Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer


Abstract
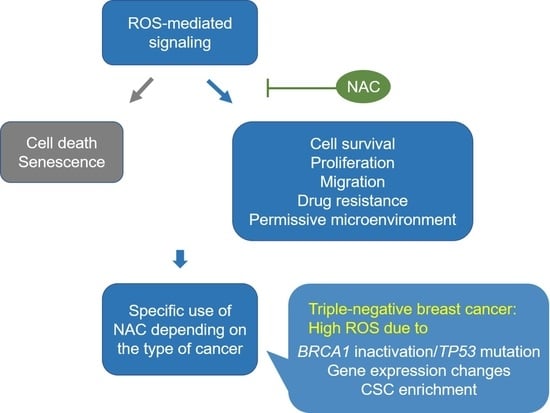
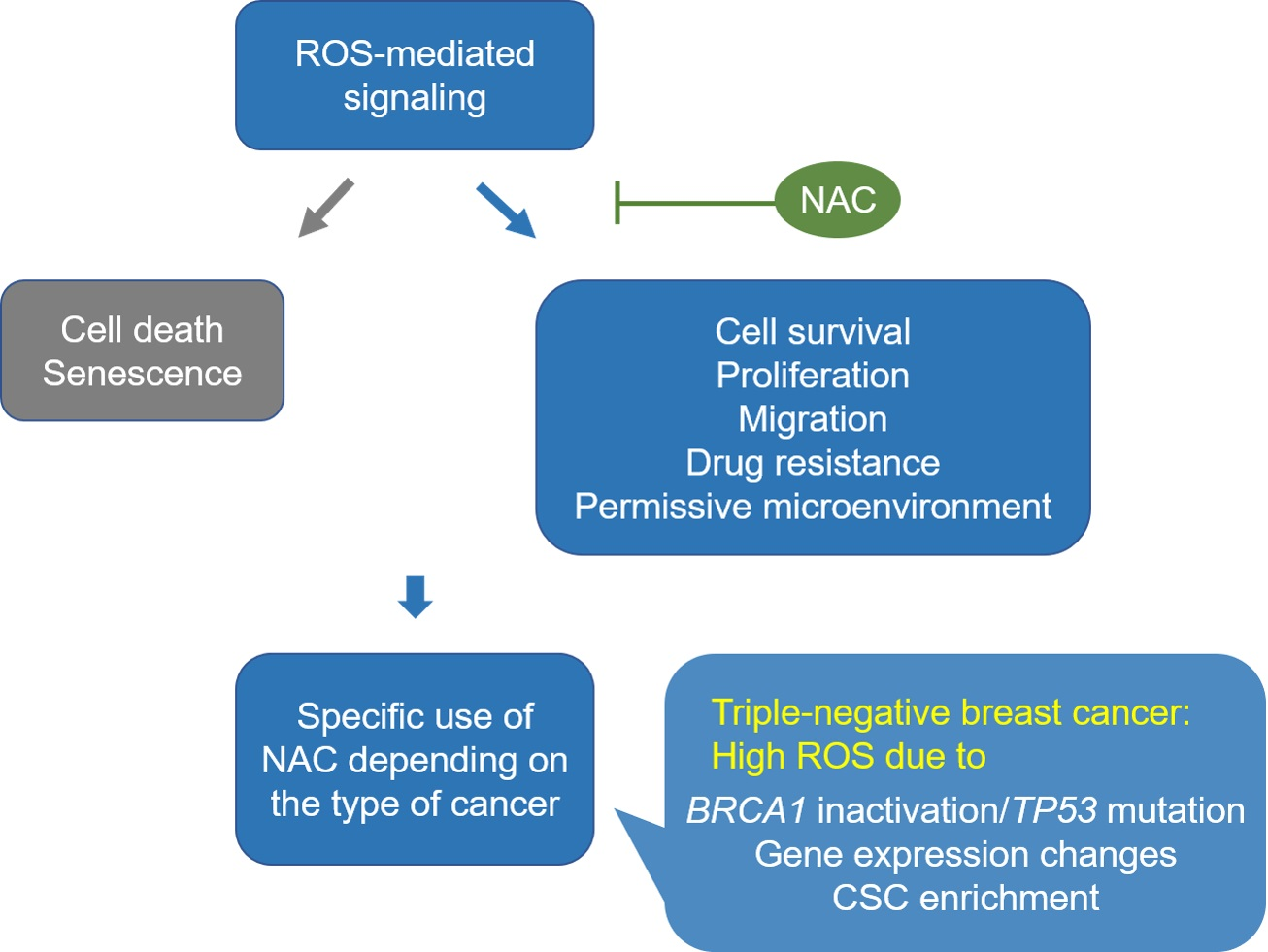{kind=link}
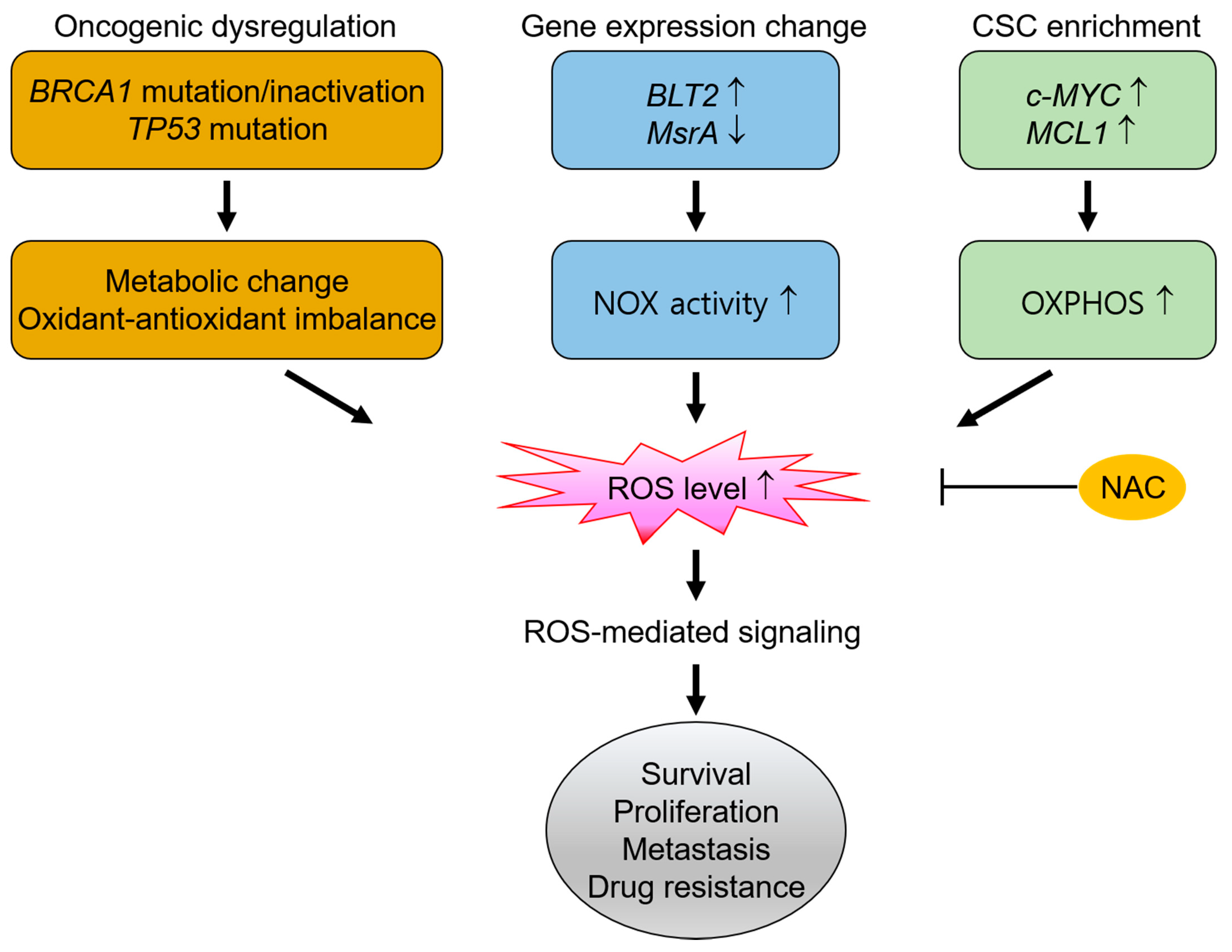{kind=link}
{kind=link}
1. Introduction
2. The Formation and Elimination of ROS
3. The Function of ROS as Signaling Molecules
4. The Effect of Antioxidant Supplementation upon Cancer Development and Progression
5. The Therapeutic Potetial of NAC in TNBC
5.1. The Dependence on ROS for the Survival and Malignant Progression in TNBC
5.1.1. High ROS Production Derived from BRCA1 Inactivation and TP53 Mutation in TNBC
5.1.2. High ROS Production Derived from Gene Expression Changes in TNBC
5.1.3. High ROS Production by Cancer Stem-Like Cells in TNBC
5.2. The Interplay between Cancer Cells and the Tumor Microenvironment via ROS
5.2.1. Conversion to the Cancer-Associated Fibroblast Phenotype via ROS
5.2.2. Conversion to Tumor-associated Macrophage Phenotype via ROS
5.2.3. Activation of Hypoxic Responses via ROS-Mediated Tumor-Stromal Interaction
6. Conclusions
Funding
Conflicts of Interest
References
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.O.; Shay, K.P.; Kelley, A.R.; Butler, J.A.; Hagen, T.M. Glutathione maintenance mitigates age-related susceptibility to redox cycling agents. Redox Biol. 2016, 10, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Coles, L.; Kartha, R.; Terpstra, M.; Oz, G.; Hovde, L.; Cloyd, J.; Tuite, P. Effect of High-Dose Oral NAC on Systemic Measures of Oxidative Stress in Individuals with Parkinson Disease and Healthy Elderly Subjects. Mov. Disord. 2016, 31, E13. [Google Scholar]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljsak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef]
- Altomare, A.; Baron, G.; Brioschi, M.; Longoni, M.; Butti, R.; Valvassori, E.; Tremoli, E.; Carini, M.; Agostoni, P.; Vistoli, G.; et al. N-Acetyl-Cysteine Regenerates Albumin Cys34 by a Thiol-Disulfide Breaking Mechanism: An Explanation of Its Extracellular Antioxidant Activity. Antioxidants 2020, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef]
- Monti, D.; Sotgia, F.; Whitaker-Menezes, D.; Tuluc, M.; Birbe, R.; Berger, A.; Lazar, M.; Cotzia, P.; Draganova-Tacheva, R.; Lin, Z.; et al. Pilot study demonstrating metabolic and anti-proliferative effects of in vivo anti-oxidant supplementation with N-Acetylcysteine in Breast Cancer. Semin. Oncol. 2017, 44, 226–232. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.R.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; de Nicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y. Food-derived polyphenols inhibit the growth of ovarian cancer cells irrespective of their ability to induce antioxidant responses. Heliyon 2018, 4, e00753. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef]
- Dedon, P.C.; Tannenbaum, S.R. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004, 423, 12–22. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; de Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 1–19. [Google Scholar] [CrossRef]
