto, A.; Kim, K.R.; Oshima, G.; Kunimoto, M.; Okawa, K.; Iwamatsu, A.; Nakagawa, Y. Nitric oxide inactivates glyoxalase I in cooperation with glutathione. J. Biochem. 2000, 128, 647–654. [Google Scholar] [CrossRef]
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteoblasts via a novel mechanism driven by H2O2 and entailing Glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-κB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17. [Google Scholar] [CrossRef]
- Antognelli, C.; Gambelunghe, A.; Talesa, V.N.; Muzi, G. Reactive oxygen species induce apoptosis in bronchial epithelial BEAS-2B cells by inhibiting the antiglycation glyoxalase I defence: Involvement of superoxide anion, hydrogen peroxide and NF-κB. Apoptosis 2014, 19, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Gambelunghe, A.; Muzi, G.; Talesa, V.N. Peroxynitrite-mediated glyoxalase I epigenetic inhibition drives apoptosis in airway epithelial cells exposed to crystalline silica via a novel mechanism involving argpyrimidine-modified Hsp70, JNK, and NF-κB. Free Radic. Biol. Med. 2015, 84, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Palumbo, I.; Aristei, C.; Talesa, V.N. Glyoxalase I inhibition induces apoptosis in irradiated MCF-7 cells via a novel mechanism involving Hsp27, p53 and NF-κB. Br. J. Cancer 2014, 111, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Gabreanu, G.R.; Angelescu, S. Erythrocyte membrane in type 2 diabetes mellitus. Discoveries Craiova 2016, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Morabito, R.; Remigante, A.; Spinelli, S.; Vitale, G.; Trichilo, V.; Loddo, S.; Marino, A. High glucose concentrations affect band 3 protein in human erythrocytes. Antioxidants 2020, 9, 365. [Google Scholar] [CrossRef]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef] [PubMed]
- Bora, S.; Adole, P.S.; Motupalli, N.; Pandi, V.R.T.; Vinod, K.V. Association between carbonyl stress markers and the risk of acute coronary syndrome in patients with type 2 diabetes mellitus-A pilot study. Diabetes Metab. Syndr. 2020, 14, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.; Cuny, J.; Nawroth, G.; Djuric, Z.; Humpert, P.M.; Zeier, M.; Bierhaus, A.; Nawroth, P.P. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia 2012, 55, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wei, G.Y.; Guo, C.S.; Deng, Y.F.; Jiang, Y.M.; Gao, C.; Jian, C.D. 20S proteasome and glyoxalase 1 activities decrease in erythrocytes derived from Alzheimer’s disease patients. Neural Regen. Res. 2020, 15, 178–183. [Google Scholar] [CrossRef]
- Morresi, C.; Cianfruglia, L.; Sartini, D.; Cecati, M.; Fumarola, S.; Emanuelli, M.; Armeni, T.; Ferretti, G.; Bacchetti, T. Effect of high glucose-induced oxidative stress on paraoxonase 2 expression and activity in Caco-2 cells. Cells 2019, 8, 1616. [Google Scholar] [CrossRef] [PubMed]
- Karga, E.; Pappa, F.; Tassia, N.; Janákyb, T.; Wittmanna, G.; Túria, S. Enhanced methylglyoxal formation in the erythrocytes of hemodialyzed patients. Metab. Clin. Exp. 2009, 58, 976–982. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J. Glyoxalase activity in human red blood cells fractioned by age. Mech. Ageing Dev. 1989, 48, 63–71. [Google Scholar] [CrossRef]
- Miranda, H.V.; Szego, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Vulnerability of human erythrocytes to persistent high glycemic index diets: Implications for ageing and neurodegeneration: Possible amelioration by carnosine. J. Aging Sci. 2019, 7, 3. [Google Scholar] [CrossRef]
