
Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration

 ,
,  ,
,  , and
, and 
Abstract
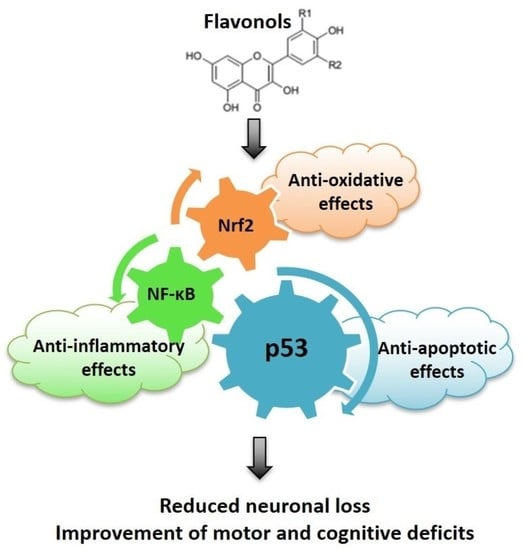
1. Introduction
2. Oxidative Stress in Neurodegenerative Diseases: The Role of Nrf2 Pathway
3. Neuroinflammation in Neurodegenerative Diseases: The Role of NF-κB Pathway
4. The Role of p53 in Neurodegenerative Diseases
4.1. Biological Functions and Structure of p53
4.2. Regulation of p53 Activity
4.3. Role of p53 in Neuronal Death
4.4. Role of p53 in Neurodegenerative Diseases
4.4.1. p53 Functions in Oxidative Stress
p53 and Mitochondrial Functions in Neurodegenerative Conditions
4.4.2. p53 and Neuroinflammation
5. Flavonols as Therapeutic Option in Neurodegenerative Diseases
5.1. Quercetin
5.2. Rutin
5.3. Myricetin
Dihydromyricetin
5.4. Myricitrin
5.5. Fisetin
5.6. Kaempferol
5.7. Morin
5.8. Isorhamnetin
5.9. Rhamnazin
5.10. Azaleatin
5.11. Gossypetin
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maresova, P.; Hruska, J.; Klimova, B.; Barakovic, S.; Krejcar, O. Activities of daily living and associated costs in the most widespread neurodegenerative diseases: A systematic review. Clin. Interv. Aging 2020, 15, 1841–1862. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s disease: Key mediators of neuronal apoptosis induced by oxidative stress and Aβ accumulation. Oxid. Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of oxidative stress and misfolded proteins in the mechanism of neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Michalska, P.; León, R. When it comes to an end: Oxidative stress crosstalk with protein aggregation and neuroinflammation induce neurodegeneration. Antioxidants 2020, 9, 740. [Google Scholar] [CrossRef]
- Behl, T.; Makkar, R.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Bungau, S.; Andronie-Cioara, F.L.; Munteanu, M.A.; Brisc, M.C.; et al. Current trends in neurodegeneration: Cross talks between oxidative stress, cell death, and inflammation. Int. J. Mol. Sci. 2021, 22, 7432. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Zhang, M.; Zhou, M.; Ye, L.; Xia, H. Mitophagy regulates neurodegenerative diseases. Cells 2021, 10, 1876. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, J.; Shin, C.; Lee, S. Intersection between redox homeostasis and autophagy: Valuable insights into neurodegeneration. Antioxidants 2021, 10, 694. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Slade, N.; Hof, P.R.; Šimić, G. The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-r.; Liu, R.-t. The toxicity and polymorphism of β-amyloid oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef]
- Sciaccaluga, M.; Megaro, A.; Bellomo, G.; Ruffolo, G.; Romoli, M.; Palma, E.; Costa, C. An unbalanced synaptic transmission: Cause or consequence of the amyloid oligomers neurotoxicity? Int. J. Mol. Sci. 2021, 22, 5991. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Esteves, A.R.; Arduíno, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Oxidative stress involvement in alpha-synuclein oligomerization in Parkinson’s disease cybrids. Antioxid. Redox Signal. 2009, 11, 439–448. [Google Scholar] [CrossRef]
- Menges, S.; Minakaki, G.; Schaefer, P.M.; Meixner, H.; Prots, I.; Schlötzer-Schrehardt, U.; Friedland, K.; Winner, B.; Outeiro, T.F.; Winklhofer, K.F.; et al. Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci. Rep. 2017, 7, 42942. [Google Scholar] [CrossRef] [PubMed]
- Scudamore, O.; Ciossek, T. Increased oxidative stress exacerbates α-synuclein aggregation in vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Du, X.-y.; Xie, X.-x.; Liu, R.-t. The role of α-synuclein oligomers in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef] [PubMed]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Long. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Haque, M.M.; Murale, D.P.; Kim, Y.K.; Lee, J.-S. Crosstalk between oxidative stress and tauopathy. Int. J. Mol. Sci. 2019, 20, 1959. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Šimić, G.; Stanić, G.; Mladinov, M.; Jovanov Milošević, N.; Kostović, I.; Hof, P.R. Does Alzheimer’s disease begin in the brainstem? Neuropathol. Appl. Neurobiol. 2009, 35, 532–554. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Španić, E.; Langer Horvat, L.; Hof, P.R.; Šimić, G. Role of microglial cells in Alzheimer’s disease tau propagation. Front. Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative links between protein aggregation and oxidative stress: A review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [PubMed]
- Sonninen, T.-M.; Goldsteins, G.; Laham-Karam, N.; Koistinaho, J.; Lehtonen, Š. Proteostasis disturbances and inflammation in neurodegenerative diseases. Cells 2020, 9, 2183. [Google Scholar] [CrossRef]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cué, C.; Rueda, N. Signalling pathways implicated in Alzheimer′s disease neurodegeneration in Individuals with and without Down syndrome. Int. J. Mol. Sci. 2020, 21, 6906. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The role of mitochondrial calcium homeostasis in Alzheimer’s and related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef]
- Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.; Yousefian-Jazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power failure of mitochondria and oxidative stress in neurodegeneration and its computational models. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Cecchi, C.; Pensalfini, A.; Bonini, S.A.; Ferrari-Toninelli, G.; Liguri, G.; Memo, M.; Uberti, D. Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids 2010, 38, 1101–1106. [Google Scholar] [CrossRef]
- Lee, K.H.; Lee, S.J.; Lee, H.J.; Choi, G.E.; Jung, Y.H.; Kim, D.I.; Gabr, A.A.; Ryu, J.M.; Han, H.J. Amyloid β1-42 (Aβ1-42) induces the CDK2-mediated phosphorylation of tau through the activation of the mTORC1 signaling pathway while promoting neuronal cell death. Front. Mol. Neurosci. 2017, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Lucassen, P.J.; Krsnik, Ž.; Krušlin, B.; Kostović, I.; Winblad, B.; Bogdanović, N. nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Exp. Neurol. 2000, 165, 12–26. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Hansson Petersen, C.; Glaser, E.; Ankarcrona, M. Mitochondrial accumulation of APP and Abeta: Significance for Alzheimer disease pathogenesis. J. Cell. Mol. Med. 2009, 13, 4137–4145. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Vlainić, J.; Radovanović, V.; Erhardt, J.; Oršolić, N. Effects of copper overload in P19 neurons: Impairment of glutathione redox homeostasis and crosstalk between caspase and calpain protease systems in ROS-induced apoptosis. Biometals 2014, 27, 1303–1322. [Google Scholar] [CrossRef] [PubMed]
