
The KEAP1-NRF2 System in Healthy Aging and Longevity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
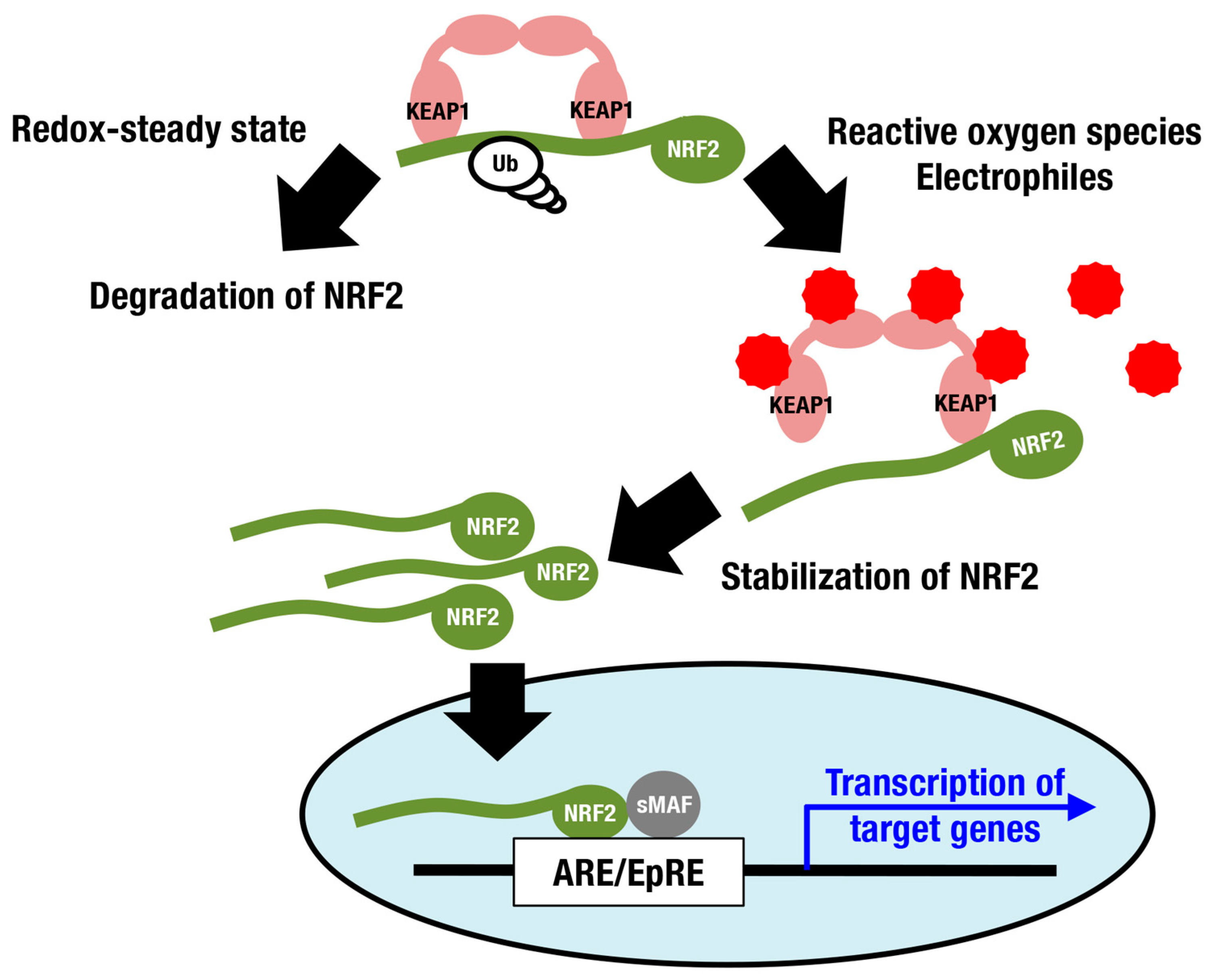
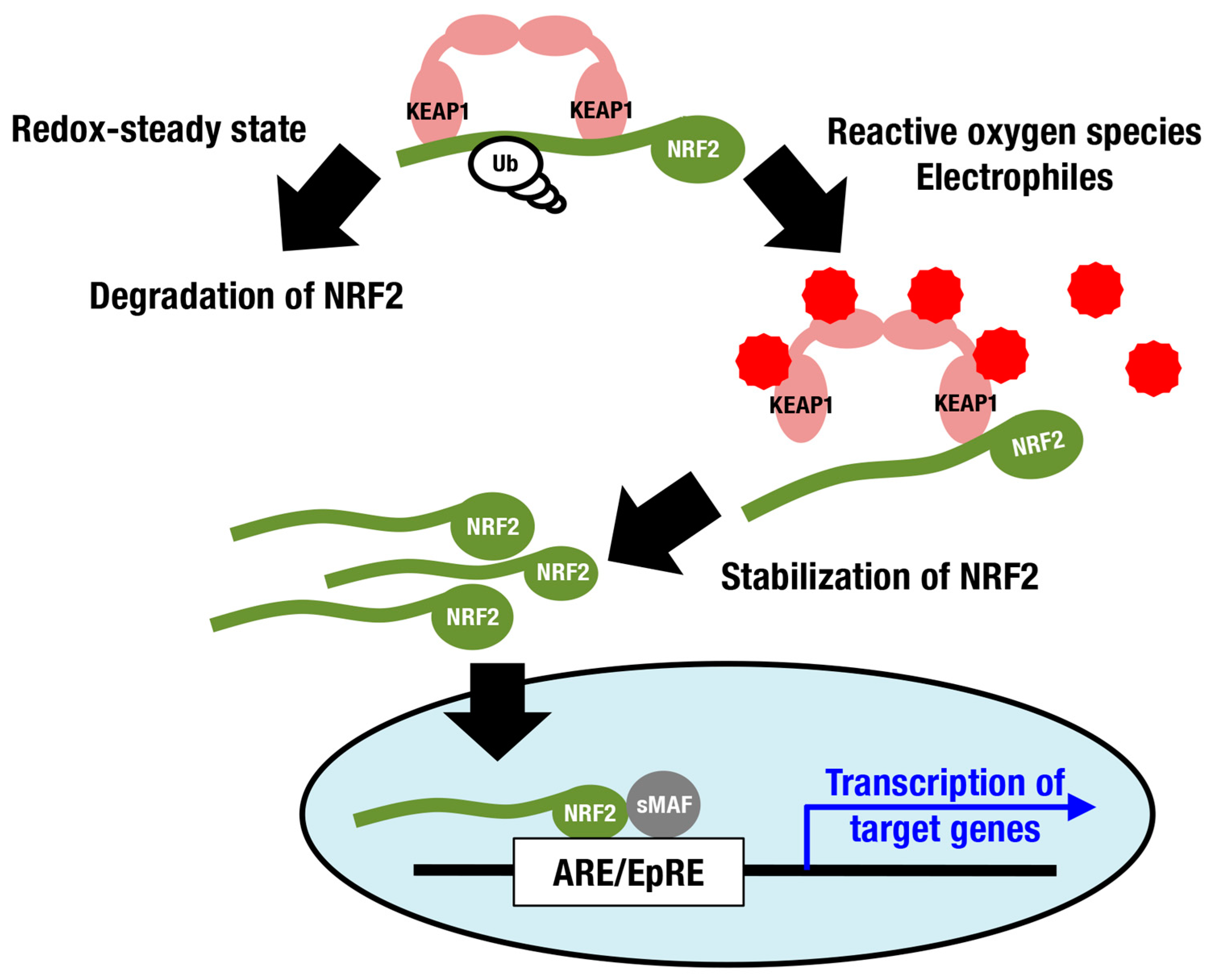
:1. Introduction
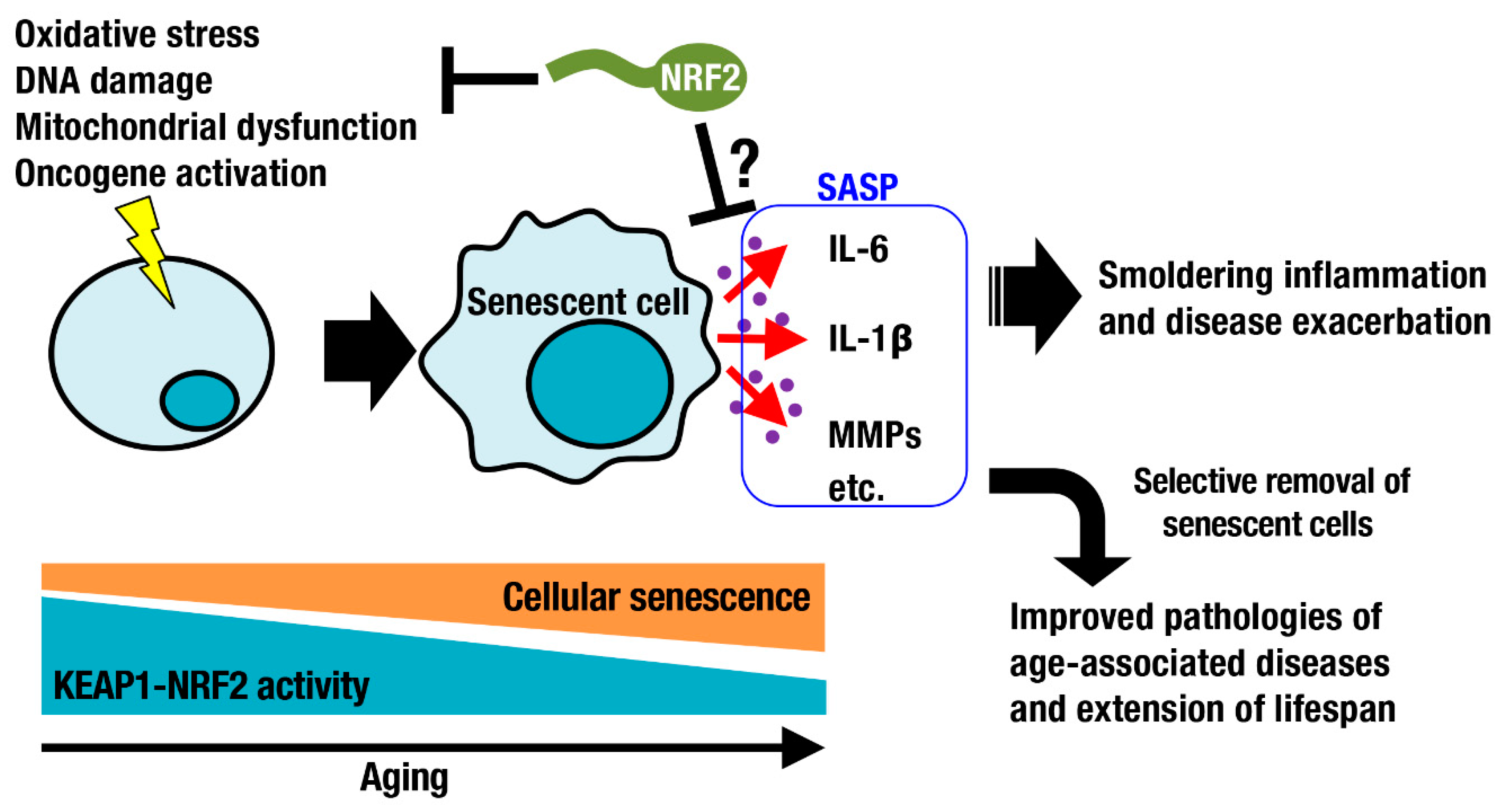
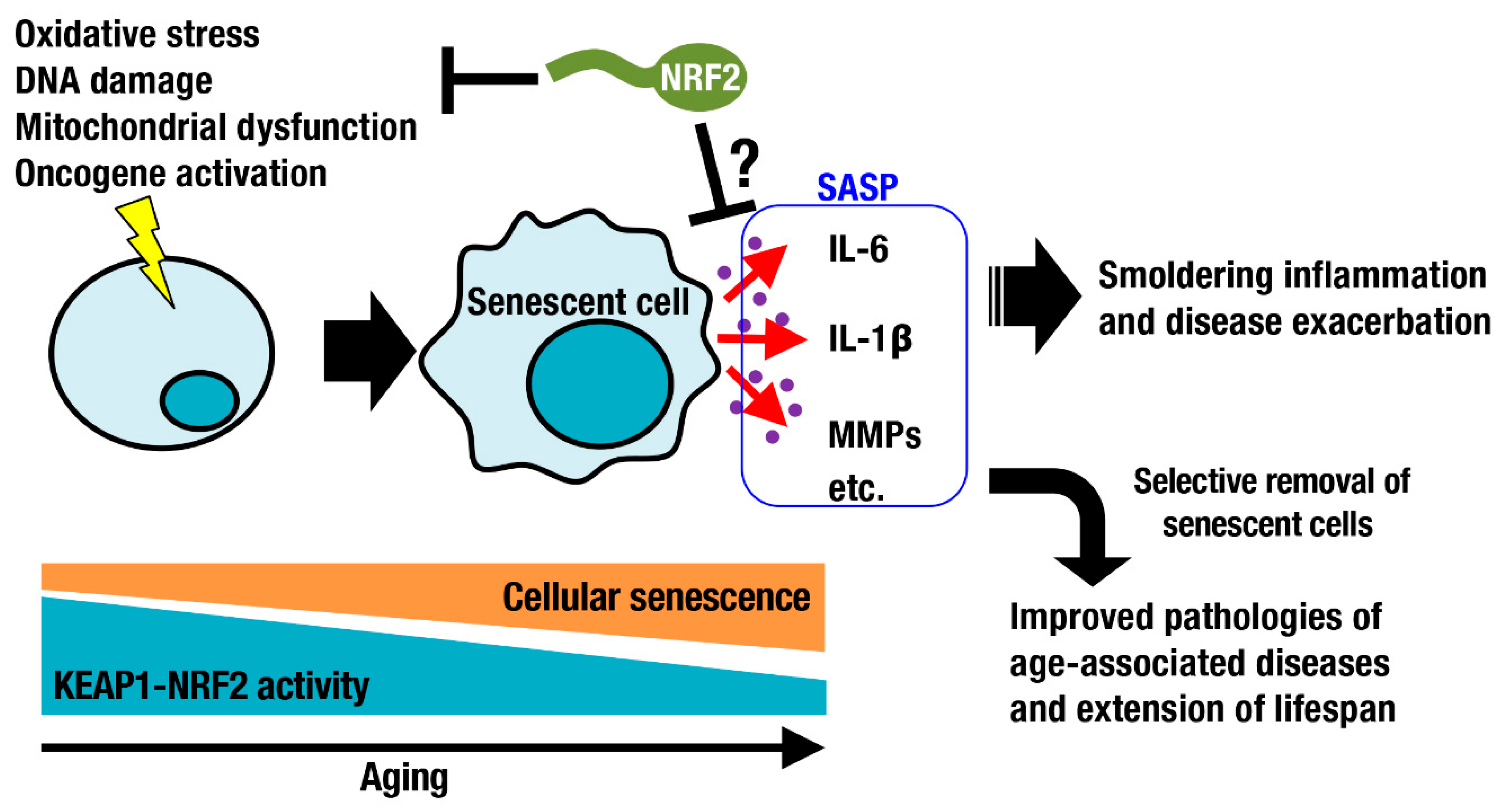
2. Cellular Senescence and the KEAP1-NRF2 System
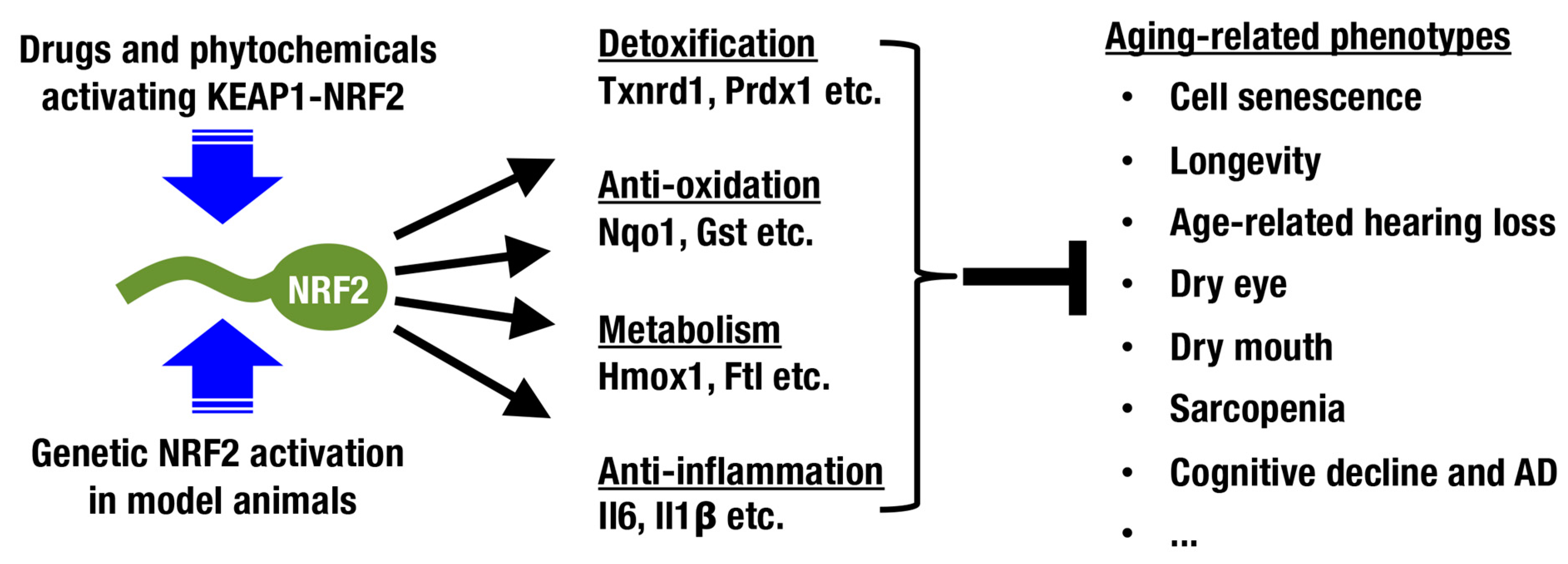
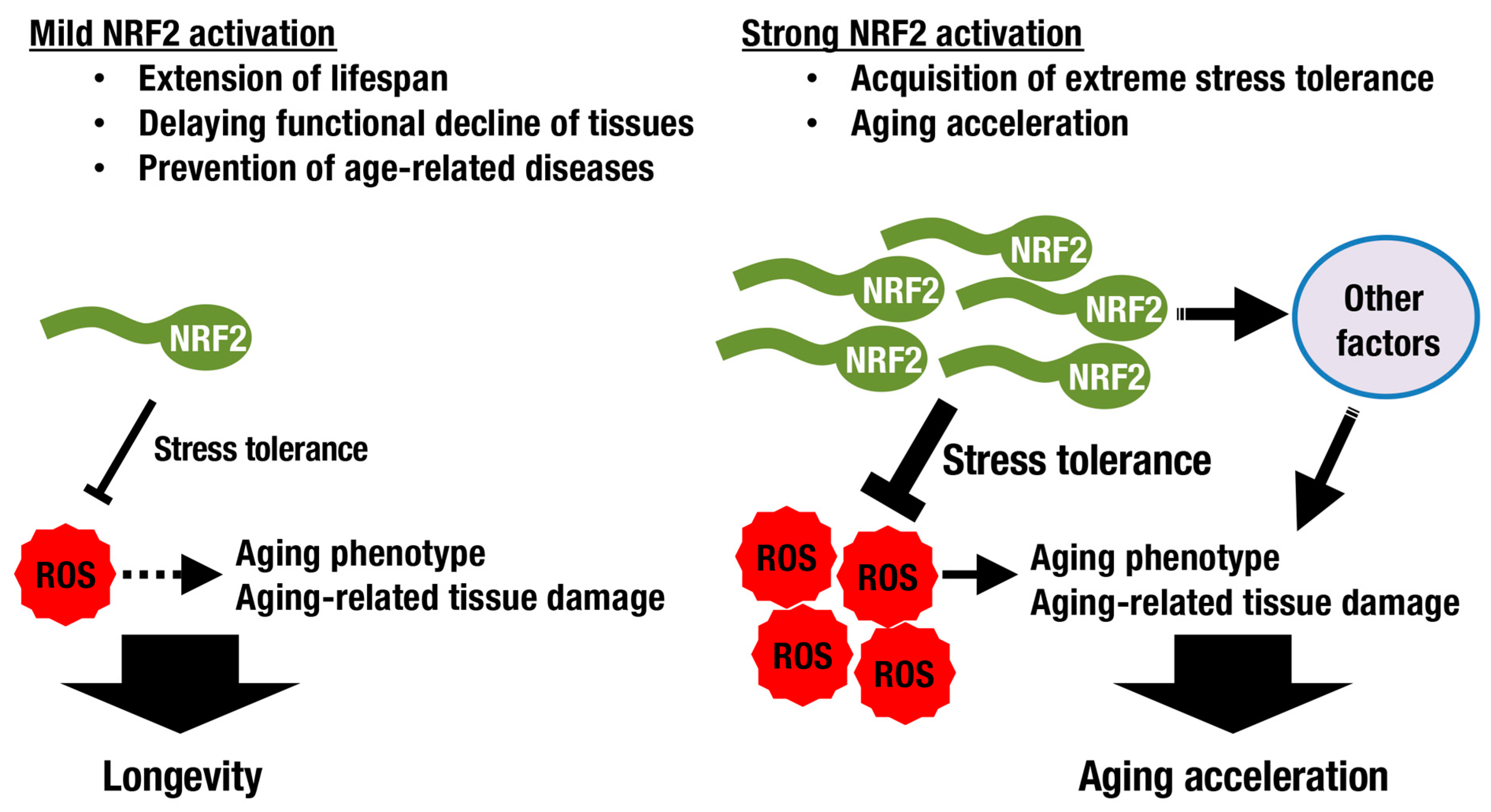
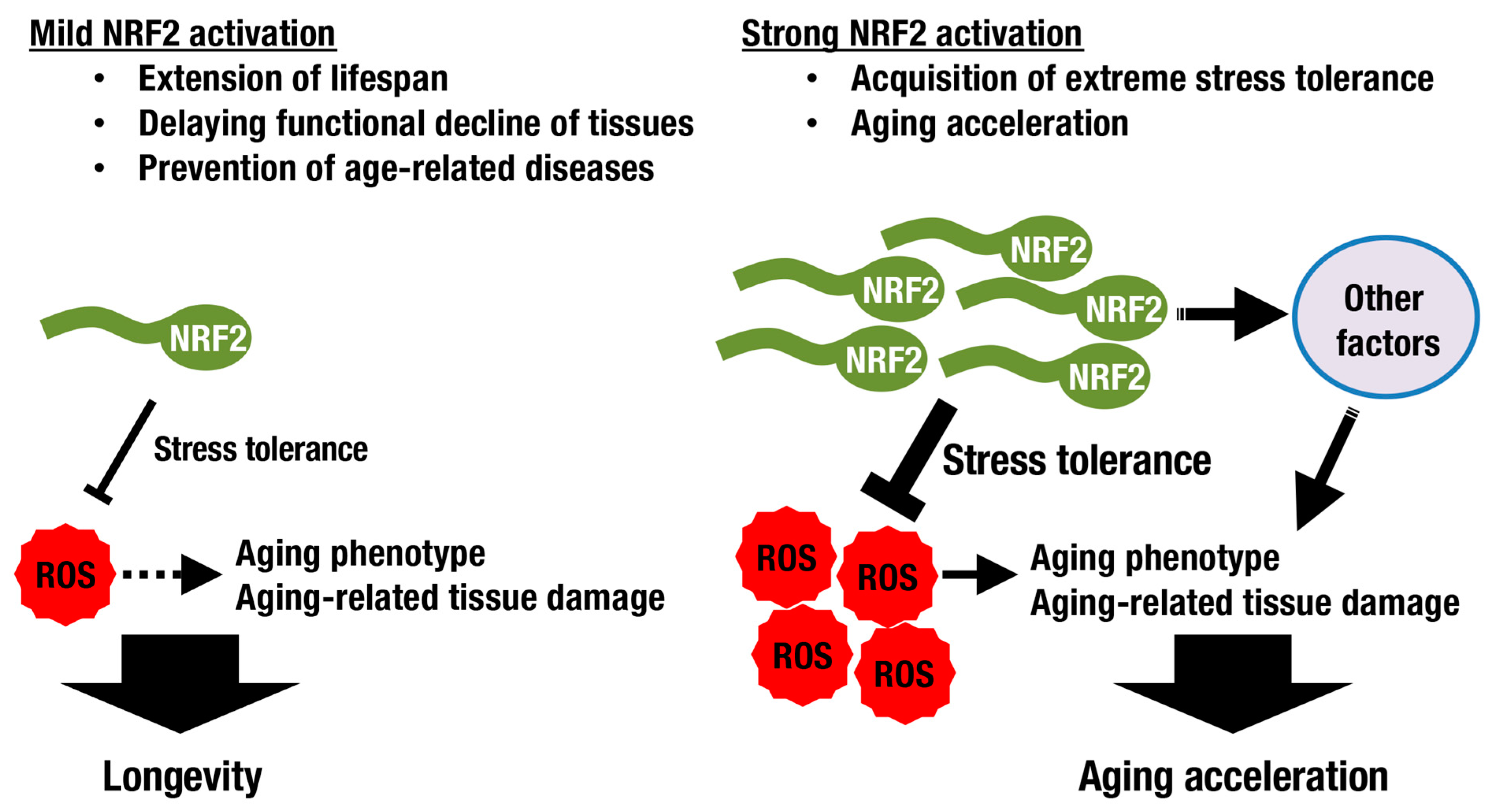
3. Longevity and the KEAP1-NRF2 System
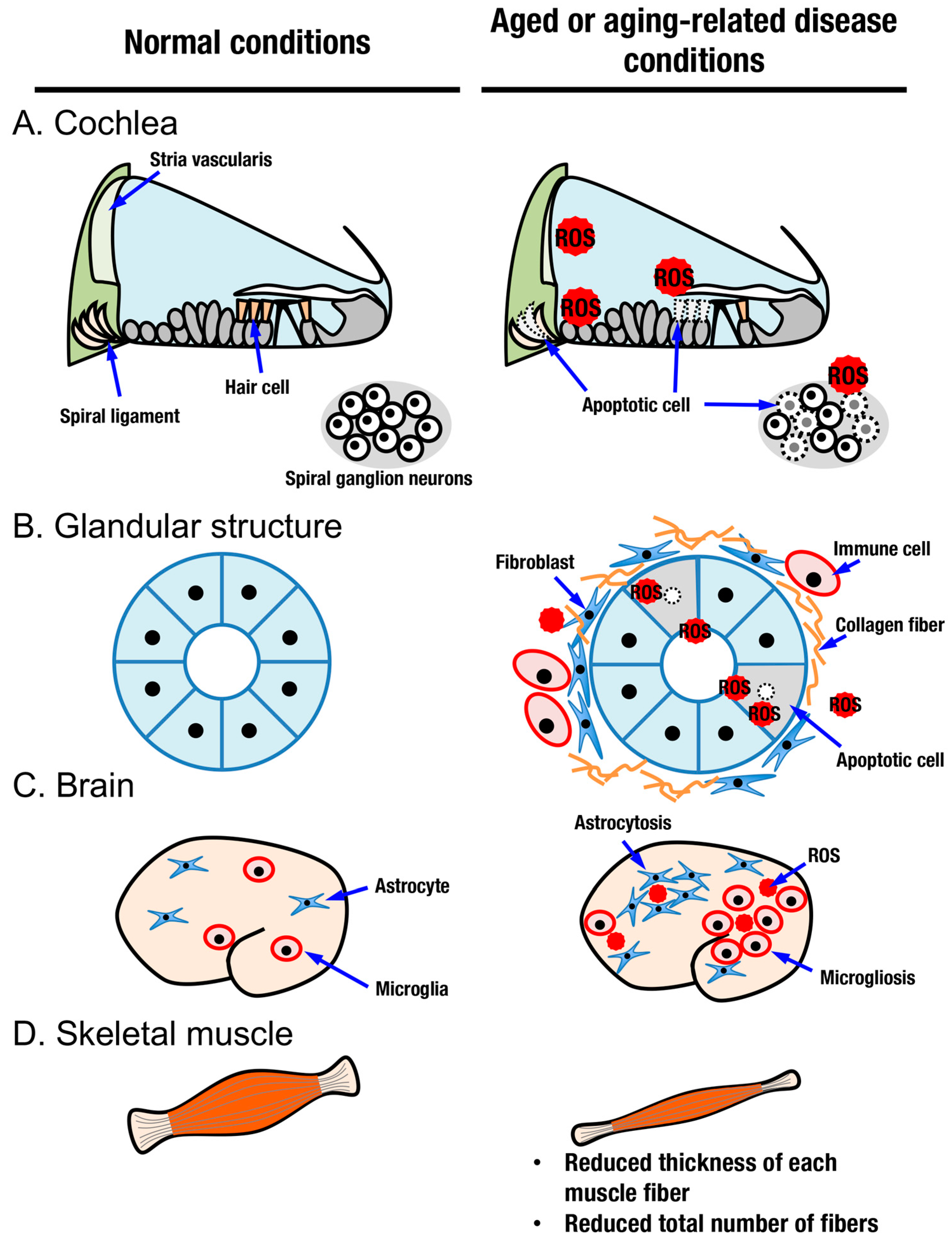
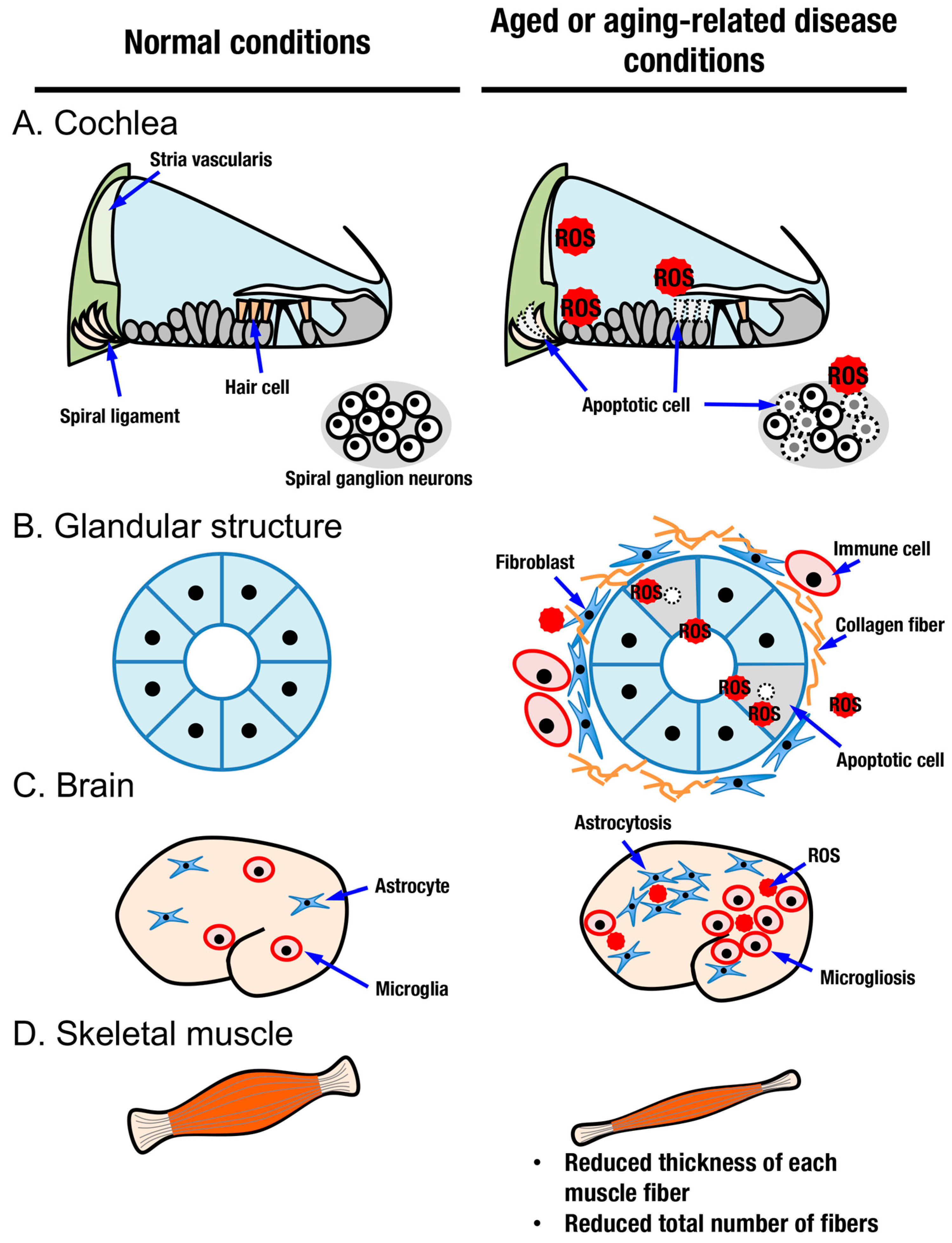
4. Tissue Aging and the KEAP1-NRF2 System
4.1. Aging in Sensory Organs and the KEAP1-NRF2 System
4.2. Aging in Glandular Structures and the KEAP1-NRF2 System
4.3. Aging in the Brain, Neurodegenerative Diseases and the KEAP1-NRF2 System
4.4. Aging in Skeletal Muscle and the KEAP1-NRF2 System
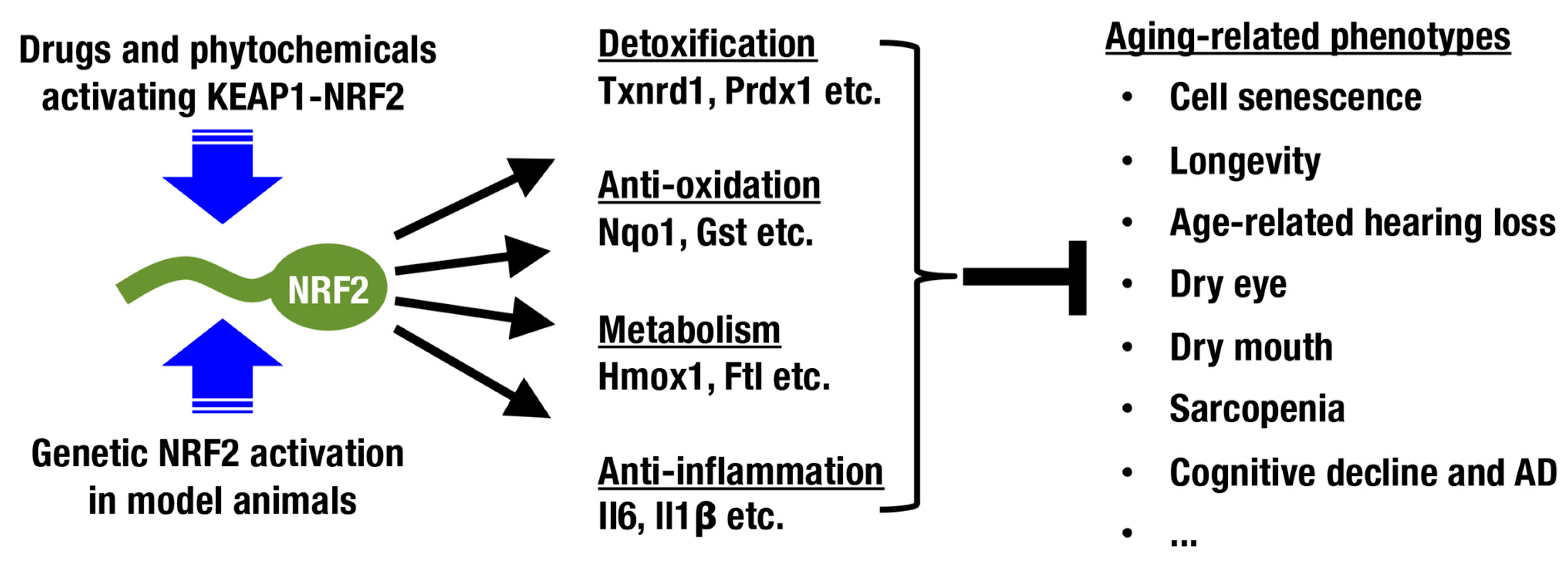
5. Pharmacological Intervention for Increasing NRF2 Activity
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Golden, T.R.; Hinerfeld, D.A.; Melov, S. Oxidative stress and aging: Beyond correlation. Aging Cell 2002, 1, 117–123. [Google Scholar] [CrossRef]
- Pérez, V.I.; Bokov, A.; Van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, A.; Idelchik, M.D.P.S.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2016, 11, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Buffenstein, R.; Jarvis, J.U. The Naked Mole Rat–A New Record for the Oldest Living Rodent. Sci. Aging Knowl. Environ. 2002, 2002, pe7. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of Antioxidants Supplementation on Aging and Longevity. BioMed Res. Int. 2014, 2014, 404680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Long, J.; Liu, J. Mitochondrial free radical theory of aging: Who moved my premise? Geriatr. Gerontol. Int. 2014, 14, 740–749. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Huang, H.-C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element: Degradation of Nrf2 by the 26 S proteasom. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic Evidence that Small Maf Proteins Are Essential for the Activation of Antioxidant Response Element-Dependent Genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [Green Version]
- Rushmore, T.H.; Pickett, C.B. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem. 1990, 265, 14648–14653. [Google Scholar] [CrossRef]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Murakami, S.; Biswal, S.S.; Sakaguchi, S.; Harigae, H.; Yamamoto, M.; Motohashi, H. Systemic Activation of NRF2 Alleviates Lethal Autoimmune Inflammation in Scurfy Mice. Mol. Cell. Biol. 2017, 37, e00063-17. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Suzuki, T.; Harigae, H.; Romeo, P.-H.; Yamamoto, M.; Motohashi, H. NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells. Mol. Cell. Biol. 2017, 37, e00086-17. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, E.; Suzuki, T.; Morita, M.; Taguchi, K.; Tsuchida, K.; Motohashi, H.; Doita, M.; Yamamoto, M. Hyperactivation of Nrf2 leads to hypoplasia of bone in vivo. Genes Cells 2018, 23, 386–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Onodera, Y.; Murakami, S.; Suzuki, T.; Motohashi, H. IL-11 contribution to tumorigenesis in an NRF2 addiction cancer model. Oncogene 2017, 36, 6315–6324. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Anzawa, H.; Liu, Z.; Ota, N.; Kitamura, H.; Onodera, Y.; Alam, M.M.; Matsumaru, D.; Suzuki, T.; Katsuoka, F.; et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 2020, 11, 5911. