
Aβ1–40-Induced Platelet Adhesion Is Ameliorated by Rosmarinic Acid through Inhibition of NADPH Oxidase/PKC-δ/Integrin αIIbβ3 Signaling


Abstract
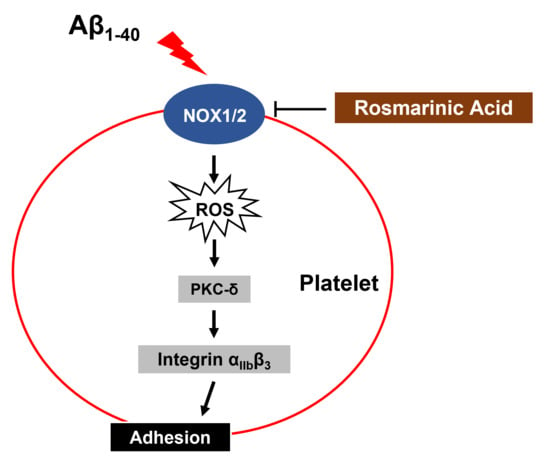
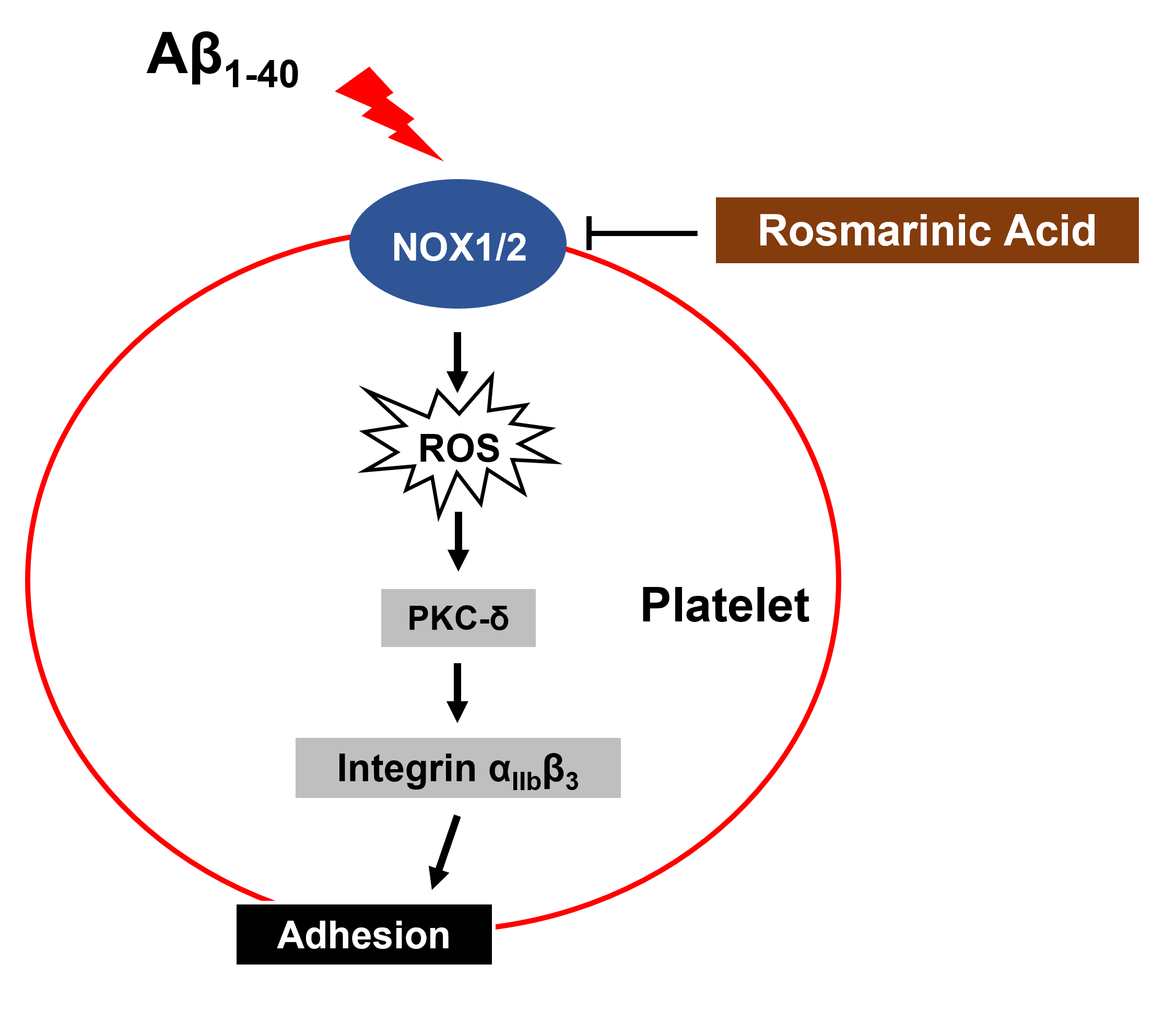{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Washed Platelet Preparation
2.3. Aβ1–40 Preparation
2.4. Platelet Adhesion to Fibronectin
2.5. Measurement of Filopodia Length and Spread Area in Platelet
2.6. Surface Expression of Integrins αIIb and β3
2.7. Measurement of Free-Radical-Scavenging Activity by DPPH Assay
2.8. ABTS Assay for Free-Radical-Scavenging Activity
2.9. ROS Measurement
2.10. Measurement of NADPH Oxidase Activity
2.11. Preparation of the PKC Membrane Fraction
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
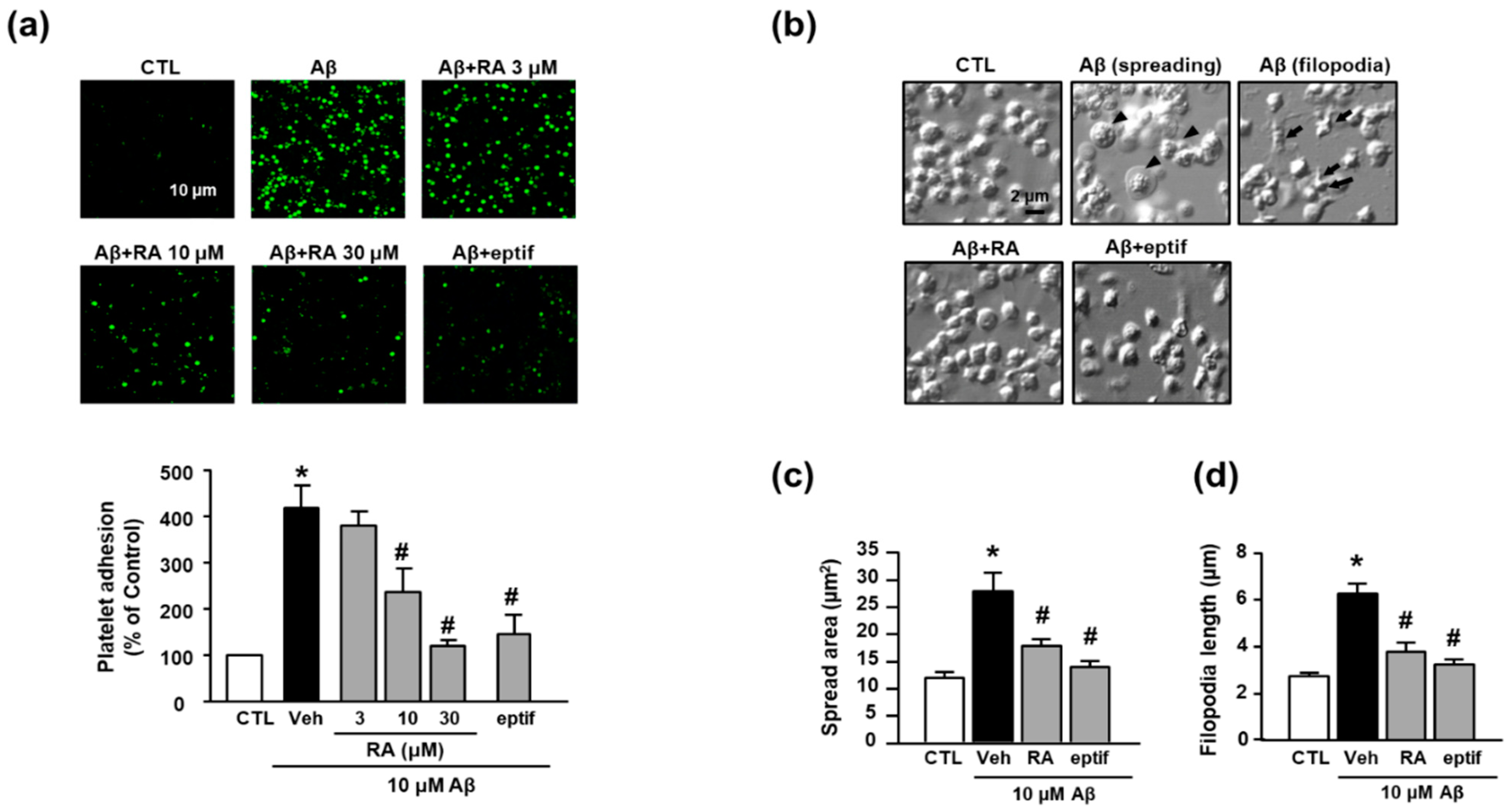
3.1. RA Reduces Aβ1–40-Induced Platelet Adhesion via Integrin αIIbβ3 Blockade
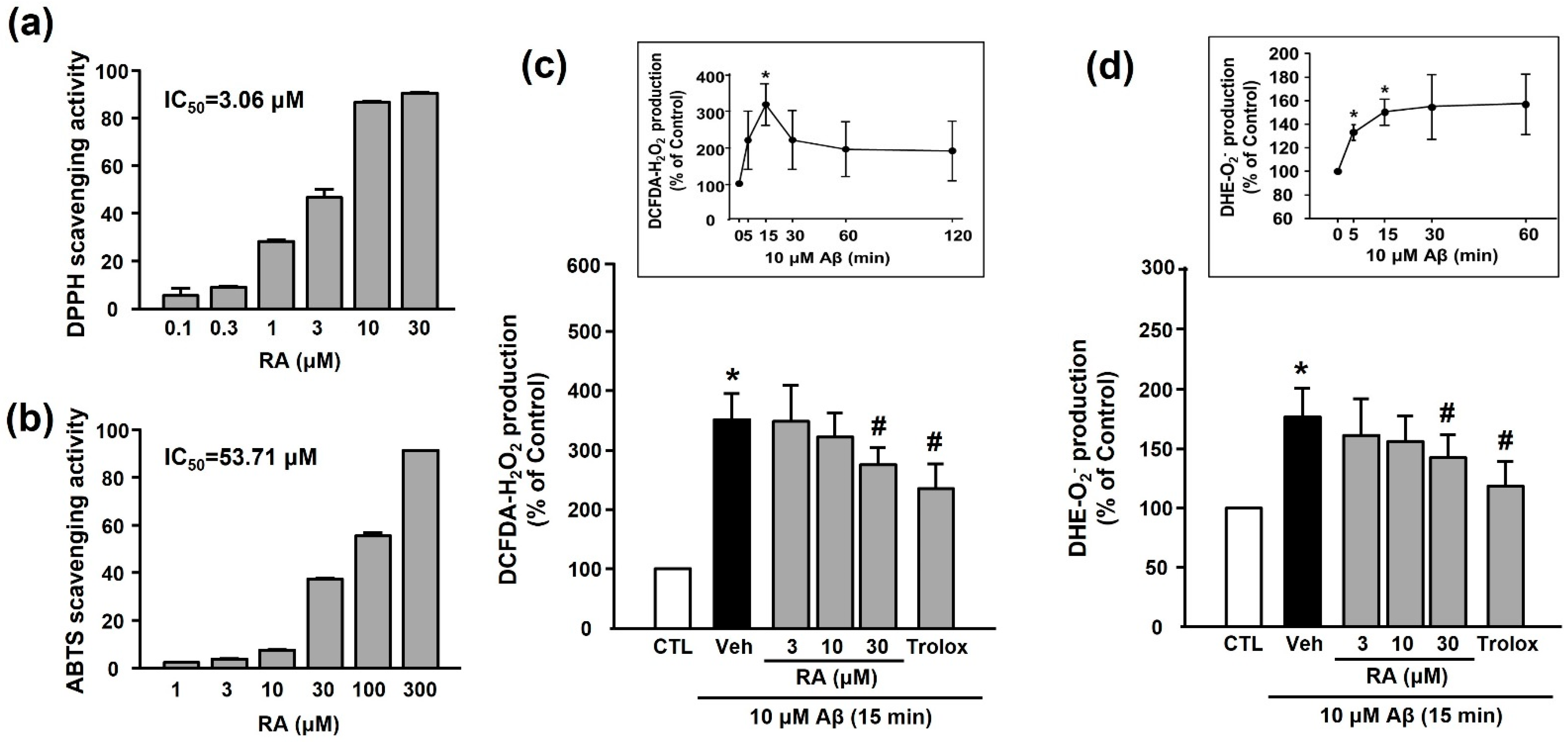
3.2. Antioxidant Activity of RA
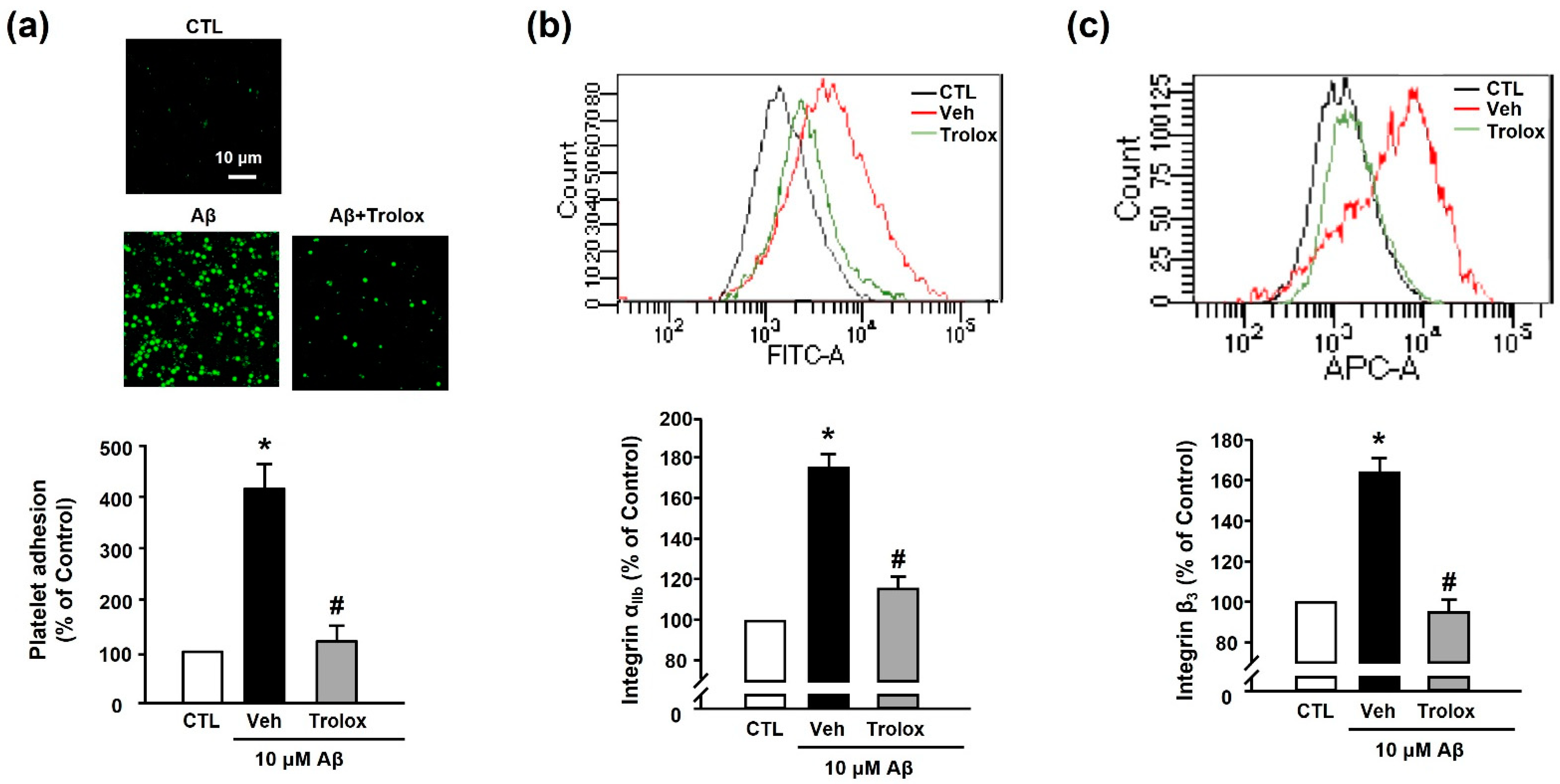
3.3. Effects of Trolox on Aβ1–40-Induced Platelet Adhesion and Integrin αIIbβ3 Activation
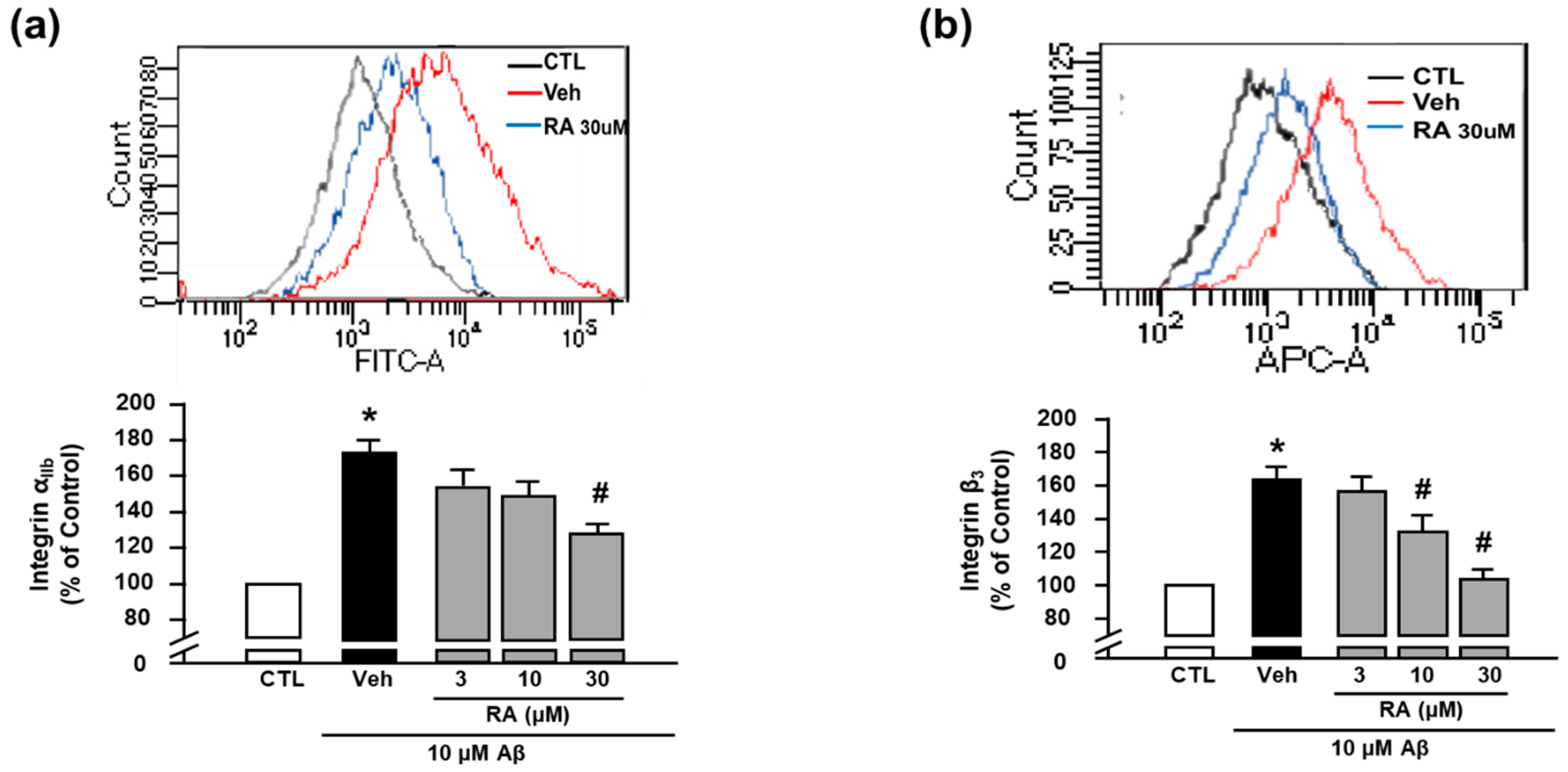
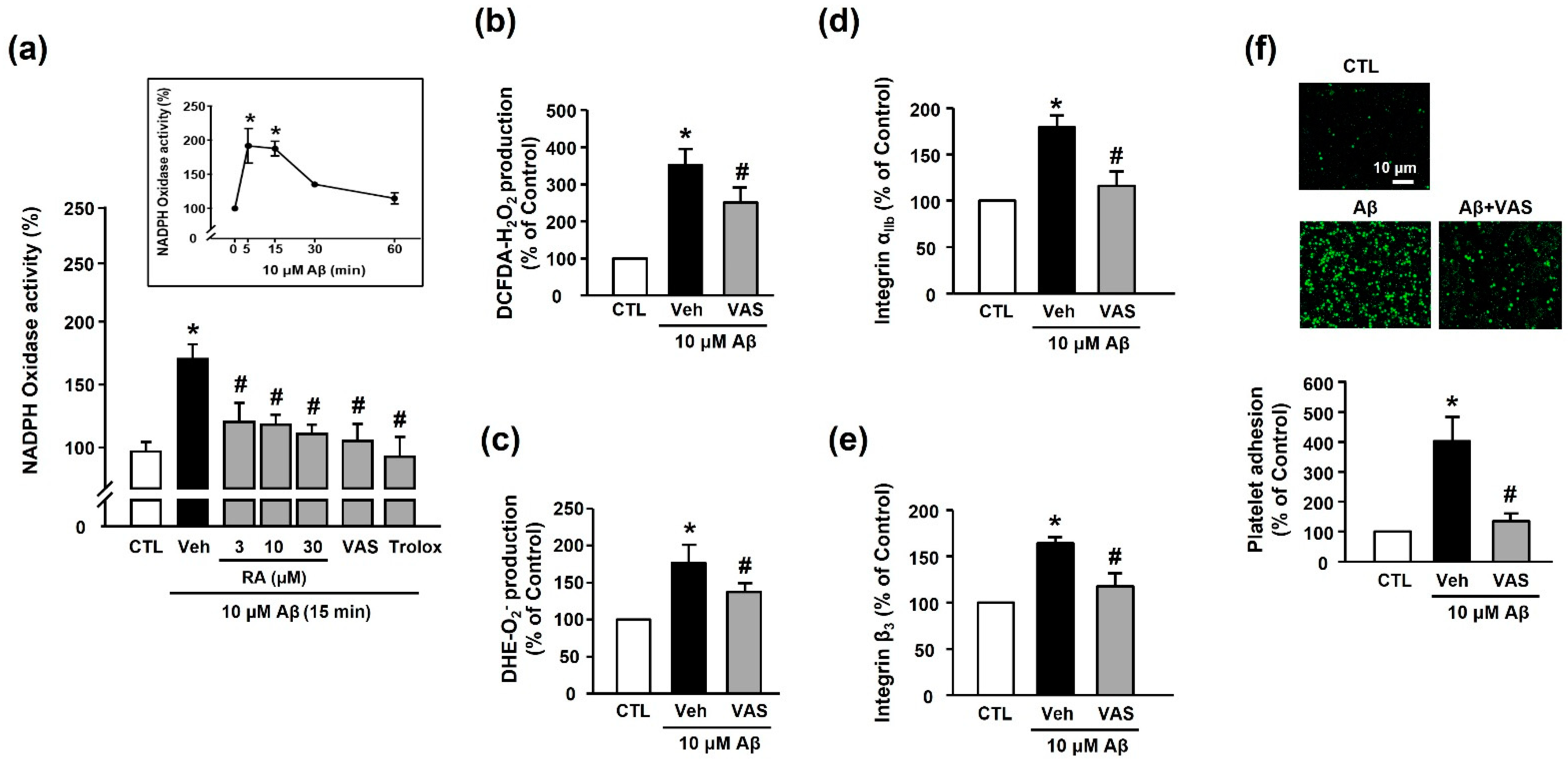
3.4. RA Decreases Aβ1–40-Induced Platelet Activation Possibly through Inhibition of NADPH Oxidase
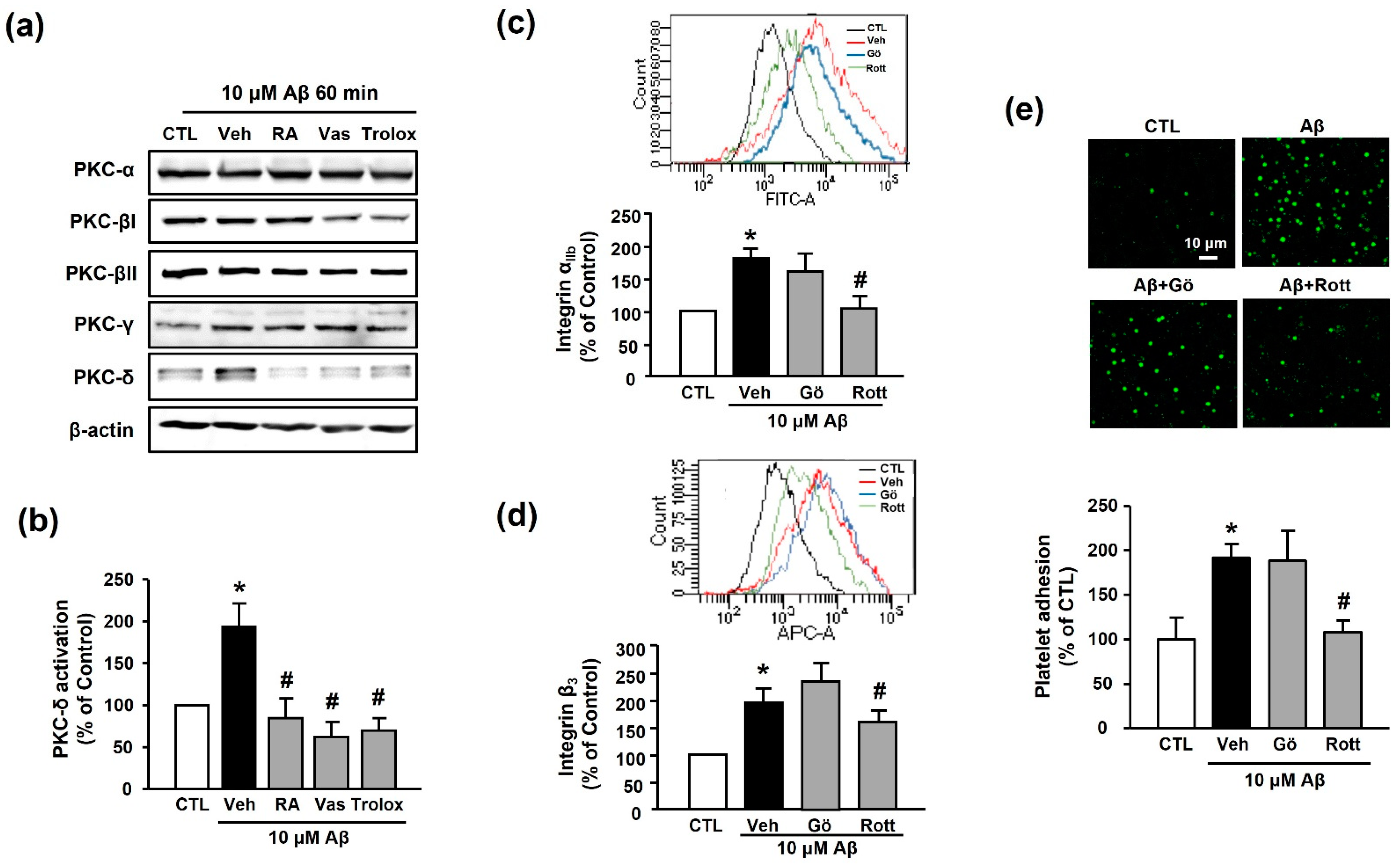