- Zubčić, K.; Hof, P.R.; Šimić, G.; Jazvinšćak Jembrek, M. The role of copper in tau-related pathology in Alzheimer’s disease. Front. Mol. Neurosci. 2020, 13, 572308. [Google Scholar] [CrossRef]
- Ejaz, H.W.; Wang, W.; Lang, M. Copper toxicity links to pathogenesis of Alzheimer’s disease and therapeutics approaches. Int. J. Mol. Sci. 2020, 21, 7660. [Google Scholar] [CrossRef]
- Gu, M.; Bode, D.C.; Viles, J.H. Copper redox cycling inhibits Aβ fibre formation and promotes fibre fragmentation, while generating a dityrosine Aβ dimer. Sci Rep. 2018, 8, 16190. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Babić, M.; Pivac, N.; Hof, P.R.; Šimić, G. Hyperphosphorylation of tau by GSK-3β in Alzheimer’s disease: The interaction of Aβ and sphingolipid mediators as a therapeutic target. Translat. Neurosci. 2013, 4, 466–476. [Google Scholar]
- Islam Khan, R.; Nirzhor, S.S.R.; Rashid, B. A closer look into the role of protein tau in the identification of promising therapeutic targets for Alzheimer’s disease. Brain Sci. 2018, 8, 162. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Pesce, M.; Patruno, A.; Moradi, S.Z.; Iranpanah, A.; Farzaei, M.H.; Sobarzo-Sánchez, E. Attenuation of Nrf2/Keap1/ARE in Alzheimer’s disease by plant secondary metabolites: A mechanistic review. Molecules 2020, 25, 4926. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Jo, M.G.; Kim, M.O. 17-β estradiol rescued immature rat brain against glutamate-induced oxidative stress and neurodegeneration via regulating Nrf2/HO-1 and MAP-kinase signaling pathway. Antioxidants 2021, 10, 892. [Google Scholar] [CrossRef] [PubMed]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 pathway on oxidative damage and mitochondrial failure in Parkinson and Alzheimer’s disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef]
- Devi, S.; Kumar, V.; Singh, S.K.; Dubey, A.K.; Kim, J.-J. Flavonoids: Potential candidates for the treatment of neurodegenerative disorders. Biomedicines 2021, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Aloi, M.S.; Su, W.; Garden, G.A. The p53 transcriptional network influences microglia behavior and neuroinflammation. Crit. Rev. Immunol. 2015, 35, 401–415. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Nosi, D.; Lana, D.; Giovannini, M.G.; Delfino, G.; Zecchi-Orlandini, S. Neuroinflammation: Integrated nervous tissue response through intercellular interactions at the “whole system” scale. Cells 2021, 10, 1195. [Google Scholar] [CrossRef]
- Kaminska, B.; Gozdz, A.; Zawadzka, M.; Ellert-Miklaszewska, A.; Lipko, M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat. Rec. 2009, 292, 1902–1913. [Google Scholar] [CrossRef]
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 2017, 31, 5–10. [Google Scholar] [CrossRef]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediators Inflamm. 2013, 2013, 342931. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Keogh, C.E.; Rude, K.M.; Gareau, M.G. Role of pattern recognition receptors and the microbiota in neurological disorders. J. Physiol. 2021, 599, 1379–1389. [Google Scholar] [CrossRef]
- Ginwala, R.; Bhavsar, R.; Chigbu, D.I.; Jain, P.; Khan, Z.K. Potential role of flavonoids in treating chronic inflammatory diseases with a special focus on the anti-inflammatory activity of apigenin. Antioxidants 2019, 8, 35. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed]
- Torreilles, F.; Salman-Tabcheh, S.; Guérin, M.; Torreilles, J. Neurodegenerative disorders: The role of peroxynitrite. Brain Res. Rev. 1999, 30, 153–163. [Google Scholar] [CrossRef]
- Bal-Price, A.; Brown, G.C. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflamm. 2005, 2, 20. [Google Scholar] [CrossRef]
- Zhou, F.; Qu, L.; Lv, K.; Chen, H.; Liu, J.; Liu, X.; Li, Y.; Sun, X. Luteolin protects against reactive oxygen species-mediated cell death induced by zinc toxicity via the PI3K-Akt-NF-κB-ERK-dependent pathway. J. Neurosci. Res. 2011, 89, 1859–1868. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Johnson, D.A.; Johnson, J.A. Nrf2-a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Heo, S.; Kim, D.-W.; Kim, I.-S.; Ahn, D.; Tae, H.-J.; Kim, M.-K.; Park, B.-Y. Ethanol extract of Maclura tricuspidata fruit protects SH-SY5Y neuroblastoma cells against H2O2-induced oxidative damage via inhibiting MAPK and NF-κB signaling. Int. J. Mol. Sci. 2021, 22, 6946. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B.E. Role of p53 in neurodegenerative diseases. Neurodegener. Dis. 2012, 9, 68–80. [Google Scholar] [CrossRef]
- Szybińska, A.; Leśniak, W. p53 dysfunction in neurodegenerative diseases—The cause or effect of pathological changes? Aging Dis. 2017, 8, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.K.; Leu, J.I.; Zhou, Y.; Devarajan, K.; Nedelko, T.; Klein-Szanto, A.; Hollstein, M.; Murphy, M.E. The codon 72 polymorphism of p53 regulates interaction with NF-κB and transactivation of genes involved in immunity and inflammation. Mol. Cell. Biol. 2011, 31, 1201–1213. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, J.; Feng, Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013, 3, 9. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Kubra, K.T.; Akhter, M.S.; Uddin, M.A.; Barabutis, N. P53 versus inflammation: An update. Cell Cycle 2020, 19, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Lanni, C.; Govoni, S.; Uberti, D.; D’Orazi, G.; Racchi, M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: Is HIPK2 the link? Aging 2010, 2, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Aprigliano, R.; Aksu, M.E.; Bradamante, S.; Mihaljevic, B.; Wang, W.; Rian, K.; Montaldo, N.P.; Grooms, K.M.; Fordyce Martin, S.L.; Bordin, D.L.; et al. Increased p53 signaling impairs neural differentiation in HUWE1-promoted intellectual disabilities. Cell Rep. Med. 2021, 2, 100240. [Google Scholar] [CrossRef]
- Maor-Nof, M.; Shipony, Z.; Lopez-Gonzalez, R.; Nakayama, L.; Zhang, Y.J.; Couthouis, J.; Blum, J.A.; Castruita, P.A.; Linares, G.R.; Ruan, K.; et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708. [Google Scholar] [CrossRef]
- Vieler, M.; Sanyal, S. p53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Anbarasan, T.; Bourdon, J.C. The emerging landscape of p53 isoforms in physiology, cancer and degenerative diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef]