- Fleming, T.H.; Theilen, T.M.; Masania, J.; Wunderle, M.; Karimi, J.; Vittas, S.; Bernauer, R.; Bierhaus, A.; Rabbani, N.; Thornalley, P.J.; et al. Aging-dependent reduction in glyoxalase 1 delays wound healing. Gerontology 2013, 59, 427–437. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Glycation research in amino acids: A place to call home. Amino Acids 2012, 42, 1087–1096. [Google Scholar] [CrossRef]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—Role in ageing and disease. Drug Metab. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy diabetes. Diabetes 2014, 63, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Beppu, M.; Kikugawa, K.; Nagai, R.; Horiuchi, S. Membrane proteins of human erythrocytes are modified by advanced glycation end products during aging in the circulation. Biochem. Biophys. Res. Commun. 1999, 258, 1123–1127. [Google Scholar] [CrossRef]
- Sibbersen, C.; Oxvig, A.-M.S.; Olesen, S.B.; Nielsen, C.B.; Galligan, J.J.; Jørgensen, K.A.; Palmfeldt, J.; Johannsen, M. Profiling of methylglyoxal blood metabolism and advanced glycation end-product proteome using a chemical probe. ACS Chem. Biol. 2018, 13, 3294–3305. [Google Scholar] [CrossRef] [PubMed]
- Semchyshyn, H.M. Reactive carbonyl species in vivo: Generation and dual biological effects. Sci. World J. 2014, 2014, 417842. [Google Scholar] [CrossRef]
- Nokin, M.-J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellachene, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed]
- Kosmachevskaya, O.V.; Shumaev, K.B.; Topunov, A.F. Signal and regulatory effects of methylglyoxal in eukaryotic cells. Appl. Biochem. Microbiol. 2017, 53, 273–289. [Google Scholar] [CrossRef]
- Kosmachevskaya, O.V.; Shumaev, K.B.; Topunov, A.F. Electrophilic signaling: The role of reactive carbonyl compounds. Biochem. Mosc. 2019, 84, S206–S224. [Google Scholar] [CrossRef]
- Chen, H.-J.C.; Chen, Y.-C.; Hsiao, C.-F.; Chen, P.-F. Mass spectrometric analysis of glyoxal and methylglyoxal-induced modifications in human hemoglobin from poorly controlled type 2 diabetes mellitus patients. Chem. Res. Toxicol. 2015, 28, 2377–2389. [Google Scholar] [CrossRef]
- Leoncini, G.; Maresca, M.; Ronchi, S.; Bonsignore, A. Studies on the inactivation of glyceraldehyde-3-phoshate dehydrogenase by methylglyoxal. Experientia 1981, 37, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Brahm, J.; Mortensen, H.B. Anion transport as related to hemoglobin A1c in erythrocytes of diabetic children. Clin. Chem. 1988, 34, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- Walder, J.A.; Chatterjee, R.; Steck, T.L.; Low, P.S.; Musso, G.F.; Kaiser, E.T.; Rogers, P.H.; Arnone, A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. J. Biol. Chem. 1984, 259, 10238–10246. [Google Scholar] [CrossRef]
- Chu, H.; Breite, A.; Ciraolo, P.; Franco, R.S.; Low, P.S. Characterization of the deoxyhemoglobin binding site on human erythrocyte band 3: Implications for O2 regulation of erythrocyte properties. Blood 2008, 111, 932–938. [Google Scholar] [CrossRef]
- Sega, M.F.; Chu, H.; Christian, J.A.; Low, P.S. Fluorescence assay of the interaction between hemoglobin and the cytoplasmic domain of erythrocyte membrane band 3. Blood Cells Mol. Dis. 2015, 55, 266–271. [Google Scholar] [CrossRef]
- Kosmachevskaya, O.V.; Nasybullina, E.I.; Blindar, V.N.; Topunov, A.F. Binding of erythrocyte hemoglobin to the membrane to realize signal-regulatory function. Appl. Biochem. Microbiol. 2019, 55, 83–98. [Google Scholar] [CrossRef]
- Li, G.; Liu, L.; Hu, H.; Zhao, Q.; Xie, F.; Chen, K.; Liu, S.; Chen, Y.; Shi, W.; Yin, D. Age-related carbonyl stress and erythrocyte membrane protein carbonylation. Clin. Hemorheol. Microcirc. 2010, 46, 305–311. [Google Scholar] [CrossRef]
- Breitling-Utzmann, C.M.; Unger, A.; Friedl, D.A.; Lederer, M.O. Identification and quantification of phosphatidylethanolamine-derived glucosylamines and aminoketoses from human erythrocytes—Influence of glycation products on lipid peroxidation. Arch. Biochem. Biophys. 2001, 391, 245–254. [Google Scholar] [CrossRef]