3.5. RA Decreases Aβ1–40-Induced Platelet Activation Possibly through Inhibition of PKC-δ
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laske, C.; Sopova, K.; Stellos, K. Platelet activation in Alzheimer’s disease: From pathophysiology to clinical value. Curr. Vasc. Pharmacol. 2012, 10, 626–630. [Google Scholar] [CrossRef]
- Song, J.; Lee, W.T.; Park, K.A.; Lee, J.E. Association between risk factors for vascular dementia and adiponectin. BioMed Res. Int. 2014, 2014, 261672. [Google Scholar] [CrossRef] [PubMed]
- Raz, L.; Knoefel, J.; Bhaskar, K. The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab. 2016, 36, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Kljajevic, V. Overestimating the effects of healthy aging. Front. Aging Neurosci. 2015, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Donner, L.; Fälker, K.; Gremer, L.; Klinker, S.; Pagani, G.; Ljungberg, L.U.; Lothmann, K.; Rizzi, F.; Schaller, M.; Gohlke, H.; et al. Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci. Signal 2016, 9, ra52. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liang, Y.; Delaney, M.K.; Zhang, Y.; Kim, K.; Li, J.; Bai, Y.; Cho, J.; Ushio-Fukai, M.; Cheng, N.; et al. Shear and integrin outside-in signaling activate NADPH-oxidase 2 to promote platelet activation. Arter. Thromb. Vasc. Biol. 2021, 41, 1638–1653. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C.; Sexton, T.; Smyth, S.S. Translational implications of platelets as vascular first responders. Circ. Res. 2018, 122, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Canobbio, I.; Abubaker, A.A.; Visconte, C.; Torti, M.; Pula, G. Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Berndt, M.C.; Bush, A.I.; Rumble, B.; Mackenzie, I.; Friedhuber, A.; Beyreuther, K.; Masters, C.L. Membrane-associated forms of the beta A4 amyloid protein precursor of Alzheimer’s disease in human platelet and brain: Surface expression on the activated human platelet. Blood 1994, 84, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Visconte, C.; Canino, J.; Vismara, M.; Guidetti, G.F.; Raimondi, S.; Pula, G.; Torti, M.; Canobbio, I. Fibrillar amyloid peptides promote platelet aggregation through the coordinated action of ITAM- and ROS-dependent pathways. J. Thromb. Haemost. 2020, 18, 3029–3042. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Davidson, B.P.; Yue, Q.; Belcik, T.; Xie, A.; Inaba, Y.; McCarty, O.J.; Tormoen, G.W.; Zhao, Y.; Ruggeri, Z.M.; et al. Molecular imaging of inflammation and platelet adhesion in advanced atherosclerosis effects of antioxidant therapy with NADPH oxidase inhibition. Circ. Cardiovasc. Imaging 