- Horvat, A.; Tadijan, A.; Vlašić, I.; Slade, N. p53/p73 protein network in colorectal cancer and other muman malignancies. Cancers 2021, 13, 2885. [Google Scholar] [CrossRef]
- Chatoo, W.; Abdouh, M.; Bernier, G. p53 pro-oxidant activity in the central nervous system: Implication in aging and neurodegenerative diseases. Antioxid. Redox Signal. 2011, 15, 1729–1737. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef]
- Eizenberg, O.; Faber-Elman, A.; Gottlieb, E.; Oren, M.; Rotter, V.; Schwartz, M. p53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol. Cell. Biol. 1996, 16, 5178–5185. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.B.; Kaplan, D.R.; Miller, F.D. The p53 family in nervous system development and disease. J. Neurochem. 2006, 97, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Di Giovanni, S. The non-apoptotic role of p53 in neuronal biology: Enlightening the dark side of the moon. EMBO Rep. 2009, 10, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Marin Navarro, A.; Pronk, R.J.; van der Geest, A.T.; Oliynyk, G.; Nordgren, A.; Arsenian-Henriksson, M.; Falk, A.; Wilhelm, M. p53 controls genomic stability and temporal differentiation of human neural stem cells and affects neural organization in human brain organoids. Cell Death Dis. 2020, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Mandir, A.S.; Simbulan-Rosenthal, C.M.; Poitras, M.F.; Lumpkin, J.R.; Dawson, V.L.; Smulson, M.E.; Dawson, T.M. A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J. Neurochem. 2002, 83, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.D.; Wu, G.; Liu, Z.R. Dynamics of posttranslational modifications of p53. Comput. Math. Methods Med. 2014, 2014, 245610. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85. [Google Scholar] [CrossRef]
- Carr, M.I.; Jones, S.N. Regulation of the Mdm2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res. 2016, 5, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Mancini, F.; Pieroni, L.; Monteleone, V.; Lucà, R.; Fici, L.; Luca, E.; Urbani, A.; Xiong, S.; Soddu, S.; Masetti, R.; et al. MDM4/HIPK2/p53 cytoplasmic assembly uncovers coordinated repression of molecules with anti-apoptotic activity during early DNA damage response. Oncogene 2016, 35, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Gu, W. SIRT1: Regulator of p53 Deacetylation. Genes Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Raz, L.; Zhang, Q.G.; Han, D.; Dong, Y.; De Sevilla, L.; Brann, D.W. Acetylation of the pro-apoptotic factor, p53 in the hippocampus following cerebral ischemia and modulation by estrogen. PLoS ONE 2011, 6, e27039. [Google Scholar] [CrossRef]
- Demyanenko, S.; Sharifulina, S. The role of post-translational acetylation and deacetylation of signaling proteins and transcription factors after cerebral ischemia: Facts and hypotheses. Int. J. Mol. Sci. 2021, 22, 7947. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Veenstra, T.D.; Morrison, R.S. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J. Neurosci. 2009, 29, 2824–2832. [Google Scholar] [CrossRef]
- Brochier, C.; Dennis, G.; Rivieccio, M.A.; McLaughlin, K.; Coppola, G.; Ratan, R.R.; Langley, B. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci. 2013, 33, 8621–8632. [Google Scholar] [CrossRef]
- Lanni, C.; Nardinocchi, L.; Puca, R.; Stanga, S.; Uberti, D.; Memo, M.; Govoni, S.; D’Orazi, G.; Racchi, M. Homeodomain interacting protein kinase 2: A target for Alzheimer’s beta amyloid leading to misfolded p53 and inappropriate cell survival. PLoS ONE 2010, 5, e10171. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, V.; Agam, G.; Lawatscheck, J.; Rutz, D.A.; Lamb, D.C.; Buchner, J. Coordinated conformational processing of the tumor suppressor protein p53 by the Hsp70 and Hsp90 chaperone machineries. Mol. Cell 2019, 74, 816–830.e7. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Lambert, J.M.; Hainaut, P.; Wiman, K.G. Mutant p53 rescue and modulation of p53 redox state. Cell Cycle 2009, 8, 2509–2517. [Google Scholar] [CrossRef] [PubMed]
- Formigari, A.; Gregianin, E.; Irato, P. The effect of zinc and the role of p53 in copper-induced cellular stress responses. J. Appl. Toxicol. 2013, 33, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Phatak, V.M.; Muller, P.A.J. Metal toxicity and the p53 protein: An intimate relationship. Toxicol. Res. 2015, 4, 576–591. [Google Scholar] [CrossRef]
- Nijboer, C.H.; Heijnen, C.J.; van der Kooij, M.A.; Zijlstra, J.; van Velthoven, C.T.; Culmsee, C.; van Bel, F.; Hagberg, H.; Kavelaars, A. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann. Neurol. 2011, 70, 255–264. [Google Scholar] [CrossRef]
- Mellett, K.; Ren, D.; Alexander, S.; Osier, N.; Beers, S.R.; Okonkwo, D.O.; Puccio, A.M.; Conley, Y.P. Genetic variation in the TP53 gene and patient outcomes following severe traumatic brain injury. Biol. Res. Nurs. 2020, 22, 334–340. [Google Scholar] [CrossRef]
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell. Oncol. 2014, 1, e955995. [Google Scholar] [CrossRef]
- Steckley, D.; Karajgikar, M.; Dale, L.B.; Fuerth, B.; Swan, P.; Drummond-Main, C.; Poulter, M.O.; Ferguson, S.S.; Strasser, A.; Cregan, S.P. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 2007, 27, 12989–12999. [Google Scholar] [CrossRef]
- Hikisz, P.; Kiliańska, Z.M. PUMA, a critical mediator of cell death—One decade on from its discovery. Cell. Mol. Biol. Lett. 2012, 17, 646–669. [Google Scholar] [CrossRef]
- Tuffy, L.P.; Concannon, C.G.; D’Orsi, B.; King, M.A.; Woods, I.; Huber, H.J.; Ward, M.W.; Prehn, J.H. Characterization of Puma-dependent and Puma-independent neuronal cell death pathways following prolonged proteasomal inhibition. Mol. Cell. Biol. 2010, 30, 5484–5501. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Radovanović, V.; Vlainić, J.; Vuković, L.; Hanžić, N. Neuroprotective effect of zolpidem against glutamate-induced toxicity is mediated via the PI3K/Akt pathway and inhibited by PK11195. Toxicology 2018, 406–407, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zubčić, K.; Radovanović, V.; Vlainić, J.; Hof, P.R.; Oršolić, N.; Šimić, G.; Jazvinšćak Jembrek, M. PI3K/Akt and ERK1/2 signalling are involved in quercetin-mediated neuroprotection against copper-induced injury. Oxid. Med. Cell. Longev. 2020, 2020, 9834742. [Google Scholar] [CrossRef] [PubMed]
- Li, M. The role of P53 up-regulated modulator of apoptosis (PUMA) in ovarian development, cardiovascular and neurodegenerative diseases. Apoptosis 2021, 26, 235–247. [Google Scholar] [CrossRef]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/miR-34 axis in development and disease. J. Mol. Cell Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef]