- Wert, K.J.; Velez, G.; Cross, M.R.; Wagner, B.A.; Teoh-Fitzgerald, M.L.; Buettner, G.R.; McAnany, J.J.; Olivier, A.; Tsang, S.H.; Harper, M.M.; et al. Extracellular superoxide dismutase (SOD3) regulates oxidative stress at the vitreoretinal interface. Free Radic. Biol. Med. 2018, 124, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.B.; Tephly, L.A.; Venkataraman, S.; Oberley, L.W.; Zhang, Y.; Buettner, G.R.; Spitz, D.R.; Hunninghake, G.W. High Levels of Catalase and Glutathione Peroxidase Activity Dampen H2O2Signaling in Human Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2004, 31, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Berndt, C.; Lillig, C.H.; Flohã, L. Redox regulation by glutathione needs enzymes. Front. Pharmacol. 2014, 5, 168. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen Peroxide Sensing and Signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef]
- Kawasaki, T.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Aulak, K.S.; Koeck, T.; Crabb, J.W.; Stuehr, D.J. Dynamics of protein nitration in cells and mitochondria. Am. J. Physiol. Circ. Physiol. 2004, 286, H30–H38. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative Stress and Lipid Peroxidation Products in Cancer Progression and Therapy. ISRN Oncol. 2012, 2012, 1–21. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [PubMed]
- Titorenko, V.I.; Terlecky, S.R. Peroxisome Metabolism and Cellular Aging. Traffic 2010, 12, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes Are Oxidative Organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef]
- Wang, B.; van Veldhoven, P.P.; Brees, C.; Rubio, N.; Nordgren, M.; Apanasets, O.I.E.; Kunze, M.; Baes, M.; Agostinis, P.; Fransen, M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic. Biol. Med. 2013, 65, 882–894. [Google Scholar] [CrossRef]
- di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1–44. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Ogasawara, M.; Zhang, H. Redox Regulation and Its Emerging Roles in Stem Cells and Stem-Like Cancer Cells. Antioxid. Redox Signal. 2009, 11, 1107–1122. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Gross, E.; Kastner, D.B.; A Kaiser, C.; Fass, D. Structure of Ero1p, Source of Disulfide Bonds for Oxidative Protein Folding in the Cell. Cell 2004, 117, 601–610. [Google Scholar] [CrossRef]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative Innate Immune Defenses by Nox/Duox Family NADPH Oxidases. Infect. Inflamm. Impacts Oncog. 2008, 15, 164–187. [Google Scholar] [CrossRef]
- Zana, M.; Péterfi, Z.; Kovács, H.A.; Tóth, Z.E.; Enyedi, B.; Morel, F.; Paclet, M.-H.; Morand, S.; Leto, T.L. Interaction between p22phox and Nox4 in the endoplasmic reticulum suggests a unique mechanism of NADPH oxidase complex formation. Free Radic. Biol. Med. 2018, 116, 41–49. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2009, 5, 47–62. [Google Scholar] [CrossRef]