- Emelianov, V.V.; Leontev, D.V.; Ishchenko, A.V.; Bulavintseva, T.S.; Savateeva, E.A.; Danilova, I.G. Atomic force microscopy imaging of red blood cells and metabolic disorders in experimental diabetes mellitus and its correction with lipoic acid. Biophysics 2016, 61, 906–910. [Google Scholar] [CrossRef]
- Iwata, H.; Ukeda, H.; Maruyama, T.; Fujino, T.; Sawamura, M. Effect of carbonyl compounds on red blood cells deformability. Biochem. Biophys. Res. Commun. 2004, 321, 700–706. [Google Scholar] [CrossRef]
- Singh, M.; Shin, S. Changes in erythrocyte aggregation and deformability in diabetes mellitus: A brief review. Indian J. Exp. Biol. 2009, 47, 7–15. [Google Scholar] [PubMed]
- Chen, K.; Xie, F.; Liu, S.; Li, G.; Chen, Y.; Shi, W.; Hu, H.; Liu, L.; Yin, D. Plasma reactive carbonyl species: Potential risk factor for hypertension. Free Radic. Res. 2011, 45, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.; Mignemi, N.; Rose, K.; O’Rear, L.; Sarilla, S.; Hamm, H.E.; Barnett, J.V.; Verhamme, I.M.; Schoenecker, J. The hyperglycemic byproduct methylglyoxal impairs anticoagulant activity through covalent adduction of antithrombin III. Thromb. Res. 2014, 134, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Su, Y.; Cayabyab, F.S.; Liu, L. Endothelial Na+/H+ exchanger NHE1 participates in redox-sensitive leukocyte recruitment triggered by methylglyoxal. Cardiovasc. Diabetol. 2014, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Maillard, L.C. Action des acides amines sur les sucres; formation de melanoidines par voie méthodique (Action of amino acids on sugars. Formation of melanoidins in a methodical way). Compt. Rend. 1912, 154, 66–68. [Google Scholar]
- Hodge, J.E. Dehydrated foods, chemistry of browning reactions in model systems. J. Agric. Food Chem. 1953, 1, 928–943. [Google Scholar] [CrossRef]
- Allen, D.W.; Schroeder, W.A.; Balog, J. Observations on the chromatographic heterogeneity of normal adult and fetal human hemoglobin: A study of the effects of crystallization and chromatography on the heterogeneity and isoleucine content. J. Am. Chem. Soc. 1958, 80, 1628–1634. [Google Scholar] [CrossRef]
- Huisman, T.H.; Martis, E.A.; Dozy, A. Chromatography of hemoglobin types on carboxymethylcellulose. J. Lab. Clin. Med. 1958, 52, 312–327. [Google Scholar] [PubMed]
- Bookchin, R.M.; Gallop, P.M. Structure of haemoglobin A1c: Nature of the N-terminal beta chain blocking group. Biochem. Biophys. Res. Commun. 1968, 32, 86–93. [Google Scholar] [CrossRef]
- Peterson, K.P.; Pavlovich, J.G.; Goldstein, D.; Little, R.; England, J.; Peterson, C.M. What is hemoglobin A1c? An analysis of glycated hemoglobins by electrospray ionization mass spectrometry. Clin. Chem. 1998, 44, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, S. An abnormal hemoglobin in red cells of diabetics. Clin. Chim. Acta 1968, 22, 296–298. [Google Scholar] [CrossRef]
- Rahbar, S.; Blumenfeld, O.; Ranney, H.M. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem. Biophys. Res. Commun. 1969, 36, 838–843. [Google Scholar] [CrossRef]
- Bunn, H.F.; Haney, D.N.; Gabbay, K.H.; Gallop, P.M. Further identification of the nature and linkage of the carbohydrate in haemoglobin A1c. Biochem. Biophys. Res. Commun. 1975, 67, 103–109. [Google Scholar] [CrossRef]
- Koenig, R.J.; Peterson, C.M.; Jones, R.L.; Saudek, C.; Lehrman, M.; Cerami, A. Correlation of glucose regulation and hemoglobin AIc in diabetes mellitus. N. Engl. J. Med. 1976, 295, 417–420. [Google Scholar] [CrossRef]
- Gopalkrishnapillai, B.; Nadanathangam, V.; Karmakar, N.; Anand, S.; Misra, A. Evaluation of autofluorescent property of hemoglobin-advanced glycation end product as a long-term glycemic index of diabetes. Diabetes 2003, 52, 1041–1046. [Google Scholar] [CrossRef][Green Version]