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; Von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear Erythroid Factor 2-mediated Proteasome Activation Delays Senescence in Human Fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. GeroScience 2018, 40, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, L.; Liu, H.; Wu, K.; Liu, Y.; Bai, L.; Wang, Q.; Qi, B.; Zhang, L. Reduced NRF2 expression suppresses endothelial progenitor cell function and induces senescence during aging. Aging 2019, 11, 7021–7035. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, P.; Wietecha, M.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.-D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Shibata, T.; Takaya, K.; Shiraishi, K.; Kohno, T.; Kunitoh, H.; Tsuta, K.; Furuta, K.; Goto, K.; Hosoda, F.; et al. Regulatory Nexus of Synthesis and Degradation Deciphers Cellular Nrf2 Expression Levels. Mol. Cell. Biol. 2013, 33, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Linna-Kuosmanen, S.M.; Sihvola, V.; Kansanen, E.; Kaikkonen, M.U.; Levonen, A.-L. MicroRNAs mediate the senescence-associated decline of NRF2 in endothelial cells. Redox Biol. 2018, 18, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Podlutsky, A.J.; Khritankov, A.M.; Ovodov, N.D.; Austad, S.N. A New Field Record for Bat Longevity. Journals Gerontol. Ser. A: Boil. Sci. Med. Sci. 2005, 60, 1366–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef]
- Ladiges, W.; Van Remmen, H.; Strong, R.; Ikeno, Y.; Treuting, P.; Rabinovitch, P.; Richardson, A. Lifespan extension in genetically modified mice. Aging Cell 2009, 8, 346–352. [Google Scholar] [CrossRef]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Kohler, W.J.; Guo, A.H.; Gendron, C.; Han, M.; Ding, W.; Lyu, Y.; Ching, T.-T.; Wang, F.-Y.; Chakraborty, T.S.; et al. High-throughput small molecule screening reveals Nrf2-dependent and -independent pathways of cellular stress resistance. Sci. Adv. 2020, 6, eaaz7628. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, G.M.; Xu, Z.; Zhou, L.; Duh, E.J. Adaptation of the master antioxidant response connects metabolism, lifespan and feather development pathways in birds. Nat. Commun. 2020, 11, 2476. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Keap1/Nrf2 Signaling Regulates Oxidative Stress Tolerance and Lifespan in Drosophila. Dev. Cell 2008, 14, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiers, J.G.; Breda, C.; Robinson, S.; Giorgini, F.; Steinert, J.R. Drosophila Nrf2/Keap1 Mediated Redox Signaling Supports Synaptic Function and Longevity and Impacts on Circadian Activity. Front. Mol. Neurosci. 2019, 12, 86. [Google Scholar] [CrossRef]
- Tullet, J.M.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct Inhibition of the Longevity-Promoting Factor SKN-1 by Insulin-like Signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tullet, J.M.; Green, J.W.; Au, C.; Benedetto, A.; Thompson, M.A.; Clark, E.; Gilliat, A.F.; Young, A.; Schmeisser, K.; Gems, D. The SKN-1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell 2017, 16, 1191–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakiri, E.N.; Gumeni, S.; Iliaki, K.K.; Benaki, D.; Vougas, K.; Sykiotis, G.P.; Gorgoulis, V.G.; Mikros, E.; Scorrano, L.; Trougakos, I.P. Hyperactivation of Nrf2 increases stress tolerance at the cost of aging acceleration due to metabolic deregulation. Aging Cell 2019, 18, e12845. [Google Scholar] [CrossRef]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Hellyer, J.A.; Stehr, H.; Hoang, N.T.; Niu, X.; Das, M.; Padda, S.K.; Ramchandran, K.; Neal, J.W.; Wakelee, H.A.; et al. Role of KEAP1/NFE2L2 Mutations in the Chemotherapeutic Response of Patients with Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.C.; Jordan, E.; Kim, H.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, K.; Papagiannakopoulos, T.; Motohashi, H. Metabolic features of cancer cells in NRF2 addiction status. Biophys. Rev. 2020, 12, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Saigusa, D.; Motoike, I.N.; Saito, S.; Zorzi, M.; Aoki, Y.; Kitamura, H.; Suzuki, M.; Katsuoka, F.; Ishii, H.; Kinoshita, K.; et al. Impacts of NRF2 activation in non–small-cell lung cancer cell lines on extracellular metabolites. Cancer Sci. 2020, 111, 667–678. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Howes, R.M. The Free Radical Fantasy: A Panoply of Paradoxes. Ann. N. Y. Acad. Sci. 2006, 1067, 22–26. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Bowl, M.R.; Dawson, S.J. Age-Related Hearing Loss. Cold Spring Harb. Perspect. Med. 2019, 9, a033217. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.-Z.; O’Malley, J.T.; De Gruttola, V.; Liberman, M.C. Age-Related Hearing Loss Is Dominated by Damage to Inner Ear Sensory Cells, Not the Cellular Battery That Powers Them. J. Neurosci. 2020, 40, 6357–6366. [Google Scholar] [CrossRef] [PubMed]
- Hequembourg, S.; Liberman, M.C. Spiral Ligament Pathology: A Major Aspect of Age-Related Cochlear Degeneration in C57BL/6 Mice. J. Assoc. Res. Otolaryngol. 2001, 2, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, M.; Harrison, R.V. Degeneration of stria vascularis in age-related hearing loss; a corrosion cast study in a mouse model. Acta Oto-Laryngol. 2016, 136, 385–390. [Google Scholar] [CrossRef]
- Fujimoto, C.; Yamasoba, T. Oxidative Stresses and Mitochondrial Dysfunction in Age-Related Hearing Loss. Oxidative Med. Cell. Longev. 2014, 2014, 582849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawa, S.G.; Liberman, M.C. Acceleration of Age-Related Hearing Loss by Early Noise Exposure: Evidence of a Misspent Youth. J. Neurosci. 2006, 26, 2115–2123. [Google Scholar] [CrossRef]
- Alvarado, J.C.; Fuentes-Santamaría, V.; Gabaldón-Ull, M.C.; Juiz, J.M. Age-Related Hearing Loss Is Accelerated by Repeated Short-Duration Loud Sound Stimulation. Front. Neurosci. 2019, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.; Cruickshanks, K.J.; Klein, B.E.K.; Klein, R.; Hong, O.; Wallhagen, M.I. The Contribution of Ototoxic Medications to Hearing Loss Among Older Adults. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2020, 75, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.; Gopinath, B.; McMahon, C.M.; Rochtchina, E.; Wang, J.J.; Boyages, S.C.; Leeder, S.R. Relationship of Type 2 diabetes to the prevalence, incidence and progression of age-related hearing loss. Diabet. Med. 2009, 26, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Wattamwar, K.; Qian, Z.J.; Otter, J.; Leskowitz, M.J.; Caruana, F.F.; Siedlecki, B.; Spitzer, J.B.; Lalwani, A.K. Association of Cardiovascular Comorbidities with Hearing Loss in the Older Old. JAMA Otolaryngol.-Head Neck Surg. 2018, 144, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Vuckovic, D.; Krishnamoorthy, N.; Rubinato, E.; Ambrosetti, U.; Castorina, P.; Franzè, A.; Vozzi, D.; La Bianca, M.; Cappellani, S.; et al. Next-generation sequencing identified SPATC1L as a possible candidate gene for both early-onset and age-related hearing loss. Eur. J. Hum. Genet. 2019, 27, 70–79. [Google Scholar] [CrossRef]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.R.; Yu, H.; Ding, D.; Jiang, H.; Gagnon, L.H.; Salvi, R.J. Separate and combined effects of Sod1 and Cdh23 mutations on age-related hearing loss and cochlear pathology in C57BL/6J mice. Hear. Res. 2010, 268, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.; Kim, M.-J.; Han, C.; Park, H.-J.; Ding, D.; Boyd, K.; Walker, L.; Linser, P.; Meneses, Z.; Slade, C.; et al. Loss of IDH2 Accelerates Age-related Hearing Loss in Male Mice. Sci. Rep. 2018, 8, 5039. [Google Scholar] [CrossRef]