- Allen, E.M.; Mieyal, J.J. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Benov, L.T. How superoxide radical damages the cell. Protoplasma 2001, 217, 33–36. [Google Scholar] [CrossRef]
- Pani, G.; Bedogni, B.; Colavitti, R.; Anzevino, R.; Borrello, S.; Galeotti, T. Cell Compartmentalization in Redox Signaling. IUBMB Life 2001, 52, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, Y.; Loscalzo, J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10813–10817. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Brees, C.; Knoops, B.; van Veldhoven, P.P.; Fransen, M. Peroxisomes as Modulators of Cellular Protein Thiol Oxidation: A New Model System. Antioxid. Redox Signal. 2019, 30, 22–39. [Google Scholar] [CrossRef]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Barygina, V.; Becatti, M.; Prignano, F.; Lotti, T.; Taddei, N.; Fiorillo, C. Fibroblasts to Keratinocytes Redox Signaling: The Possible Role of ROS in Psoriatic Plaque Formation. Antioxidants 2019, 8, 566. [Google Scholar] [CrossRef]
- Hawkins, B.J.; Madesh, M.; Kirkpatrick, C.J.; Fisher, A.B. Superoxide Flux in Endothelial Cells via the Chloride Channel-3 Mediates Intracellular Signaling. Mol. Biol. Cell 2007, 18, 2002–2012. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Nishino, H.; Tokuda, H.; Satomi, Y.; Masuda, M.; Osaka, Y.; Yogosawa, S.; Wada, S.; Mou, X.Y.; Takayasu, J.; Murakoshi, M.; et al. Cancer prevention by antioxidants. BioFactors 2004, 22, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Wada, S. Cancer preventive effects of vitamin E. Curr. Pharm. Biotechnol. 2012, 13, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Collins, A. Antioxidant intervention as a route to cancer prevention. Eur. J. Cancer 2005, 41, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Kim, Y.-W.; Ju, W.; Choi, H.J.; Bae, W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol. 2009, 21, 166–179. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.; Tangen, C.M.; Lucia, M.S.; Goodman, P.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; Karp, D.D.; et al. Vitamin E and the risk of prostate cancer: Updated results of the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J. Clin. Oncol. 2012, 30, 7. [Google Scholar] [CrossRef]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.-C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nat. Cell Biol. 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; de Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; di Donato, M.; Pezone, A.; di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Courtney, M.J.; Laukkanen, M.O. Redox-Activated Signal Transduction Pathways Mediating Cellular Functions in Inflammation, Differentiation, Degeneration, Transformation, and Death. Oxid. Med. Cell. Longev. 2016, 2016, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2010, 24, 157–167. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2009, 220, 263–280. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.J.