- Rahbar, S. The discovery of glycated hemoglobin. A major event in the study of nonenzymatic chemistry in biological systems. Ann. N. Y. Acad. Sci. 2005, 1043, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Nirala, B.K.; Saluja, S.; Mishra, A.; Gohil, N.K. Erythrocyte ATPase activity in relation to hyperglycemia and hyperlipidemia in diabetic urban Indian population. Int. J. Diabetes Metab. 2012, 20, 75–79. [Google Scholar]
- Monnier, V.M. Nonenzymatic glycosylation, the Maillard reaction and aging process. J. Gerontol. 1990, 45, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Golubev, A.G. The other side of metabolism. Biochem. Mosc. 1996, 61, 1443–1460. [Google Scholar]
- Ahmed, M.U.; Frye, E.B.; Degenhardt, T.P.; Thorpe, S.R.; Baynes, J.W. Nε-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem. J. 1997, 324, 565–570. [Google Scholar] [CrossRef]
- Severin, F.F.; Feniouk, B.A.; Skulachev, V.P. Advanced glycation of cellular proteins as a possible basic component of the “master biological clock”. Biochem. Mosc. 2013, 78, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Nasybullina, E.I.; Nikitaev, V.G.; Pronichev, A.N.; Blindar, V.N.; Kosmachevskaya, O.V.; Topunov, A.F. Expert diagnostic system for hemoglobinopathies using the data on blood, erythrocyte, and hemoglobin state. Bull. Lebedev Phys. Inst. 2015, 42, 206–208. [Google Scholar] [CrossRef]
- Amendolia, S.R.; Brunetti, A.; Carta, P.; Cossu, G.; Ganadu, M.L.; Golosio, B.; Mura, G.M.; Pirastru, M.G. A real-time classification system of thalassemic pathologies based on artificial neural networks. Med. Decis. Making 2002, 22, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Plebani, M.J. Recent developments and innovations in red blood cells diagnostics. J. Lab. Precis. Med. 2018, 3, 68. [Google Scholar] [CrossRef]
- Stefanovic, A.; Jeremic, K.; Kadija, S.; Mitrovic, M.; Filimonovic, D.; Jankovic-Raznatovic, S.; Tavcar, J. Uterine tumor resembling ovarian sex cord tumor. Case report and review of literature. Eur. J. Gynaecol. Oncol. 2013, 34, 275–277. [Google Scholar]
- Rifkind, J.M.; Nagababu, E. Hemoglobin redox reactions and red blood cell aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef] [PubMed]
- Bryszewska, M. Interaction of normal and glycated human haemoglobin with erythrocyte membranes from normal and diabetic individuals. J. Clin. Chem. Clin. Biochem. 1988, 26, 809–813. [Google Scholar] [CrossRef]
- Marschner, J.P.; Rietbrock, N. Oxygen release kinetics in healthy subjects and diabetic patients. I: The role of 2,3-diphosphoglycerate. Int. J. Clin. Pharmacol. Ther. 1994, 32, 533–535. [Google Scholar] [PubMed]
- Antonelou, M.H.; Kriebardis, A.G.; Papassideri, I.S. Aging and death signalling in mature red cells: From basic science to transfusion practice. Blood Transfus. 2010, 8, 39–47. [Google Scholar] [CrossRef]
- Doctor, A.; Spinella, P. Effect of processing and storage on red blood cell function in vivo. Semin. Perinatol. 2012, 36, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Koltai, K.; Feher, G.; Kesmarky, G.; Keszthelyi, Z.; Czopf, L.; Toth, K. The effect of blood glucose levels on hemorheological parameters, platelet activation and aggregation in oral glucose tolerance tests. Clin. Hemorheol. Microcirc. 2006, 35, 517–525. [Google Scholar] [CrossRef]
- Buehler, P.W.; D’Agnillo, F. Toxicological consequences of extracellular hemoglobin: Biochemical and physiological perspectives. Antioxid. Redox. Signal. 2010, 12, 275–291. [Google Scholar] [CrossRef]
- Saleh, J. Glycated hemoglobin and its spinoffs: Cardiovascular disease markers or risk factors? World J. Cardiol. 2015, 7, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.J.; Basu, A. Biomarkers in diabetes: Hemoglobin A1c, vascular and tissue markers. Transl. Res. 2012, 159, 303–312. [Google Scholar] [CrossRef]