2013, 6, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Cifuentes-Pagano, E.; Pagano, P.J.; Pula, G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica 2019, 104, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arter. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb. Haemost. 2014, 111, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.P.; de Castro, A.A.; Soares, F.V.; da Cunha, E.F.F.; Ramalho, T.C. Future therapeutic perspectives into the Alzheimer’s disease targeting the oxidative stress hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, parkinson’s disease, and huntington’s disease: A mini review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Petersen, M.; Simmonds, M.S.J. Rosmarinic acid. Phytochemistry 2003, 62, 121–125. [Google Scholar] [CrossRef]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Toyoda, T.; Abe, K.; Hamaguchi, T.; Ono, K.; et al. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid beta aggregation by increasing monoamine secretion. Sci. Rep. 2019, 9, 8711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, J.; Zhu, H.; Jiang, X.; Liu, J.; Xu, W.; Ma, H.; Feng, Q.; Wu, J.; Zhao, M.; et al. Aqueous extract of rabdosia rubescens leaves: Forming nanoparticles, targeting P-selectin, and inhibiting thrombosis. Int. J. Nanomed. 2015, 10, 6905–6918. [Google Scholar] [CrossRef]
- Chapado, L.; Linares-Palomino, P.J.; Salido, S.; Altarejos, J.; Rosado, J.A.; Salido, G.M. Synthesis and evaluation of the platelet antiaggregant properties of phenolic antioxidants structurally related to rosmarinic acid. Bioorg. Chem. 2010, 38, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Jung, S.Y.; Choi, S.H.; Yoo, S.Y.; Baek, S.H.; Kwon, S.M. Modulation of human cardiac progenitors via hypoxia-ERK circuit improves their functional bioactivities. Biomol. Ther. 2013, 21, 196–203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yacoub, D.; Théorêt, J.F.; Villeneuve, L.; Abou-Saleh, H.; Mourad, W.; Allen, B.G.; Merhi, Y. Essential role of protein kinase C delta in platelet signaling, alpha IIb beta 3 activation, and thromboxane A2 release. J. Biol. Chem. 2006, 281, 30024–30035. [Google Scholar] [CrossRef] [PubMed]
- Catricala, S.; Torti, M.; Ricevuti, G. Alzheimer disease and platelets: How’s that relevant. Immun. Ageing 2012, 9, 20. [Google Scholar] [CrossRef]
- Hampel, H.; Shen, Y.; Walsh, D.M.; Aisen, P.; Shaw, L.M.; Zetterberg, H.; Trojanowski, J.Q.; Blennow, K. Biological markers of amyloid beta-related mechanisms in Alzheimer’s disease. Exp. Neurol. 