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; André, F.; Bergh, J.; et al. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Ellis, P.; Tutt, A.N. Molecular heterogeneity of triple-negative breast cancer and its clinical implications. Curr. Opin. Oncol. 2011, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Guideline Update. Arch. Pathol. Lab. Med. 2020, 144, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, F.; Fontanella, C.; Amoroso, V.; Bianchi, G.V.; Bisagni, G.; Falci, C.; Fontana, A.; Generali, D.; Gianni, L.; Grassadonia, A.; et al. Current challenges in HER2-positive breast cancer. Crit. Rev. Oncol. 2016, 98, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Artacho-Cordón, A.; Artacho-Cordon, F.; Ríos-Arrabal, S.; Calvente, I.; Núñez, M.I. Tumor microenvironment and breast cancer progression. Cancer Biol. Ther. 2012, 13, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Smith, B.D.; Zhou, Y.; Kaufman, M.D.; Godwin, A.K. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene 2013, 34, 144–153. [Google Scholar] [CrossRef]
- Kwon, Y.; Godwin, A.K. Regulation of HGF and c-MET Interaction in Normal Ovary and Ovarian Cancer. Reprod. Sci. 2016, 24, 494–501. [Google Scholar] [CrossRef]
- Ouyang, L.; Chang, W.; Fang, B.; Qin, J.; Qu, X.; Cheng, F. Estrogen-induced SDF-1α production promotes the progression of ER-negative breast cancer via the accumulation of MDSCs in the tumor microenvironment. Sci. Rep. 2016, 6, 39541. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Balliet, R.M.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Hereditary ovarian cancer and two-compartment tumor metabolism: Epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFkappaB activation in the tumor stroma. Cell Cycle 2012, 11, 4152–4166. [Google Scholar] [CrossRef][Green Version]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Göbl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle 2010, 9, 3276–3296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.L.; Linnoila, I.; Liu, Z.-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- de Berardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef]
- le Bleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman-Kocher, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Hubalek, M.; Czech, T.; Müller, H. Biological Subtypes of Triple-Negative Breast Cancer. Breast Care 2017, 12, 8–14. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Santonja, A.; Sánchez-Muñoz, A.; Lluch, A.; Chica-Parrado, M.R.; Albanell, J.; Chacón, J.I.; Antolín, S.; Jerez, J.M.; de la Haba, J.; de Luque, V.; et al. Triple negative breast cancer subtypes and pathologic complete response rate to neoadjuvant chemotherapy. Oncotarget 2018, 9, 26406–26416. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Salinas, F.L.; Delgado-Magallón, A.; Montes-Alvarado, J.B.; Ramírez-Ramírez, D.; Flores-Alonso, J.C.; Cortés-Hernández, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast Cancer Subtypes Present a Differential Production of Reactive Oxygen Species (ROS) and Susceptibility to Antioxidant Treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast 2015, 24, S36–S40. [Google Scholar] [CrossRef]