- Lundholm, M.D.; Ashraf, A.; Nadeem, S. Applications and pitfalls of hemoglobin A1C and alternative methods of glycemic monitoring. J. Diabetes Complicat. 2020, 34, 8. [Google Scholar] [CrossRef] [PubMed]
- Suo, M.; Wen, D.; Wang, W.; Zhang, T. Comparative study on hemoglobin A1c, glycated albumin and glycosylated serum protein in aplastic anemia patients with Type 2 diabetes mellitus. Biosci. Rep. 2020, 40, 5. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J. Glycation and biomarkers of vascular complications of diabetes. Amino Acids 2012, 42, 1171–1183. [Google Scholar] [CrossRef]
- Shamsaldeen, Y.A.; Mackenzie, L.S.; Lione, L.A.; Benham, C.D. Methylglyoxal, a metabolite increased in diabetes is associated with insulin resistance, vascular dysfunction and neuropathies. Curr. Drug Metab. 2016, 17, 359–367. [Google Scholar] [CrossRef]
- Kamble, P.S.; Pujari, K.N.; Jadkar, S.P. Estimation of serum methylglyoxal level in type 2 diabetes mellitus. Int. J. Biotech. Biochem. 2018, 14, 45–49. [Google Scholar]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Beisswenger, P.J. Methylglyoxal in diabetes: Link to treatment, glycaemic control and biomarkers of complications. Biochem. Soc. Trans. 2014, 42, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Sell, D.R.; Genuth, S. Glycation products as markers and predictors of the progression of diabetic complications. Ann. N. Y. Acad. Sci. 2005, 1043, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, E.G.; Lutgers, H.L.; Kleefstra, N.; Graaff, R.; Groenier, K.H.; Smit, A.J.; Gans, R.O.; Bilo, H.J. Skin autofluorescence: A tool to identify type 2 diabetic patients at risk for developing microvascular complications. Diabetes Care 2008, 31, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Shilton, B.H.; Walton, D.J. Sites of glycation of human and horse liver alcohol dehydrogenase in vivo. J Biol. Chem. 1991, 266, 5587–5592. [Google Scholar] [CrossRef]
- Johansen, M.B.; Kiemer, L.; Brunak, S. Analisis and prediction of mammalian protein glycation. Glycobiology 2006, 16, 844–853. [Google Scholar] [CrossRef]
- Kosmachevskaya, O.V.; Topunov, A.F. Hemoglobins: Diversity of structures and functions. Appl. Biochem. Microbiol. 2009, 45, 563–587. [Google Scholar] [CrossRef]
- Bhat, L.R.; Vedantham, S.; Krishnan, U.M.; Rayappan, J.B.B. Methylglyoxal—An emerging biomarker for diabetes mellitus diagnosis and its detection methods. Biosens. Bioelectron. 2019, 133, 107–124. [Google Scholar] [CrossRef]
- Ogawa, S.; Nakayama, K.; Nakayama, M.; Mori, T.; Matsushima, M.; Okamura, M.; Senda, M.; Nako, K.; Miyata, T.; Ito, S. Methylglyoxal is a predictor in type 2 diabetic patients of intima-media thickening and elevation of blood pressure. Hypertension 2010, 56, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Douglass, E.F., Jr.; Fitzgerald, K.J.; Spiegel, D.A. A “turn-on” fluorescent sensor for methylglyoxal. J. Am. Chem. Soc. 2013, 135, 12429–12433. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Wen, J.; Chen, A. Electrochemical determination of methylglyoxal as a biomarker in human plasma. Biosens. Bioelectron. 2013, 42, 349–354. [Google Scholar] [CrossRef]
- Wu, X.; Dai, Y.; Wang, L.; Peng, Y.; Lu, L.; Zhu, Y.; Shi, Y.; Zhuang, S. Diagnosis of methylglyoxal in blood by using far-infrared spectroscopy and o-phenylenediamine derivation. Biomed. Opt. Express 2020, 11, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Nwose, E.U.; Richards, R.S.; McDonald, S.; Jelinek, H.F. Assessment of diabetic macrovascular complications: A prediabetes model. Brit. J. Biomed. Sci. 2010, 67, 59–66. [Google Scholar] [CrossRef]