- Frye, M.D.; Yang, W.; Zhang, C.; Xiong, B.; Hu, B.H. Dynamic activation of basilar membrane macrophages in response to chronic sensory cell degeneration in aging mouse cochleae. Hear. Res. 2017, 344, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschuur, C.; Agyemang-Prempeh, A.; Newman, T.A. Inflammation is associated with a worsening of presbycusis: Evidence from the MRC national study of hearing. Int. J. Audiol. 2014, 53, 469–475. [Google Scholar] [CrossRef]
- Noben-Trauth, K.; Zheng, Q.Y.; Johnson, K.R. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat. Genet. 2003, 35, 21–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spongr, V.P.; Flood, D.G.; Frisina, R.D.; Salvi, R.J. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J. Acoust. Soc. Am. 1997, 101, 3546–3553. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Y.; Johnson, K.R.; Erway, L.C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear. Res. 1999, 130, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Henry, K.R. Males lose hearing earlier in mouse models of late-onset age-related hearing loss; females lose hearing earlier in mouse models of early-onset hearing loss. Hear. Res. 2004, 190, 141–148. [Google Scholar] [CrossRef]
- Hosokawa, K.; Hosokawa, S.; Ishiyama, G.; Ishiyama, A.; Lopez, I.A. Immunohistochemical localization of Nrf2 in the human cochlea. Brain Res. 2018, 1700, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Tabuchi, K.; Nishimura, B.; Tanaka, S.; Nakayama, M.; Ishii, T.; Warabi, E.; Yanagawa, T.; Shimizu, R.; Yamamoto, M.; et al. Protective role of Nrf2 in age-related hearing loss and gentamicin ototoxicity. Biochem. Biophys. Res. Commun. 2011, 415, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Oishi, T.; Matsumaru, D.; Ota, N.; Kitamura, H.; Zhang, T.; Honkura, Y.; Katori, Y.; Motohashi, H. Activation of the NRF2 pathway in Keap1-knockdown mice attenuates progression of age-related hearing loss. NPJ Aging Mech. Dis. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Honkura, Y.; Matsuo, H.; Murakami, S.; Sakiyama, M.; Mizutari, K.; Shiotani, A.; Yamamoto, M.; Morita, I.; Shinomiya, N.; Kawase, T.; et al. NRF2 Is a Key Target for Prevention of Noise-Induced Hearing Loss by Reducing Oxidative Damage of Cochlea. Sci. Rep. 2016, 6, 19329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhao, H.; Cui, Z.-K.; Tian, G. The Role of Nrf2 in Hearing Loss. Front. Pharmacol. 2021, 12, 620921. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res. 2014, 119, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, E.M.; Alves, M.; Rios, J.D.; Dartt, D.A. The Aging Lacrimal Gland: Changes in Structure and Function. Ocul. Surf. 2008, 6, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Uchino, Y.; Kawakita, T.; Miyazawa, M.; Ishii, T.; Onouchi, H.; Yasuda, K.; Ogawa, Y.; Shimmura, S.; Ishii, N.; Tsubota, K. Oxidative Stress Induced Inflammation Initiates Functional Decline of Tear Production. PLoS ONE 2012, 7, e45805. [Google Scholar] [CrossRef] [Green Version]
- Van Haeringen, N.J. Aging and the lacrimal system. Br. J. Ophthalmol. 1997, 81, 824–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, H.; Yamamoto, S.; Horiuchi, H.; Machinami, R. Histopathologic Study of Human Lacrimal Gland. Ophthalmology 1995, 102, 678–686. [Google Scholar] [CrossRef]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [Green Version]
- Cano, M.; Thimmalappula, R.; Fujihara, M.; Nagai, N.; Sporn, M.; Wang, A.L.; Neufeld, A.H.; Biswal, S.; Handa, J.T. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res. 2010, 50, 652–664. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Dogru, M.; Higuchi, A.; Nagata, T.; Ibrahim, O.M.; Inaba, T.; Tsubota, K. The effect of Nrf2 knockout on ocular surface protection from acute tobacco smoke exposure: Evidence from Nrf2 knockout mice. Am. J. Pathol. 2015, 185, 776–785. [Google Scholar] [CrossRef]