- Boukerroucha, M.; Josse, C.; El-Guendi, S.; Boujemla, B.; Frères, P.; Marée, R.; Wenric, S.; Segers, K.; Collignon, J.; Jerusalem, G.; et al. Evaluation of BRCA1-related molecular features and microRNAs as prognostic factors for triple negative breast cancers. BMC Cancer 2015, 15, 1–10. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Wei, M.; Grushko, T.A.; Dignam, J.; Hagos, F.; Nanda, R.; Sveen, L.; Xu, J.; Fackenthal, J.; Tretiakova, M.; Das, S.; et al. BRCA1 Promoter Methylation in Sporadic Breast Cancer Is Associated with Reduced BRCA1 Copy Number and Chromosome 17 Aneusomy. Cancer Res. 2005, 65, 10692–10699. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Bae, I. BRCA1 and Oxidative Stress. Cancers 2014, 6, 771–795. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Rih, J.K.; Rosen, E.M. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS Lett. 2009, 583, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Yu, X. Tissue-Specific Carcinogens as Soil to Seed BRCA1/2-Mutant Hereditary Cancers. Trends Cancer 2020, 6, 559–568. [Google Scholar] [CrossRef]
- Li, M.; Chen, Q.; Yu, X. Chemopreventive Effects of ROS Targeting in a Murine Model of BRCA1-Deficient Breast Cancer. Cancer Res. 2017, 77, 448–458. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-Induced Up-Regulation of MnSOD and GPx but not Catalase Increases Oxidative Stress and Apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef]
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.-P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev. 2018, 32, 230–243. [Google Scholar] [CrossRef]
- Dhar, S.K.; Xu, Y.; Chen, Y.; Clair, D.K.S. Specificity Protein 1-dependent p53-mediated Suppression of Human Manganese Superoxide Dismutase Gene Expression. J. Biol. Chem. 2006, 281, 21698–21709. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 3010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-A.; Lee, J.-W.; Kim, H.; Kim, E.-Y.; Seo, J.-M.; Ko, J.; Kim, J.-H. Pro-survival of estrogen receptor-negative breast cancer cells is regulated by a BLT2–reactive oxygen species-linked signaling pathway. Carcinogenesis 2009, 31, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Uchida, T.; Yoshie, T.; Mizote, Y.; Ishikawa, F.; Katsuyama, M.; Shibanuma, M. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS -activated cancer cells. FEBS J. 2019, 286, 459–478. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jang, J.-H.; Park, G.-S.; Chung, Y.; You, H.J.; Kim, J.H. BLT2, a leukotriene B4 receptor 2, as a novel prognostic biomarker of triple-negative breast cancer. BMB Rep. 2018, 51, 373–377. [Google Scholar] [CrossRef]