- Desai, K.M.; Chang, T.; Wang, H.; Banigesh, A.; Dhar, A.; Liu, J.; Untereiner, A.; Wu, L. Oxidative stress and aging: Is methylglyoxal the hidden enemy? Can. J. Physiol. Pharmacol. 2010, 88, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.H.; Isbell, T.S.; Huang, Z.; Shiva, S.; Chacko, B.K.; Schechter, A.N.; Darley-Usmar, V.M.; Kerby, J.D.; Lang, J.D., Jr.; Kraus, D.; et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 2006, 107, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Kosmachevskaya, O.V.; Shumaev, K.B.; Nasybullina, E.I.; Topunov, A.F. Formation of nitri- and nitrosylhemoglobin in systems modeling the Maillard reaction. Clin. Chem. Lab. Med. 2014, 52, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kosmachevskaya, O.V.; Topunov, A.F. Alternate and additional functions of erythrocyte hemoglobin. Biochem. Mosc. 2018, 83, 1575–1593. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Moriyama, H. Mechanisms of hemoglobin adaptation to high altitude hypoxia. High Alt. Med. Biol. 2008, 9, 148–157. [Google Scholar] [CrossRef]
- McMahon, T.J.; Stone, A.E.; Bonaventura, J.; Stamler, J.S. S-nitrosylation of hemoglobin increases its oxygen affinity. Am. J. Respir. Crit. Care Med. 1999, 159, 352. [Google Scholar]
- Stepuro, T.L.; Zinchuk, V.V. Nitric oxide effect on the hemoglobin-oxygen affinity. J. Physiol. Pharmacol. 2006, 57, 29–38. [Google Scholar]
- Kuhn, V.; Diederich, L.; Keller, T.C.S., IV; Kramer, C.M.; Lückstädt, W.; Panknin, C.; Suvorava, T.; Isakson, B.E.; Kelm, M.; Cortese-Krott, M.M. Red blood cell function and dysfunction: Redox regulation, nitric oxide metabolism, anemia. Antioxid. Redox Signal. 2017, 26, 718–742. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, M.; Sharma, T.K.; Singh, I.; Singh, N.; Ghalaut, V.S.; Vardey, S.K.; Shankar, V. Antioxidant enzymes and lipid peroxidation in type 2 diabetes mellitus patients with and without nephropathy. N. Am. J. Med. Sci. 2013, 5, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Aouacheri, O.; Saka, S.; Krim, M.; Messaadia, A.; Maidi, I. The investigation of the oxidative stress-related parameters in type 2 diabetes mellitus. Can. J. Diabetes. 2015, 39, 44–49. [Google Scholar] [CrossRef]
- Waggiallah, H.; Alzohairy, M. The effect of oxidative stress on human red cells glutathione peroxidase, glutathione reductase level, and prevalence of anemia among diabetics. N. Am. J. Med. Sci. 2011, 3, 344–347. [Google Scholar] [CrossRef]
- Kondo, T. Impaired glutathione metabolism in hemolytic anemia. Rinsho Byori 1990, 38, 355–359. [Google Scholar]
- Hailu, N.A.; Tolessa, T.; Gufue, Z.H.; Tsegay, E.W.; Tekola, K.B. The magnitude of anemia and associated factors among adult diabetic patients in tertiary teaching hospital, Northern Ethiopia, 2019, cross-sectional study. PLoS ONE 2020, 15, 0240678. [Google Scholar] [CrossRef]
- Dincer, Y.; Akcay, T.; Alademir, Z.; Ilkova, H. Effect of oxidative stress on glutathione pathway in red blood cells from patients with insulin-dependent diabetes mellitus. Metabolism 2002, 51, 1360–1362. [Google Scholar] [CrossRef]
- Beard, K.M.; Shangari, N.; Wu, B.; O’Brien, P.J. Metabolism, not autoxidation, plays a role in α-oxoaldehyde- and reducing sugar-induced erythrocyte GSH depletion: Relevance for diabetes mellitus. Mol. Cell. Biochem. 2003, 252, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Whillier, S.; Raftos, J.E.; Kuchel, P.W. Glutathione synthesis by red blood cells in type 2 diabetes mellitus. Redox Rep. 2008, 13, 277–282. [Google Scholar] [CrossRef]