- Kawai, M.; Ogawa, Y.; Shimmura, S.; Ohta, S.; Suzuki, T.; Kawamura, N.; Kuwana, M.; Kawakami, Y.; Tsubota, K. Expression and localization of aging markers in lacrimal gland of chronic graft-versus-host disease. Sci. Rep. 2013, 3, 2455. [Google Scholar] [CrossRef] [Green Version]
- de Souza, R.G.; Yu, Z.; Hernandez, H.; Trujillo-Vargas, C.M.; Lee, A.; Mauk, K.E.; Cai, J.; Alves, M.R.; de Paiva, C.S. Modulation of Oxidative Stress and Inflammation in the Aged Lacrimal Gland. Am. J. Pathol. 2020, 191, 294–308. [Google Scholar] [CrossRef]
- Affoo, R.H.; Foley, N.; Garrick, R.; Siqueira, W.L.; Martin, R.E. Meta-Analysis of Salivary Flow Rates in Young and Older Adults. J. Am. Geriatr. Soc. 2015, 63, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Astor, F.C.; Hanft, K.L.; Ciocon, J.O. Xerostomia: A Prevalent Condition in the Elderly. Ear Nose Throat J. 1999, 78, 476–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J. Quantitative age changes in the histological structure of human submandibular salivary glands. Arch. Oral Biol. 1977, 22, 221–227. [Google Scholar] [CrossRef]
- Scott, J.; Flower, E.A.; Burns, J. A quantitative study of histological changes in the human parotid gland occurring with adult age. J. Oral Pathol. Med. 1987, 16, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Matsuno, T.; Omata, K.; Satoh, T. Relationship between hyposalivation and oxidative stress in aging mice. J. Clin. Biochem. Nutr. 2017, 61, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wati, S.M.; Matsumaru, D.; Motohashi, H. NRF2 pathway activation by KEAP1 inhibition attenuates the manifestation of aging phenotypes in salivary glands. Redox Biol. 2020, 36, 101603. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Nakane, T.; Kimura, T.; Arisawa, K.; Yoneda, K.; Yamamoto, T.; Osaki, T. Treatment of Xerostomia with the Bile Secretion-Stimulating Drug Anethole Trithione: A Clinical Trial. Am. J. Med. Sci. 1999, 318, 146–151. [Google Scholar] [CrossRef]
- Smith, E.J.; Shay, K.P.; Thomas, N.O.; Butler, J.A.; Finlay, L.F.; Hagen, T.M. Age-related loss of hepatic Nrf2 protein homeostasis: Potential role for heightened expression of miR-146a. Free Radic. Biol. Med. 2015, 89, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, P. Astaxanthin, cell membrane nutrient with diverse clinical benefits and anti-aging potential. Altern. Med. Rev. 2011, 16, 355–364. [Google Scholar]
- Kuraji, M.; Matsuno, T.; Satoh, T. Astaxanthin affects oxidative stress and hyposalivation in aging mice. J. Clin. Biochem. Nutr. 2016, 59, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandlur, A.; Satyamoorthy, K.; Gangadharan, G. Oxidative Stress in Cognitive and Epigenetic Aging: A Retrospective Glance. Front. Mol. Neurosci. 2020, 13, 41. [Google Scholar] [CrossRef] [Green Version]
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging: Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Perrig, W.J.; Perrig, P.; Stähelin, H.B. The Relation Between Antioxidants and Memory Performance in the Old and Very Old. J. Am. Geriatr. Soc. 1997, 45, 718–724. [Google Scholar] [CrossRef]
- Berr, C.; Balansard, B.; Arnaud, J.; Roussel, A.-M.; Alpérovitch, A. Cognitive decline is associated with systemic oxidative stress: The EVA study. J. Am. Geriatr. Soc. 2000, 48, 1285–1291. [Google Scholar] [CrossRef]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidatives stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.; Chauhan, A. Oxidative stress in Alzheimer’s disease. Pathophysiology 2006, 13, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Saito, T.; Saido, T.C. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative Stress in the Progression of Alzheimer Disease in the Frontal Cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutachan, J.-J.; Casas, Z.; Albarracin, S.L.; Stab, B.R.; Samudio, I.; Gonzalez, J.; Morales, L.; Barreto, G.E. Cellular and molecular mechanisms of antioxidants in Parkinson’s disease. Nutr. Neurosci. 2012, 15, 120–126. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, J.; Luber-Narod, J.; Styren, S.D.; Civin, W.H. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar] [CrossRef]