- Speidel, D. Transcription-independent p53 apoptosis: An alternative route to death. Trends Cell Biol. 2010, 20, 14–24. [Google Scholar] [CrossRef]
- Park, J.H.; Ko, J.; Hwang, J.; Koh, H.C. Dynamin-related protein 1 mediates mitochondria-dependent apoptosis in chlorpyrifos-treated SH-SY5Y cells. Neurotoxicology 2015, 51, 145–157. [Google Scholar] [CrossRef]
- Goiran, T.; Duplan, E.; Rouland, L.; El Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Alves da Costa, C. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef]
- Yang, Y.; Karsli-Uzunbas, G.; Poillet-Perez, L.; Sawant, A.; Hu, Z.S.; Zhao, Y.; Moore, D.; Hu, W.; White, E. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev. 2020, 34, 688–700. [Google Scholar] [CrossRef]
- Park, J.H.; Zhuang, J.; Li, J.; Hwang, P.M. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016, 590, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.N.; Yang, L.Y.; Greig, N.H.; Wang, Y.C.; Lai, C.C.; Wang, J.Y. Neuroprotective effects of pifithrin-α against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 2018, 8, 2368. [Google Scholar] [CrossRef]
- Plesnila, N.; von Baumgarten, L.; Retiounskaia, M.; Engel, D.; Ardeshiri, A.; Zimmermann, R.; Hoffmann, F.; Landshamer, S.; Wagner, E.; Culmsee, C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-ҡB transcriptional activity. Cell Death Differ. 2007, 14, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, X.X.; Cao, Y.; Liang, Z.Q.; Han, R.; Wu, J.C.; Gu, Z.L.; Qin, Z.H. p53 induction contributes to excitotoxic neuronal death in rat striatum through apoptotic and autophagic mechanisms. Eur. J. Neurosci. 2009, 30, 2258–2270. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Sakhi, S.; Sun, N.; Wing, L.L.; Mehta, P.; Schreiber, S.S. Nuclear accumulation of p53 protein following kainic acid-induced seizures. Neuroreport 1996, 7, 493–496. [Google Scholar] [CrossRef]
- Jordán, J.; Galindo, M.F.; Prehn, J.H.; Weichselbaum, R.R.; Beckett, M.; Ghadge, G.D.; Roos, R.P.; Leiden, J.M.; Miller, R.J. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J. Neurosci. 1997, 17, 1397–1405. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, X.; Yu, Q.S.; Chan, S.L.; Camandola, S.; Guo, Z.; Greig, N.H.; Mattson, M.P. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001, 77, 220–228. [Google Scholar] [CrossRef]
- Rachmany, L.; Tweedie, D.; Rubovitch, V.; Yu, Q.S.; Li, Y.; Wang, J.Y.; Pick, C.G.; Greig, N.H. Cognitive impairments accompanying rodent mild traumatic brain injury involve p53-dependent neuronal cell death and are ameliorated by the tetrahydrobenzothiazole PFT-α. PLoS ONE 2013, 8, e79837. [Google Scholar] [CrossRef]
- Neema, M.; Navarro-Quiroga, I.; Chechlacz, M.; Gilliams-Francis, K.; Liu, J.; Lamonica, K.; Lin, S.L.; Naegele, J.R. DNA damage and nonhomologous end joining in excitotoxicity: Neuroprotective role of DNA-PKcs in kainic acid-induced seizures. Hippocampus 2005, 15, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1997, 232, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y.; Asahara, H.; Chui, D.H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Abeta42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 1, 255–257. [Google Scholar]
- Hooper, C.; Meimaridou, E.; Tavassoli, M.; Melino, G.; Lovestone, S.; Killick, R. p53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci. Lett. 2007, 418, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Cenini, G.; Sultana, R.; Perluigi, M.; Uberti, D.; Memo, M.; Butterfield, D.A. Glutathionylation of the pro-apoptotic protein p53 in Alzheimer’s disease brain: Implications for AD pathogenesis. Neurochem. Res. 2009, 34, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. P44, the ‘longevity-assurance’ isoform of P53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef]
- Lapresa, R.; Agulla, J.; Sánchez-Morán, I.; Zamarreño, R.; Prieto, E.; Bolaños, J.P.; Almeida, A. Amyloid-ß promotes neurotoxicity by Cdk5-induced p53 stabilization. Neuropharmacology 2019, 146, 19–27. [Google Scholar] [CrossRef]
- Karunakaran, S.; Saeed, U.; Mishra, M.; Valli, R.K.; Joshi, S.D.; Meka, D.P.; Seth, P.; Ravindranath, V. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J. Neurosci. 2008, 28, 12500–12509. [Google Scholar] [CrossRef]
- Ho, D.H.; Kim, H.; Kim, J.; Sim, H.; Ahn, H.; Kim, J.; Seo, H.; Chung, K.C.; Park, B.J.; Son, I.; et al. Leucine-rich repeat kinase 2 (LRRK2) phosphorylates p53 and induces p21(WAF1/CIP1) expression. Mol. Brain 2015, 8, 54. [Google Scholar] [CrossRef]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef]
- Merlo, P.; Frost, B.; Peng, S.; Yang, Y.J.; Park, P.J.; Feany, M. p53 prevents neurodegeneration by regulating synaptic genes. Proc. Natl. Acad. Sci. USA 2014, 111, 18055–18060. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Knights, C.D.; Rao, M.; Yakovlev, A.; Beers, J.; Catania, J.; Avantaggiati, M.L.; Faden, A.I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006, 25, 4084–4096. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, S.; Rathore, K. p53-dependent pathways in neurite outgrowth and axonal regeneration. Cell Tissue Res. 2012, 349, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Nguyen, T.; Puttagunta, R.; Gaub, P.; Di Giovanni, S. A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 2009, 16, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Vecino, R.; Burguete, M.C.; Jover-Mengual, T.; Agulla, J.; Bobo-Jiménez, V.; Salom, J.B.; Almeida, A.; Delgado-Esteban, M. The MDM2-p53 pathway is involved in preconditioning-induced neuronal tolerance to ischemia. Sci Rep. 2018, 8, 1610. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Cenini, G.; Sultana, R.; Di Domenico, F.; Fiorini, A.; Perluigi, M.; Noel, T.; Wang, C.; Mancuso, C.; St Clair, D.K.; et al. Lack of p53 decreases basal oxidative stress levels in the brain through upregulation of thioredoxin-1, biliverdin reductase-A, manganese superoxide dismutase, and nuclear factor kappa-B. Antioxid. Redox Signal. 2012, 16, 1407–1420. [Google Scholar] [CrossRef]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.; Zhang, J.; Dong, W.; Wu, J.; Wang, T.; Xie, Z. P53 and Parkin co-regulate mitophagy in bone marrow mesenchymal stem cells to promote the repair of early steroid-induced osteonecrosis of the femoral head. Cell Death Dis. 2020, 11, 42. [Google Scholar] [CrossRef]