2010, 223, 334–346. [Google Scholar] [CrossRef]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Sturchler-Pierrat, C.; Burki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.A.; Long, H.J.; Eisenhauer, P.B.; Hastey, R.; Cribbs, D.H.; Fine, R.E.; Simons, E.R. Beta amyloid fragments derived from activated platelets deposit in cerebrovascular endothelium: Usage of a novel blood brain barrier endothelial cell model system. Amyloid 2000, 7, 153–165. [Google Scholar] [CrossRef]
- Lee, B.K.; Kim, M.H.; Lee, S.Y.; Son, S.J.; Hong, C.H.; Jung, Y.S. Downregulated platelet miR-1233-5p in patients with Alzheimer’s pathologic change with mild cognitive impairment is associated with abeta-induced platelet activation via P-selectin. J. Clin. Med. 2020, 9, 1642. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and beta-amyloid protein in Alzheimer’s disease. Neuromol. Med. 2011, 13, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Baldeiras, I.; Santana, I.; Proença, M.T.; Garrucho, M.H.; Pascoal, R.; Rodrigues, A.; Duro, D.; Oliveira, C.R. Peripheral oxidative damage in mild cognitive impairment and mild Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 15, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Greilberger, J.; Koidl, C.; Greilberger, M.; Lamprecht, M.; Schroecksnadel, K.; Leblhuber, F.; Fuchs, D.; Oettl, K. Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer’s disease. Free Radic. Res. 2008, 42, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, A.A.; Vara, D.; Eggleston, I.; Canobbio, I.; Pula, G. A novel flow cytometry assay using dihydroethidium as redox-sensitive probe reveals NADPH oxidase-dependent generation of superoxide anion in human platelets exposed to amyloid peptide β. Platelets 2019, 30, 181–189. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Klann, E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 2007, 9, 233–244. [Google Scholar] [CrossRef]
- Narayan, P.; Holmstrom, K.M.; Kim, D.H.; Whitcomb, D.J.; Wilson, M.R.; St George-Hyslop, P.; Wood, N.W.; Dobson, C.M.; Cho, K.; Abramov, A.Y.; et al. Rare individual amyloid-beta oligomers act on astrocytes to initiate neuronal damage. Biochemistry 2014, 53, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, A.A.; Vara, D.; Visconte, C.; Eggleston, I.; Torti, M.; Canobbio, I.; Pula, G. Amyloid peptide beta1-42 induces integrin alphaIIbbeta3 activation, platelet adhesion, and thrombus formation in a NADPH oxidase-dependent manner. Oxid. Med. Cell. Longev. 2019, 2019, 1050476. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S.; Jee, Y.; Lee, S.H.; Kim, S. Characterization of integrin alphaIIbbeta3-mediated outside-in signaling by protein kinase cdelta in platelets. Int. J. Mol. Sci. 2020, 21, 6563. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Habtemariam, S. Molecular pharmacology of rosmarinic and salvianolic acids: Potential seeds for Alzheimer’s and vascular dementia drugs. Int. J. Mol. Sci. 2018, 19, 458. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.K.; Jee, H.J.; Jung, Y.-S. Aβ1–40-Induced Platelet Adhesion Is Ameliorated by Rosmarinic Acid through Inhibition of NADPH Oxidase/PKC-δ/Integrin αIIbβ3 Signaling. Antioxidants 2021, 10, 1671. https://doi.org/10.3390/antiox10111671
Lee BK, Jee HJ, Jung Y-S. Aβ1–40-Induced Platelet Adhesion Is Ameliorated by Rosmarinic Acid through Inhibition of NADPH Oxidase/PKC-δ/Integrin αIIbβ3 Signaling. Antioxidants. 2021; 10(11):1671. https://doi.org/10.3390/antiox10111671
Chicago/Turabian StyleLee, Bo Kyung, Hye Jin Jee, and Yi-Sook Jung. 2021. "Aβ1–40-Induced Platelet Adhesion Is Ameliorated by Rosmarinic Acid through Inhibition of NADPH Oxidase/PKC-δ/Integrin αIIbβ3 Signaling" Antioxidants 10, no. 11: 1671. https://doi.org/10.3390/antiox10111671
APA StyleLee, B. K., Jee, H. J., & Jung, Y.-S. (2021). Aβ1–40-Induced Platelet Adhesion Is Ameliorated by Rosmarinic Acid through Inhibition of NADPH Oxidase/PKC-δ/Integrin αIIbβ3 Signaling. Antioxidants, 10(11), 1671. https://doi.org/10.3390/antiox10111671