- Hendrickx, J.O.; Martinet, W.; Van Dam, D.; De Meyer, G.R.Y. Inflammation, Nitro-Oxidative Stress, Impaired Autophagy, and Insulin Resistance as a Mechanistic Convergence Between Arterial Stiffness and Alzheimer’s Disease. Front. Mol. Biosci. 2021, 8, 651215. [Google Scholar] [CrossRef]
- Herrero, M.T.; Estrada, C.; Maatouk, L.; Vyas, S. Inflammation in Parkinson′s disease: Role of glucocorticoids. Front. Neuroanat. 2015, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, H.; Katsuoka, F.; Toide, K.; Shimizu, Y.; Furusako, S.; Yamamoto, M. Nrf2 deficiency leads to behavioral, neurochemical and transcriptional changes in mice. Genes Cells 2013, 18, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear Factor E2-Related Factor 2-Dependent Antioxidant Response Element Activation by tert-Butylhydroquinone and Sulforaphane Occurring Preferentially in Astrocytes Conditions Neurons against Oxidative Insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Calingasan, N.Y.; Thomas, B.; Chaturvedi, R.K.; Kiaei, M.; Wille, E.J.; Liby, K.T.; Williams, C.; Royce, D.; Risingsong, R.; et al. Neuroprotective Effects of the Triterpenoid, CDDO Methyl Amide, a Potent Inducer of Nrf2-Mediated Transcription. PLoS ONE 2009, 4, e5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 Improves Leptin and Insulin Resistance Provoked by Hypothalamic Oxidative Stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, K.F.; Al-Mubarak, B.; Martel, M.-A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef] [Green Version]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the Antioxidant Glutathione in Neurons: Supply by Astrocytes of CysGly as Precursor for Neuronal Glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Resch, J.M.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2–ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 2007, 207, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A. Brain-Protective Mechanisms of Transcription Factor NRF2: Toward a Common Strategy for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2021, 62. [Google Scholar] [CrossRef] [PubMed]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, E.; Leon, J.; Mazzei, G.; Abolhassani, N.; Haruyama, N.; Saito, T.; Saido, T.; Hokama, M.; Iwaki, T.; Ohara, T.; et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 2017, 7, 17762. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s disease–like pathology in the APP/ PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging 2015, 36, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uruno, A.; Matsumaru, D.; Ryoke, R.; Saito, R.; Kadoguchi, S.; Saigusa, D.; Saito, T.; Saido, T.C.; Kawashima, R.; Yamamoto, M. Nrf2 Suppresses Oxidative Stress and Inflammation in App Knock-In Alzheimer’s Disease Model Mice. Mol. Cell. Biol. 2020, 40, e00467-19. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.-K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.-L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.-L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, M.; Wille, E.; Calingasan, N.Y.; Tampellini, D.; Williams, C.; Gouras, G.K.; Liby, K.; Sporn, M.; Beal, M.F.; Lin, M.T. Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2009, 109, 502–512. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic Activation of Nrf2 Pathway in Parkinson’s Disease. Mov. Disord. 2020, 35, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.-X.; Dai, S.-X.; Guo, Y.C.; Han, F.-F.; Zheng, J.-J.; Li, G.-H.; Huang, J.