- Geden, M.J.; Romero, S.E.; Deshmukh, M. p53 is required for nuclear but not mitochondrial DNA damage-induced degeneration. Cell Death Dis. 2021, 12, 104. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S253–S262. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sesaki, H.; Qi, X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem. J. 2014, 461, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Invest. 2013, 123, 5371–5388. [Google Scholar] [CrossRef]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging. 2012, 33, 767–785. [Google Scholar] [CrossRef]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The close encounter between alpha-synuclein and mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865. [Google Scholar] [CrossRef]
- Wilkaniec, A.; Lenkiewicz, A.M.; Babiec, L.; Murawska, E.; Jęśko, H.M.; Cieślik, M.; Culmsee, C.; Adamczyk, A. Exogenous alpha-synuclein evoked parkin downregulation promotes mitochondrial dysfunction in neuronal cells. Implications for Parkinson’s disease pathology. Front. Aging Neurosci. 2021, 13, 591475. [Google Scholar] [CrossRef]
- Alves Da Costa, C.; Paitel, E.; Vincent, B.; Checler, F. Alpha-synuclein lowers p53-dependent apoptotic response of neuronal cells. Abolishment by 6-hydroxydopamine and implication for Parkinson’s disease. J. Biol. Chem. 2002, 277, 50980–50984. [Google Scholar] [CrossRef]
- Duplan, E.; Giordano, C.; Checler, F.; Alves da Costa, C. Direct α-synuclein promoter transactivation by the tumor suppressor p53. Mol. Neurodegener. 2016, 11, 13. [Google Scholar] [CrossRef]
- Jayadev, S.; Nesser, N.K.; Hopkins, S.; Myers, S.J.; Case, A.; Lee, R.J.; Seaburg, L.A.; Uo, T.; Murphy, S.P.; Morrison, R.S.; et al. Transcription factor p53 influences microglial activation phenotype. Glia 2011, 59, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Jebelli, J.; Hooper, C.; Pocock, J.M. Microglial p53 activation is detrimental to neuronal synapses during activation-induced inflammation: Implications for neurodegeneration. Neurosci. Lett. 2014, 583, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Hopkins, S.; Nesser, N.K.; Sopher, B.; Silvestroni, A.; Ammanuel, S.; Jayadev, S.; Möller, T.; Weinstein, J.; Garden, G.A. The p53 transcription factor modulates microglia behavior through microRNA-dependent regulation of c-Maf. J. Immunol. 2014, 192, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011, 7, e1001360. [Google Scholar] [CrossRef] [PubMed]
- Davenport, C.M.; Sevastou, I.G.; Hooper, C.; Pocock, J.M. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. J. Neurochem. 2010, 112, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Yu, Q.; Chen, F.S.; Tan, W.F.; Zhang, Z.L.; Ma, H. Inhibiting aberrant p53-PUMA feedback loop activation attenuates ischaemia reperfusion-induced neuroapoptosis and neuroinflammation in rats by downregulating caspase 3 and the NF-κB cytokine pathway. J. Neuroinflamm. 2018, 15, 250. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A perspective on multi-target drugs for Alzheimer’s disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as prospective neuroprotectants and their therapeutic propensity in aging associated neurological disorders. Front. Aging Neurosci. 2019, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Atrahimovich, D.; Avni, D.; Khatib, S. Flavonoids-macromolecules interactions in human diseases with focus on Alzheimer, atherosclerosis and cancer. Antioxidants 2021, 10, 423. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Silva, R.F.M.; Pogačnik, L. Polyphenols from food and natural products: Neuroprotection and safety. Antioxidants 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Vuković, L.; Puhović, J.; Erhardt, J.; Oršolić, N. Neuroprotective effect of quercetin against hydrogen peroxide-induced oxidative injury in P19 neurons. J. Mol. Neurosci. 2021, 47, 286–299. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Čipak Gašparović, A.; Vuković, L.; Vlainić, J.; Žarković, N.; Oršolić, N. Quercetin supplementation: Insight into the potentially harmful outcomes of neurodegenerative prevention. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Vlainić, J.; Čadež, V.; Šegota, S. Atomic force microscopy reveals new biophysical markers for monitoring subcellular changes in oxidative injury: Neuroprotective effects of quercetin at the nanoscale. PLoS ONE 2018, 13, e0200119. [Google Scholar] [CrossRef]
- Maher, P. The potential of flavonoids for the treatment of neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef]
- Maan, G.; Sikdar, B.; Kumar, A.; Shukla, R.; Mishra, A. Role of flavonoids in neurodegenerative diseases: Limitations and future perspectives. Curr. Top. Med. Chem. 2020, 20, 1169–1194. [Google Scholar] [CrossRef]
- Novak, V.; Rogelj, B.; Župunski, V. Therapeutic potential of polyphenols in amyotrophic lateral sclerosis and frontotemporal dementia. Antioxidants 2021, 10, 1328. [Google Scholar] [CrossRef]
- Prasanna, P.; Upadhyay, A. Flavonoid-based nanomedicines in Alzheimer’s disease therapeutics: Promises made, a long way to go. Pharmacol. Transl. Sci. 2021, 4, 74–95. [Google Scholar] [CrossRef]
- Davinelli, S.; Maes, M.; Corbi, G.; Zarrelli, A.; Willcox, D.C.; Scapagnini, G. Dietary phytochemicals and neuro-inflammaging: From mechanistic insights to translational challenges. Immun. Ageing 2016, 13, 16. [Google Scholar] [CrossRef]
- Kim, H.P.; Son, K.H.; Chang, H.W.; Kang, S.S. Anti-inflammatory plant flavonoids and cellular action mechanisms. J. Pharmacol. Sci. 2004, 96, 229–245. [Google Scholar] [CrossRef]
- Hoensch, H.P.; Oertel, R. The value of flavonoids for the human nutrition: Short review and perspectives. Clin. Nutr. Exp. 2015, 3, 8–14. [Google Scholar] [CrossRef]
- Radovanović, V.; Vlainić, J.; Hanžić, N.; Ukić, P.; Oršolić, N.; Baranović, G.; Jazvinšćak Jembrek, M. Neurotoxic effect of ethanolic extract of propolis in the presence of copper ions is mediated through enhanced production of ROS and stimulation of caspase-3/7 activity. Toxins 2019, 11, 273. [Google Scholar] [CrossRef]
- Sadžak, A.; Vlašić, I.; Kiralj, Z.; Batarelo, M.; Oršolić, N.; Jazvinšćak Jembrek, M.; Kušen, I.; Šegota, S. Neurotoxic effect of flavonol myricetin in the presence of excess copper. Molecules 2021, 26, 845. [Google Scholar] [CrossRef]