- Zavodnik, L.; Zavodnik, I.B.; Niekurzak, A.; Szosland, K. Activation of red blood cell glutathione peroxidase and morphological transformation of erythrocytes under the action of tert-butyl hydroperoxide. Biochem. Mol. Biol. Int. 1998, 44, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Giugliano, D.; Pizza, G.; Gambardella, A.; Varricchio, M.; D’Onofrio, F. Glutathione infusion potentiates glucose-induced insulin secretion in aged patients with impaired glucose tolerance. Diabetes Care 1992, 15, 1–7. [Google Scholar] [CrossRef]
- Prestes, A.S.; Dos Santos, M.M.; Ecker, A.; Zanini, D.; Schetinger, M.R.; Rosemberg, D.B.; da Rocha, J.B.; Barbosa, N.V. Evaluation of methylglyoxal toxicity in human erythrocytes, leukocytes and platelets. Toxicol. Mech. Method. 2017, 27, 307–317. [Google Scholar] [CrossRef]
- Turpin, C.; Catan, A.; Guerin-Dubourg, A.; Debussche, X.; Bravo, S.B.; Álvarez, E.; Van Den Elsen, J.; Meilhac, O.; Rondeau, P.; Bourdon, E. Enhanced oxidative stress and damage in glycated erythrocytes. PLoS ONE 2020, 15, 0235335. [Google Scholar] [CrossRef]
- Anandan, S.; Mahadevamurthy, M.; Ansari, M.A.; Alzohairy, M.A.; Alomary, M.N.; Farha Siraj, S.; Halugudde Nagaraja, S.; Chikkamadaiah, M.; Thimappa Ramachandrappa, L.; Naguvanahalli Krishnappa, H.K.; et al. Biosynthesized ZnO-NPs from Morus indica attenuates methylglyoxal-induced protein glycation and RBC damage: In-vitro, in-vivo and molecular docking study. Biomolecules 2019, 9, 882. [Google Scholar] [CrossRef] [PubMed]
- Aarushi, B.; Shankar, H.; Swati, S.A. Comparative analysis of haematological parameters in diabetics and non diabetics and their correlation with fasting blood sugar levels and glycated haemoglobin. Indian J. Pathol. Res. Pract. 2020, 9, 1. [Google Scholar] [CrossRef]
- Adane, T.; Getaneh, Z.; Asrie, F. Red blood cell parameters and their correlation with renal function tests among diabetes mellitus patients: A comparative cross-sectional study. Diabetes Metab. Syndr. Obes. 2020, 2020, 3937–3946. [Google Scholar] [CrossRef] [PubMed]
- Biadgo, B.; Melku, M.; Abebe, S.M.; Abebe, M. Hematological indices and their correlation with fasting blood glucose level and anthropometric measurements in type 2 diabetes mellitus patients in Gondar, Northwest Ethiopia. Diabetes Metab. Syndr. Obes. 2016, 2016, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal acts as a tumor-promoting factor in anaplastic thyroid cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef]
- Maessen, D.E.M.; Stehouwer, C.D.A.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef]
- Freund, M.A.; Chen, B.; Decker, E.A. The inhibition of advanced glycation end products by carnosine and other natural dipeptides to reduce diabetic and age-related complications. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Hipkiss, A.R.; Cartwright, S.P.; Bromley, C.; Gross, S.R.; Bill, R.M. Carnosine: Can understanding its actions on energy metabolism and protein homeostasis inform its therapeutic potential? Chem. Cent. J. 2013, 7, 38. [Google Scholar] [CrossRef]
- Renner, C.; Asperger, A.; Seyffarth, A.; Meixensberger, J.; Gebhardt, R.; Gaunitz, F. Carnosine inhibits ATP production in cells from malignant glioma. Neurol. Res. 2010, 32, 101–105. [Google Scholar] [CrossRef]
- Ikeda, T.; Kimura, K.; Hama, T.; Tamaki, N. Activation of rabbit muscle fructose 1,6-bisphosphatase by histidine and carnosine. J. Biochem. 1980, 87, 179–185. [Google Scholar] [CrossRef]
- Prokopieva, V.D.; Bohan, N.A.; Johnson, P.; Abe, H.; Boldyrev, A.A. Effects of carnosine and related compounds on the stability and morphology of erythrocytes from alcoholics. Alcohol Alcohol. 2000, 35, 44–48. [Google Scholar] [CrossRef]
- Aydogan, S.; Yapislar, H.; Artis, S.; Aydogan, B. Impaired erythrocytes deformability in H2O2-induced oxidative stress: Protective effect of L-carnosine. Clin. Hemorheol. Microcirc. 2008, 39, 93–98. [Google Scholar] [CrossRef]