- de Luca, A.; Sanna, F.; Sallese, M.; Ruggiero, C.; Grossi, M.; Sacchetta, P.; Rossi, C.; de Laurenzi, V.; di Ilio, C.; Favaloro, B. Methionine sulfoxide reductase A down-regulation in human breast cancer cells results in a more aggressive phenotype. Proc. Natl. Acad. Sci. USA 2010, 107, 18628–18633. [Google Scholar] [CrossRef]
- Svineng, G.; Ravuri, C.; Rikardsen, O.; Huseby, N.-E.; Winberg, J.-O. The Role of Reactive Oxygen Species in Integrin and Matrix Metalloproteinase Expression and Function. Connect. Tissue Res. 2008, 49, 197–202. [Google Scholar] [CrossRef]
- Yin, H.-L.; Wu, C.-C.; Lin, C.-H.; Chai, C.-Y.; Hou, M.-F.; Chang, S.-J.; Tsai, H.-P.; Hung, W.-C.; Pan, M.-R.; Luo, C.-W. β1 Integrin as a Prognostic and Predictive Marker in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1432. [Google Scholar] [CrossRef]
- Valdivia, C.D.A.; Duran, C.; Martin, A.S. The role of Nox-mediated oxidation in the regulation of cytoskeletal dynamics. Curr. Pharm. Des. 2015, 21, 6009–6022. [Google Scholar] [CrossRef]
- Honoré, S.; Kovacic, H.; Pichard, V.; Briand, C.; Rognoni, J.-B. α2β1-integrin signaling by itself controls G1/S transition in a human adenocarcinoma cell line (Caco-2): Implication of NADPH oxidase-dependent production of ROS. Exp. Cell Res. 2003, 285, 59–71. [Google Scholar] [CrossRef]
- Zhu, P.; Tan, M.J.; Huang, R.-L.; Tan, C.K.; Chong, H.C.; Pal, M.; Lam, C.R.I.; Boukamp, P.; Pan, J.Y.; Tan, S.H.; et al. Angiopoietin-like 4 Protein Elevates the Prosurvival Intracellular O2−:H2O2 Ratio and Confers Anoikis Resistance to Tumors. Cancer Cell 2011, 19, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lee, S.H.; Wu, S. Effects of N-acetyl-L-cysteine on adhesive strength between breast cancer cell and extracellular matrix proteins after ionizing radiation. Life Sci. 2013, 93, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Qiao, J.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Bao, B.; Mitrea, C.; Wijesinghe, P.; Marchetti, L.; Girsch, E.; Farr, R.L.; Boerner, J.L.; Mohammad, R.; Dyson, G.; Terlecky, S.R.; et al. Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity. Sci. Rep. 2017, 7, 44125. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276. [Google Scholar] [CrossRef]
- Qian, X.; Nie, X.; Yao, W.; Klinghammer, K.; Sudhoff, H.; Kaufmann, A.M.; Albers, A.E. Reactive oxygen species in cancer stem cells of head and neck squamous cancer. Semin. Cancer Biol. 2018, 53, 248–257. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Balliet, R.; Lin, Z.; Whitaker-Menezes, D.; Birbe, R.C.; Bombonati, A.; Pavlides, S.; Lamb, R.; Sneddon, S.; Howell, A.; et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: Implications for breast cancer prevention with antioxidant therapies. Cell Cycle 2012, 11, 4402–4413. [Google Scholar] [CrossRef]