- Batiha, G.E.-S.; Beshbishy, A.M.; Ikram, M.; Mulla, Z.S.; El-Hack, M.E.A.; Taha, A.E.; Algammal, A.M.; Elewa, Y.H.A. The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: Quercetin. Foods 2020, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of neuroprotection by quercetin: Counteracting oxidative stress and more. Oxid. Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective effect of quercetin against the detrimental effects of LPS in the adult mouse brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective effects of quercetin in Alzheimer’s disease. Biomolecules 2020, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Amanzadeh, E.; Esmaeili, A.; Abadi, R.; Kazemipour, N.; Pahlevanneshan, Z.; Beheshti, S. Quercetin conjugated with superparamagnetic iron oxide nanoparticles improves learning and memory better than free quercetin via interacting with proteins involved in LTP. Sci. Rep. 2019, 9, 6876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yi, P.; Yi, M.; Tong, X.; Cheng, X.; Yang, J.; Hu, Y.; Peng, W. Protective effect of quercetin against H2O2-induced oxidative damage in PC-12 cells: Comprehensive analysis of a lncRNA-associated ceRNA network. Oxid. Med. Cell. Longev. 2020, 2020, 6038919. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sun, L.; Liu, Z.; Wang, H.; Xu, C. Protection afforded by quercetin against H2O2-induced apoptosis on PC12 cells via activating PI3K/Akt signal pathway. J. Recept. Signal Transduct. 2016, 36, 98–102. [Google Scholar] [CrossRef]
- Arredondo, F.; Echeverry, C.; Abin-Carriquiry, J.A.; Blasina, F.; Antúnez, K.; Jones, D.P.; Go, Y.-M.; Liang, Y.-L.; Dajas, F. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free. Radic. Biol. Med. 2010, 49, 738–747. [Google Scholar] [CrossRef]
- Bao, D.; Wang, J.; Pang, X.; Liu, H. Protective effect of quercetin against qxidative stress-induced cytotoxicity in rat pheochromocytoma (PC-12) cells. Molecules 2017, 22, 1122. [Google Scholar] [CrossRef]
- Sharma, D.R.; Wani, W.Y.; Sunkaria, A.; Kandimalla, R.J.; Sharma, R.K.; Verma, D.; Bal, A.; Gill, K.D. Quercetin attenuates neuronal death against aluminum-induced neurodegeneration in the rat hippocampus. Neuroscience 2016, 324, 163–176. [Google Scholar] [CrossRef]
- Rishitha, N.; Muthuraman, A. Therapeutic evaluation of solid lipid nanoparticle of quercetin in pentylenetetrazole induced cognitive impairment of zebrafish. Life Sci. 2019, 199, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-M.; Li, S.-Q.; Wu, W.-L.; Zhu, X.-Y.; Wang, Y.; Yuan, H.-Y. Effects of long-term treatment with quercetin on cognition and mitochondrial function in a mouse model of Alzheimer’s disease. Neurochem. Res. 2014, 39, 1533–1543. [Google Scholar] [CrossRef]
- Vargas-Restrepo, F.; Sabogal-Guáqueta, A.M.; Cardona-Gómez, G.P. Quercetin ameliorates inflammation in CA1 hippocampal region in aged triple transgenic Alzheimer’ s disease mice model. Biomédica 2018, 38, 62–69. [Google Scholar] [CrossRef]
- Paris, D.; Mathura, V.; Ait-Ghezala, G.; Beaulieu-Abdelahad, D.; Patel, N.; Bachmeier, C.; Mullan, M. Flavonoids lower Alzheimer’s Aβ production via an NFκB dependent mechanism. Bioinformation 2011, 6, 229. [Google Scholar] [CrossRef]
- Yu, X.; Li, Y.; Mu, X. Effect of quercetin on PC12 Alzheimer’s disease cell model induced by Aβ25-35 and its mechanism based on sirtuin1/Nrf2/HO-1 pathway. Biomed. Res. Int. 2020, 2020, 8210578. [Google Scholar] [CrossRef]
- Jiang, W.; Luo, T.; Li, S.; Zhou, Y.; Shen, X.-Y.; He, F.; Xu, J.; Wang, H.-Q. Quercetin protects against okadaic acid-induced injury via MAPK and PI3K/Akt/GSK3β signaling pathways in HT22 hippocampal neurons. PLoS ONE 2016, 11, e0152371. [Google Scholar] [CrossRef]
- Kumar, S.; Krishnakumar, V.G.; Morya, V.; Gupta, S.; Datta, B. Nanobiocatalyst facilitated aglycosidic quercetin as a potent inhibitor of tau protein aggregation. Int. J. Biol. Macromol. 2019, 138, 168–180. [Google Scholar] [CrossRef]
- Shen, X.Y.; Luo, T.; Li, S.; Ting, O.Y.; He, F.; Xu, J.; Wang, H.Q. Quercetin inhibits okadaic acid-induced tau protein hyperphosphorylation through the Ca2+-calpain-p25-CDK5 pathway in HT22 cells. Int. J. Mol. Med. 2018, 41, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Krishnamurthy, P.K.; Johnson, G.V. Cdk5 phosphorylates p53 and regulates its activity. J. Neurochem. 2002, 81, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-W.; Chen, J.-Y.; Ouyang, D.; Lu, J.-H. Quercetin in animal models of Alzheimer’s disease: A systematic review of preclinical studies. Int. J. Mol. Sci. 2020, 21, 493. [Google Scholar] [CrossRef] [PubMed]
- Paula, P.C.; Angelica Maria, S.G.; Luis, C.H.; Gloria Patricia, C.G. Preventive effect of quercetin in a triple transgenic Alzheimer’s disease mice model. Molecules 2019, 24, 2287. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Li, K.; Fu, T.; Wan, C.; Zhang, D.; Song, H.; Zhang, Y.; Liu, N.; Gan, Z.; Yuan, L. Quercetin ameliorates Aβ toxicity in Drosophila AD model by modulating cell cycle-related protein expression. Oncotarget 2016, 7, 67716–67731. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, Q.; Li, Z.; Dang, M.; Lin, Y.; Hou, X. Activation of Nrf2 signaling by sitagliptin and quercetin combination against β-amyloid induced Alzheimer’s disease in rats. Drug Dev. Res. 2019, 80, 837–845. [Google Scholar] [CrossRef]
- Shal, B.; Ding, W.; Ali, H.; Kim, Y.S.; Khan, S. Anti-neuroinflammatory potential of natural products in attenuation of Alzheimer’s disease. Front. Pharmacol. 2018, 9, 548. [Google Scholar] [CrossRef]
- El-Horany, H.E.; El-Latif, R.N.; ElBatsh, M.M.; Emam, M.N. Ameliorative effect of quercetin on neurochemical and behavioral deficits in rotenone rat model of Parkinson’s disease: Modulating autophagy (Quercetin on experimental Parkinson’s disease). J. Biochem. Mol. Toxicol. 2016, 30, 360–369. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, T.; Dong, S.Y.; Guo, Y.J.; Jankovic, J.; Xu, H.; Wu, Y.C. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef]
- Zhang, M.; Lu, P.; Terada, T.; Sui, M.; Furuta, H.; Iida, K.; Katayama, Y.; Lu, Y.; Okamoto, K.; Suzuki, M.; et al. Quercetin 3,5,7,3’,4’-pentamethyl ether from Kaempferia parviflora directly and effectively activates human SIRT1. Commun. Biol. 2021, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Ay, M.; Luo, J.; Langley, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Molecular mechanisms underlying protective effects of quercetin against mitochondrial dysfunction and progressive dopaminergic neurodegeneration in cell culture and MitoPark transgenic mouse models of Parkinson’s Disease. J. Neurochem. 2017, 141, 766–782. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.-Y.; Yoon, H. Quercetin attenuates manganese-induced neuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-κB and HO-1/Nrf2 pathways. Int. J. Mol. Sci. 2017, 18, 1989. [Google Scholar] [CrossRef]