- Yapislar, H.; Aydogan, S. Effect of carnosine on erythrocyte deformability in diabetic rats. Arch. Physiol. Biochem. 2012, 118, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yapislar, H.; Taskin, E. L-carnosine alters some hemorheologic and lipid peroxidation parameters in nephrectomized rats. Med. Sci. Monit. 2014, 20, 399–405. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Misiti, F. Erythrocytes as potential link between diabetes and alzheimer’s disease. Front. Aging Neurosci. 2017, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.-H.; Kim, C.-B.; Shin, S. The effect of L-carnosine on the rheological characteristics of erythrocytes incubated in glucose media. Korea Aust. Rheol. J. 2009, 21, 103–108. [Google Scholar]
- Shumaev, K.B.; Kosmachevskaya, O.V.; Timoshin, A.A.; Vanin, A.F.; Topunov, A.F. Dinitrosyl iron complexes bound with haemoglobin as markers of oxidative stress. Methods Enzymol. 2008, 436, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Shumaev, K.B.; Gubkin, A.A.; Serezhenkov, V.A.; Lobysheva, I.I.; Kosmachevskaya, O.V.; Ruuge, E.K.; Lankin, V.Z.; Topunov, A.F.; Vanin, A.F. Interaction of reactive oxygen and nitrogen species with albumin- and hemoglobin-bound dinitrosyl iron complexes. Nitric Oxide 2008, 18, 37–46. [Google Scholar] [CrossRef]
- Shumaev, K.B.; Gorudko, I.V.; Kosmachevskaya, O.V.; Grigoryeva, D.V.; Panasenko, O.M.; Vanin, A.F.; Topunov, A.F.; Terekhova, M.S.; Sokolov, A.V.; Cherenkevich, S.N.; et al. Effect of dinitrosyl iron complexes with glutathione on red blood cell lysis by hypochlorous acid. Oxid. Med. Cell. Longev. 2019, 2019, 2798154. [Google Scholar] [CrossRef]
- Shumaev, K.B.; Kosmachevskaya, O.V.; Nasybullina, E.I.; Gromov, S.V.; Novikov, A.A.; Topunov, A.F. New dinitrosyl iron complexes bound with physiologically active dipeptide carnosine. J. Biol. Inorg. Chem. 2017, 22, 153–160. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Galiniak, S.; Bartosz, G. Polyphenols protect against protein glycoxidation. Free Radic. Biol. Med. 2014, 1, S47. [Google Scholar] [CrossRef] [PubMed]
- Maruf, A.A.; Lip, H.Y.; Wong, H.; O’Brien, P.J. Protective effects of ferulic acid and related polyphenols against glyoxal- or methylglyoxal-induced cytotoxicity and oxidative stress in isolated rat hepatocytes. Chem. Biol. Interact. 2015, 234, 96–105. [Google Scholar] [CrossRef]
- Yeh, W.J.; Hsia, S.M.; Lee, W.H.; Wu, C.H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Cianfruglia, L.; Morresi, C.; Bacchetti, T.; Armeni, T.; Ferretti, G. Protection of polyphenols against glyco-oxidative stress: Involvement of glyoxalase pathway. Antioxidants 2020, 9, 1006. [Google Scholar] [CrossRef] [PubMed]
- Iofrida, C.; Daniele, S.; Pietrobono, D.; Fusi, J.; Galetta, F.; Trincavelli, M.L.; Bonuccelli, U.; Franzoni, F.; Martini, C. Influence of physical exercise on β-amyloid, α-synuclein and tau accumulation: An in vitro model of oxidative stress in human red blood cells. Arch. Ital. Biol. 2017, 155, 33–42. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosmachevskaya, O.V.; Novikova, N.N.; Topunov, A.F. Carbonyl Stress in Red Blood Cells and Hemoglobin. Antioxidants 2021, 10, 253. https://doi.org/10.3390/antiox10020253
Kosmachevskaya OV, Novikova NN, Topunov AF. Carbonyl Stress in Red Blood Cells and Hemoglobin. Antioxidants. 2021; 10(2):253. https://doi.org/10.3390/antiox10020253
Chicago/Turabian StyleKosmachevskaya, Olga V., Natalia N. Novikova, and Alexey F. Topunov. 2021. "Carbonyl Stress in Red Blood Cells and Hemoglobin" Antioxidants 10, no. 2: 253. https://doi.org/10.3390/antiox10020253
APA StyleKosmachevskaya, O. V., Novikova, N. N., & Topunov, A. F. (2021). Carbonyl Stress in Red Blood Cells and Hemoglobin. Antioxidants, 10(2), 253. https://doi.org/10.3390/antiox10020253