- Alili, L.; Sack, M.; Puschmann, K.; Brenneisen, P. Fibroblast-to-myofibroblast switch is mediated by NAD(P)H oxidase generated reactive oxygen species. Biosci. Rep. 2014, 34, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.S.K.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2018, 8, e2562. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Park, G.-Y.; Pathak, H.B.; Godwin, A.K.; Kwon, Y. Epithelial-stromal communication via CXCL1-CXCR2 interaction stimulates growth of ovarian cancer cells through p38 activation. Cell. Oncol. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J. Natl. Cancer Inst. 2018, 110, 109–120. [Google Scholar] [CrossRef]
- Liu, L.; Liu, L.; Yao, H.H.; Zhu, Z.Q.; Ning, Z.L.; Huang, Q. Stromal Myofibroblasts Are Associated with Poor Prognosis in Solid Cancers: A Meta-Analysis of Published Studies. PLoS ONE 2016, 11, e0159947. [Google Scholar] [CrossRef]
- Tsujino, T.; Seshimo, I.; Yamamoto, H.; Ngan, C.Y.; Ezumi, K.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; Matsuura, N.; Monden, M. Stromal Myofibroblasts Predict Disease Recurrence for Colorectal Cancer. Clin. Cancer Res. 2007, 13, 2082–2090. [Google Scholar] [CrossRef]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2010, 19, 170–176. [Google Scholar] [CrossRef]
- Tobar, N.; Toyos, M.; Urra, C.; Méndez, N.; Arancibia, R.; Smith, P.C.; Martínez, J. c-Jun N terminal kinase modulates NOX-4 derived ROS production and myofibroblasts differentiation in human breast stromal cells. BMC Cancer 2014, 14, 640. [Google Scholar] [CrossRef]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Dürr, P.; Berger, P.B. ROS Signaling by NOX4 Drives Fibroblast-to-Myofibroblast Differentiation in the Diseased Prostatic Stroma. Mol. Endocrinol. 2011, 25, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Whitaker-Menezes, D.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Witkiewicz, A.K.; Heiden, M.G.V.; Migneco, G.; Chiavarina, B.; et al. The reverse Warburg Effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010, 9, 1960–1971. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Dasgupta, A.; Sammons, S.; Er, O.; Potoczek, M.B.; Guiles, F.; Sotgia, F.; Brody, J.R.; Mitchell, E.P.; Lisanti, M.P. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol. Ther. 2010, 10, 135–143. [Google Scholar] [CrossRef]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Dou, H.; Bagi, Z.; Han, W.; Su, Y.; Fulton, D.J.R. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Macharia, L.W.; Wanjiru, C.M.; Mureithi, M.W.; Pereira, C.M.; Ferrer, V.P.; Neto, V.M. MicroRNAs, Hypoxia and the Stem-Like State as Contributors to Cancer Aggressiveness. Front. Genet. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia—A mecha-nism of O-2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Tan, E.Y.; Yan, M.; Campo, L.; Han, C.; Takano, E.; Turley, H.; Candiloro, I.; Pezzella, F.; Gatter, K.C.; Millar, E.K.A.; et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br. J. Cancer 2009, 100, 405–411. [Google Scholar] [CrossRef]