- Sandhir, R.; Mehrotra, A. Quercetin supplementation is effective in improving mitochondrial dysfunctions induced by 3-nitropropionic acid: Implications in Huntington’s disease. Biochim. Biophys. Acta 2013, 1832, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Joseph, D.; Muralidhara, K.M. Enhanced neuroprotective effect of fish oil in combination with quercetin against 3-nitropropionic acid induced oxidative stress in rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 40, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener. Dis. 2012, 9, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, 5, 339–350. [Google Scholar] [CrossRef]
- Dong, Y.S.; Wang, J.L.; Feng, D.Y.; Qin, H.Z.; Wen, H.; Yin, Z.M.; Gao, G.D.; Li, C. Protective effect of quercetin against oxidative stress and brain edema in an experimental rat model of subarachnoid hemorrhage. Int. J. Med. Sci. 2014, 11, 282–290. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, C. Protective effects of quercetin against brain injury in a rat model of lipopolysaccharide-induced fetal brain injury. Int. J. Dev. Neurosci. 2018, 71, 175–180. [Google Scholar] [CrossRef]
- Benameur, T.; Soleti, R.; Porro, C. The potential neuroprotective role of free and encapsulated quercetin mediated by miRNA against neurological diseases. Nutrients 2021, 13, 1318. [Google Scholar] [CrossRef]
- Koda, T.; Kuroda, Y.; Imai, H. Rutin supplementation in the diet has protective effects against toxicant-induced hippocampal injury by suppression of microglial activation and pro-inflammatory cytokines: Protective effect of rutin against toxicant-induced hippocampal injury. Cell. Mol. Neurobiol. 2009, 29, 523–531. [Google Scholar] [CrossRef]
- Javed, H.; Khan, M.M.; Ahmad, A.; Vaibhav, K.; Ahmad, M.E.; Khan, A.; Ashafaq, M.; Islam, F.; Siddiqui, M.S.; Safhi, M.M.; et al. Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience 2012, 210, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.Y.; Ma, J.; Kong, X.X.; Wang, X.F.; Li, S.S.; Qi, X.L.; Yan, Y.H.; Cheng, J.; Liu, Q.; Jin, W.; et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 2019, 5, eaau6328. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Ahmad, A.; Ishrat, T.; Khuwaja, G.; Srivastawa, P.; Khan, M.B.; Raza, S.S.; Javed, H.; Vaibhav, K.; Khan, A.; et al. Rutin protects the neural damage induced by transient focal ischemia in rats. Brain Res. 2009, 1292, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Çelik, H.; Kandemir, F.M.; Caglayan, C.; Özdemir, S.; Çomaklı, S.; Kucukler, S.; Yardım, A. Neuroprotective effect of rutin against colistin-induced oxidative stress, inflammation and apoptosis in rat brain associated with the CREB/BDNF expressions. Mol. Biol. Rep. 2020, 47, 2023–2034. [Google Scholar] [CrossRef] [PubMed]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A dietary molecule with diverse biological activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Taheri, Y.; Suleria, H.; Martins, N.; Sytar, O.; Beyatli, A.; Yeskaliyeva, B.; Seitimova, G.; Salehi, B.; Semwal, P.; Painuli, S.; et al. Myricetin bioactive effects: Moving from preclinical evidence to potential clinical applications. BMC Complement. Med. Ther. 2020, 20, 241. [Google Scholar] [CrossRef]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Three distinct neuroprotective functions of myricetin against glutamate-induced neuronal cell death: Involvement of direct inhibition of caspase-3. J. Neurosci. Res. 2008, 86, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Multifunction of myricetin on Aβ: Neuroprotection via a conformational change of Aβ and reduction of Aβ via the interference of secretases. J. Neurosci. Res. 2008, 86, 368–377. [Google Scholar] [CrossRef]
- DeToma, A.S.; Choi, J.S.; Braymer, J.J.; Lim, M.H. Myricetin: A naturally occurring regulator of metal-induced amyloid-β aggregation and neurotoxicity. Chembiochem 2011, 12, 1198–1201. [Google Scholar] [CrossRef]
- Ramezani, M.; Darbandi, N.; Khodagholi, F.; Hashemi, A. Myricetin protects hippocampal CA3 pyramidal neurons and improves learning and memory impairments in rats with Alzheimer’s disease. Neural Regen Res. 2016, 11, 1976–1980. [Google Scholar] [CrossRef]
- Zhang, K.; Ma, Z.; Wang, J.; Xie, A.; Xie, J. Myricetin attenuated MPP(+)-induced cytotoxicity by anti-oxidation and inhibition of MKK4 and JNK activation in MES23.5 cells. Neuropharmacology 2011, 61, 329–335. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef]
- Joshi, V.; Mishra, R.; Upadhyay, A.; Amanullah, A.; Poluri, K.M.; Singh, S.; Kumar, A.; Mishra, A. Polyphenolic flavonoid (myricetin) upregulated proteasomal degradation mechanisms: Eliminates neurodegenerative proteins aggregation. J Cell Physiol. 2019, 234, 20900–20914. [Google Scholar] [CrossRef]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef]
- Sun, L.; Xu, P.; Fu, T.; Huang, X.; Song, J.; Chen, M.; Tian, X.; Yin, H.; Han, J. Myricetin against ischemic cerebral injury in rat middle cerebral artery occlusion model. Mol. Med. Rep. 2018, 17, 3274–3280. [Google Scholar] [CrossRef] [PubMed]
- Flamini, R.; Mattivi, F.; Rosso, M.D.; Arapitsas, P.; Bavaresco, L. Advanced knowledge of three important classes of grape phenolics: Anthocyanins, stilbenes and flavonols. Int. J. Mol. Sci. 2013, 14, 19651–19669. [Google Scholar] [CrossRef] [PubMed]
- Šikuten, I.; Štambuk, P.; Andabaka, Ž.; Tomaz, I.; Marković, Z.; Stupić, D.; Maletić, E.; Kontić, J.K.; Preiner, D. Grapevine as a rich source of polyphenolic compounds. Molecules 2020, 25, 5604. [Google Scholar] [CrossRef]
- Caruana, M.; Cauchi, R.; Vassallo, N. Putative role of red wine polyphenols against brain pathology in Alzheimer’s and Parkinson’s sisease. Front. Nutr. 2016, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Shen, K.; An, Y.; Qi, S.; Dai, W.X.; Yin, Z. Ampelopsin inhibits H₂O₂-induced apoptosis by ERK and Akt signaling pathways and up-regulation of heme oxygenase-1. Phytother Res. 2012, 26, 988–994. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, J.; Zhang, H.; Zeng, T. Dihydromyricetin inhibits oxidative stress and apoptosis in oxygen and glucose deprivation/reoxygenation-induced HT22 cells by activating the Nrf2/HO-1 pathway. Mol. Med. Rep. 2021, 23, 397. [Google Scholar] [CrossRef]