-F. Meta-Analysis of Parkinson’s Disease and Alzheimer’s Disease Revealed Commonly Impaired Pathways and Dysregulation of NRF2-Dependent Genes. J. Alzheimers Dis. 2017, 56, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Tarozzi, A.; Cantelli-Forti, G.; Hrelia, P. Neuroprotection by 6-(methylsulfinyl)hexyl isothiocyanate in a 6-hydroxydopamine mouse model of Parkinson’s disease. Brain Res. 2014, 1589, 93–104. [Google Scholar] [CrossRef]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; García-Yagüe, Á.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative Stress in Alzheimer’s Disease: Why Did Antioxidant Therapy Fail? Oxidative Med. Cell. Longev. 2014, 427318, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Siparsky, P.N.; Kirkendall, D.T.; Garrett, W.E., Jr. Muscle Changes in Aging: Understanding Sarcopenia. Sports Health 2014, 6, 36–40. [Google Scholar] [CrossRef] [Green Version]
- Lexell, J.; Henriksson-Larsén, K.; Winblad, B.; Sjöström, M. Distribution of different fiber types in human skeletal muscles: Effects of aging studied in whole muscle cross sections. Muscle Nerve 1983, 6, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Martinez, P.F.; Pagan, L.U.; Damatto, R.L.; Cezar, M.D.M.; Lima, A.R.R.; Okoshi, K.; Okoshi, M.P. Skeletal muscle aging: Influence of oxidative stress and physical exercise. Oncotarget 2017, 8, 20428–20440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Coen, P.M.; Musci, R.V.; Hinkley, J.M.; Miller, B.F. Mitochondria as a Target for Mitigating Sarcopenia. Front. Physiol. 2018, 9, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanti, F.; Romano, A.D.; Buglio, A.L.; Castriotta, V.; Guglielmi, G.; Greco, A.; Serviddio, G.; Vendemiale, G. Oxidative stress is increased in sarcopenia and associated with cardiovascular disease risk in sarcopenic obesity. Maturitas 2018, 109, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Manford, A.G.; Rodríguez-Pérez, F.; Shih, K.Y.; Shi, Z.; Berdan, C.A.; Choe, M.; Titov, D.V.; Nomura, D.K.; Rape, M. A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183, 46–61. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musarò, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Can, B.; Kara, O.; Kizilarslanoglu, M.C.; Arik, G.; Aycicek, G.S.; Sumer, F.; Civelek, R.; Demirtas, C.; Ulger, Z. Serum markers of inflammation and oxidative stress in sarcopenia. Aging Clin. Exp. Res. 2017, 29, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Tamura, Y.; Takahashi, K.; Takeda, K.; Takemasa, T.; Hatta, H. Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle. Physiol. Rep. 2019, 7, e13998. [Google Scholar] [CrossRef]
- Crilly, M.J.; Tryon, L.D.; Erlich, A.T.; Hood, D.A. The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J. Appl. Physiol. 2016, 121, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Coleman, V.; Sa-Nguanmoo, P.; König, J.; Schulz, T.J.; Grune, T.; Klaus, S.; Kipp, A.P.; Ost, M. Partial involvement of Nrf2 in skeletal muscle mitohormesis as an adaptive response to mitochondrial uncoupling. Sci. Rep. 2018, 8, 2446. [Google Scholar] [CrossRef]
- Huang, D.-D.; Fan, S.-D.; Chen, X.-Y.; Yan, X.-L.; Zhang, X.-Z.; Ma, B.-W.; Yu, D.-Y.; Xiao, W.-Y.; Zhuang, C.-L.; Yu, Z. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp. Gerontol. 2019, 119, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [Green Version]