- Bernardi, R.; Gianni, L. Hallmarks of triple negative breast cancer emerging at last? Cell Res. 2014, 24, 904–905. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nat. Cell Biol. 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nat. Cell Biol. 2012, 487, 380–384. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Talia, M.; Cirillo, F.; Rigiracciolo, D.C.; Scordamaglia, D.; Guzzi, R.; Miglietta, A.M.; de Francesco, E.M.; Belfiore, A.; Sims, A.H.; et al. The IL1β-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2020, 39, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Serine Synthesis Helps Hypoxic Cancer Stem Cells Regulate Redox. Cancer Res. 2016, 76, 6458–6462. [Google Scholar] [CrossRef]
- Lebert, J.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, 142–150. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Khalefa, H.G.; Shawki, M.A.; Aboelhassan, R.; el Wakeel, L.M. Evaluation of the effect of N-acetylcysteine on the prevention and amelioration of paclitaxel-induced peripheral neuropathy in breast cancer patients: A randomized controlled study. Breast Cancer Res. Treat. 2020, 183, 117–125. [Google Scholar] [CrossRef]
- Arıca, V.; Demir, I.; Tutanc, M.; Basarslan, F.; Arıca, S.; Karcıoglu, M.; Öztürk, H.; Nacar, A. N-Acetylcysteine prevents doxorubucine-induced cardiotoxicity in rats. Hum. Exp. Toxicol. 2012, 32, 655–661. [Google Scholar] [CrossRef]
- Farshid, A.A.; Tamaddonfard, E.; Simaee, N.; Mansouri, S.; Najafi, S.; Asri-Rezaee, S.; Alavi, H. Effects of Histidine and N-Acetylcysteine on Doxorubicin-Induced Cardiomyopathy in Rats. Cardiovasc. Toxicol. 2014, 14, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Brum, G.; Carbone, T.; Still, E.; Correia, V.; Szulak, K.; Calianese, D.; Best, C.; Cammarata, G.; Higgins, K.; Ji, F.; et al. N-acetylcysteine potentiates doxorubicin-induced ATM and p53 activation in ovarian cancer cells. Int. J. Oncol. 2012, 42, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Qanungo, S.; Uys, J.D.; Manevich, Y.; Distler, A.M.; Shaner, B.; Hill, E.G.; Mieyal, J.J.; Lemasters, J.J.; Townsend, D.M.; Nieminen, A.-L. N-acetyl-l-cysteine sensitizes pancreatic cancers to gemcitabine by targeting the NFκB pathway. Biomed. Pharmacother. 2014, 68, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Lyle, P.A.; Mitsopoulos, P.; Suntres, Z.E. N-Acetylcysteine Modulates the Cytotoxic Effects of Paclitaxel. Chemotherapy 2011, 57, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X. ROS Reduction Does Not Decrease the Anticancer Efficacy of X-Ray in Two Breast Cancer Cell Lines. Oxid. Med. Cell. Longev. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, Y. Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer. Antioxidants 2021, 10, 169. https://doi.org/10.3390/antiox10020169
Kwon Y. Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer. Antioxidants. 2021; 10(2):169. https://doi.org/10.3390/antiox10020169
Chicago/Turabian StyleKwon, Youngjoo. 2021. "Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer" Antioxidants 10, no. 2: 169. https://doi.org/10.3390/antiox10020169
APA StyleKwon, Y. (2021). Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer. Antioxidants, 10(2), 169. https://doi.org/10.3390/antiox10020169