- Wafi, A.M.; Hong, J.; Rudebush, T.L.; Yu, L.; Hackfort, B.; Wang, H.; Schultz, H.D.; Zucker, I.H.; Gao, L. Curcumin improves exercise performance of mice with coronary artery ligation-induced HFrEF: Nrf2 and antioxidant mechanisms in skeletal muscle. J. Appl. Physiol. 2019, 126, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Onoki, T.; Izumi, Y.; Takahashi, M.; Murakami, S.; Matsumaru, D.; Ohta, N.; Wati, S.M.; Hatanaka, N.; Katsuoka, F.; Okutsu, M.; et al. Skeletal muscle-specific Keap1 disruption modulates fatty acid utilization and enhances exercise capacity in female mice. Redox Biol. 2021, 43, 101966. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.; Alves, I.; Singh, P.; Palade, P.T.; Carvalho, E.; Børsheim, E.; Jun, S.R.; Cheema, A.; Boerma, M.; Awasthi, S.; et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell 2020, 19, e13261. [Google Scholar] [CrossRef] [PubMed]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.J.; Odelberg, S.J.; Firpo, M.A.; Paine, R., III; Hoidal, J.R.; Abel, E.D.; et al. Impaired Transcriptional Activity of Nrf2 in Age-Related Myocardial Oxidative Stress Is Reversible by Moderate Exercise Training. PLoS ONE 2012, 7, e45697. [Google Scholar] [CrossRef]
- Yamada, M.; Iwata, M.; Warabi, E.; Oishi, H.; Lira, V.A.; Okutsu, M. p62/SQSTM1 and Nrf2 are essential for exercise-mediated enhancement of antioxidant protein expression in oxidative muscle. FASEB J. 2019, 33, 8022–8032. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Done, A.J.; Traustadóttir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Yang, C.; Xue, R.; Li, S.; Zhang, T.; Pan, L.; Ma, X.; Wang, L.; Li, D. Sulforaphane alleviates muscular dystrophy in mdx mice by activation of Nrf2. J. Appl. Physiol. 2015, 118, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Zhang, Y.; Spencer, S.R.; Wilczak, C.A.; Talalay, P. The electrophile counterattack response: Protection against neoplasia and toxicity. Adv. Enzym. Regul. 1993, 33, 281–296. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.-I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; Van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Huang, M.-T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.-N.T. Inhibition of 7,12-Dimethylbenz(a)anthracene-Induced Skin Tumorigenesis in C57BL/6 Mice by Sulforaphane Is Mediated by Nuclear Factor E2–Related Factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.-X.; Korenori, Y.; Tanigawa, S.; Yamada-Kato, T.; Nagai, M.; He, X.; He, J. Dynamics of Nrf2 and Keap1 in ARE-Mediated NQO1 Expression by Wasabi 6-(Methylsulfinyl)hexyl Isothiocyanate. J. Agric. Food Chem. 2011, 59, 11975–11982. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.C.; Chan, K.M.; Sharif, R. Zinc L-carnosine suppresses inflammatory responses in lipopolysaccharide-induced RAW 264.7 murine macrophages cell line via activation of Nrf2/HO-1 signaling pathway. Immunopharmacol. Immunotoxicol. 2017, 39, 259–267. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Hamada, N.; Tanaka, A.; Fujita, Y.; Itoh, T.; Ono, Y.; Kitagawa, Y.; Tomimori, N.; Kiso, Y.; Akao, Y.; Nozawa, Y.; et al. Involvement of heme oxygenase-1 induction via Nrf2/ARE activation in protection against H2O2-induced PC12 cell death by a metabolite of sesamin contained in sesame seeds. Bioorg. Med. Chem. 2011, 19, 1959–1965. [Google Scholar] [CrossRef]
- Jain, A.D.; Potteti, H.; Richardson, B.G.; Kingsley, L.; Luciano, J.P.; Ryuzoji, A.F.; Lee, H.; Krunic, A.; Mesecar, A.D.; Reddy, S.P.; et al. Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur. J. Med. Chem. 2015, 103, 252–268. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumaru, D.; Motohashi, H. The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929. https://doi.org/10.3390/antiox10121929
Matsumaru D, Motohashi H. The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants. 2021; 10(12):1929. https://doi.org/10.3390/antiox10121929
Chicago/Turabian StyleMatsumaru, Daisuke, and Hozumi Motohashi. 2021. "The KEAP1-NRF2 System in Healthy Aging and Longevity" Antioxidants 10, no. 12: 1929. https://doi.org/10.3390/antiox10121929
APA StyleMatsumaru, D., & Motohashi, H. (2021). The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants, 10(12), 1929. https://doi.org/10.3390/antiox10121929