- Kou, X.; Liu, X.; Chen, X.; Li, J.; Yang, X.; Fan, J.; Yang, Y.; Chen, N. Ampelopsin attenuates brain aging of D-gal-induced rats through miR-34a-mediated SIRT1/mTOR signal pathway. Oncotarget 2016, 7, 74484–74495. [Google Scholar] [CrossRef]
- Sun, P.; Yin, J.B.; Liu, L.H.; Guo, J.; Wang, S.H.; Qu, C.H.; Wang, C.X. Protective role of dihydromyricetin in Alzheimer’s disease rat model associated with activating AMPK/SIRT1 signaling pathway. Biosci. Rep. 2019, 39, BSR20180902. [Google Scholar] [CrossRef]
- Kou, X.; Li, J.; Bian, J.; Yang, Y.; Yang, X.; Fan, J.; Jia, S.; Chen, N. Ampelopsin attenuates 6-OHDA-induced neurotoxicity by regulating GSK-3/NRF2/ARE signaling. J. Funct. Food 2015, 19, 765–774. [Google Scholar] [CrossRef]
- Ren, Z.X.; Zhao, Y.F.; Cao, T.; Zhen, X.C. Dihydromyricetin protects neurons in an MPTP-induced model of Parkinson’s disease by suppressing glycogen synthase kinase-3 beta activity. Acta Pharmacol. Sin. 2016, 37, 1315–1324. [Google Scholar] [CrossRef]
- Ren, Z.; Yan, P.; Zhu, L.; Yang, H.; Zhao, Y.; Kirby, B.P.; Waddington, J.L.; Zhen, X. Dihydromyricetin exerts a rapid antidepressant-like effect in association with enhancement of BDNF expression and inhibition of neuroinflammation. Psychopharmacology 2018, 235, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, G.; Cui, L.; Wang, Q. Myricetin attenuates depressant-like behavior in mice subjected to repeated restraint stress. Int. J. Mol. Sci. 2015, 16, 28377–28385. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Jeong, K.H.; Jung, U.J.; Kim, S.R. Myricitrin ameliorates 6-hydroxydopamine-induced dopaminergic neuronal loss in the substantia nigra of mouse brain. J. Med. Food 2016, 19, 374–382. [Google Scholar] [CrossRef]
- Lei, Y. Myricitrin decreases traumatic injury of the spinal cord and exhibits antioxidant and anti-inflammatory activities in a rat model via inhibition of COX-2, TGF-β1, p53 and elevation of Bcl-2/Bax signaling pathway. Mol. Med. Rep. 2017, 16, 7699–7705. [Google Scholar] [CrossRef]
- Sun, G.B.; Qin, M.; Ye, J.X.; Pan, R.L.; Meng, X.B.; Wang, M.; Luo, Y.; Li, Z.Y.; Wang, H.W.; Sun, X.B. Inhibitory effects of myricitrin on oxidative stress-induced endothelial damage and early atherosclerosis in ApoE-/- mice. Toxicol. Appl. Pharmacol. 2013, 271, 114–126. [Google Scholar] [CrossRef]
- Velazhahan, V.; Glaza, P.; Herrera, A.I.; Prakash, O.; Zolkiewski, M.; Geisbrecht, B.V.; Schrick, K. Dietary flavonoid fisetin binds human SUMO1 and blocks sumoylation of p53. PLoS ONE 2020, 15, e0234468. [Google Scholar] [CrossRef]
- Ishige, K.; Schubert, D.; Sagara, Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001, 30, 433446. [Google Scholar] [CrossRef]
- Sagara, Y.; Vanhnasy, J.; Maher, P. Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J. Neurochem. 2004, 90, 1144–1155. [Google Scholar] [CrossRef]
- Maher, P.; Takaishi, T.; Abe, K. Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc. Natl. Acad. Sci. USA 2006, 103, 16568–16573. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T.; Morimoto, T.; Shibao, M.; Watanabe, S.; Sakai-Kato, K.; Utsunomiya-Tate, N.; Abe, K. Structural requirements for the flavonoid fisetin in inhibiting fibril formation of amyloid beta protein. Neurosci Lett. 2008, 444, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ali, T.; Park, H.Y.; Badshah, H.; Rehman, S.U.; Kim, M.O. Neuroprotective effect of fisetin against amyloid-beta-induced cognitive/synaptic dysfunction, neuroinflammation, and neurodegeneration in adult mice. Mol. Neurobiol. 2017, 54, 2269–2285. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ali, T.; Rehman, S.U.; Kim, M.O. Phytomedicine-based potent antioxidant, fisetin protects CNS-insult LPS-induced oxidative stress-mediated neurodegeneration and memory impairment. J. Clin. Med. 2019, 8, 850. [Google Scholar] [CrossRef] [PubMed]
- Prakash, D.; Sudhandiran, G. Dietary flavonoid fisetin regulates aluminium chloride-induced neuronal apoptosis in cortex and hippocampus of mice brain. J. Nutr. Biochem. 2015, 26, 1527–1539. [Google Scholar] [CrossRef]
- Chen, T.J.; Feng, Y.; Liu, T.; Wu, T.T.; Chen, Y.J.; Li, X.; Li, Q.; Wu, Y.C. Fisetin regulates gut microbiota and exerts neuroprotective effect on mouse model of Parkinson’s disease. Front. Neurosci. 2020, 14, 549037. [Google Scholar] [CrossRef]
- Patel, M.Y.; Panchal, H.V.; Ghribi, O.; Benzeroual, K.E. The neuroprotective effect of fisetin in the MPTP model of Parkinson’s disease. J. Parkinsons Dis. 2012, 2, 287–302. [Google Scholar] [CrossRef]
- Rosado-Ramos, R.; Godinho-Pereira, J.; Marques, D.; Figueira, I.; Fleming Outeiro, T.; Menezes, R.; Nunes dos Santos, C. Small molecule fisetin modulates alpha–synuclein aggregation. Molecules 2021, 26, 3353. [Google Scholar] [CrossRef]
- Singh, S.; Singh, A.K.; Garg, G.; Rizvi, S.I. Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci. 2018, 193, 171–179. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, A.; Ali, W.; Jo, M.H.; Park, J.; Ikram, M.; Kim, M.O. Fisetin rescues the mice brains against D-galactose-induced oxidative stress, neuroinflammation and memory impairment. Front. Pharmacol. 2021, 12, 612078. [Google Scholar] [CrossRef]
- Alam, W.; Khan, H.; Shah, M.A.; Cauli, O.; Saso, L. Kaempferol as a dietary anti-inflammatory agent: Current therapeutic standing. Molecules 2020, 25, 4073. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.S.; Almezgagi, M.; Zhang, Y.; Bashir, A.; Abdullah, H.M.; Gamah, M.; Wang, X.; Zhu, Q.; Shen, X.; Ma, Q.; et al. Mechanistic new insights of flavonols on neurodegenerative diseases. Biomed. Pharmacother. 2021, 137, 111253. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Schaffner, W.; Hertel, C. Phytoestrogen kaempferol (3,4’,5,7-tetrahydroxyflavone) protects PC12 and T47D cells from beta-amyloid-induced toxicity. J. Neurosci. Res. 1999, 57, 399–404. [Google Scholar] [CrossRef]
- Pan, X.; Liu, X.; Zhao, H.; Wu, B.; Liu, G. Antioxidant, anti-inflammatory and neuroprotective effect of kaempferol on rotenone-induced Parkinson’s disease model of rats and SH-S5Y5 cells by preventing loss of tyrosine hydroxylase. J. Funct. Foods 2020, 74, 104140. [Google Scholar] [CrossRef]
- Jang, Y.J.; Kim, J.; Shim, J.; Kim, J.; Byun, S.; Oak, M.H.; Lee, K.W.; Lee, H.J. Kaempferol attenuates 4-hydroxynonenal-induced apoptosis in PC12 cells by directly inhibiting NADPH oxidase. J. Pharmacol. Exp. Ther. 2011, 337, 747–754. [Google Scholar] [CrossRef]
- Rahul; Naz, F.; Jyoti, S.; Siddique, Y.H. Effect of kaempferol on the transgenic Drosophila model of Parkinson’s disease. Sci. Rep. 2020, 10, 13793. [Google Scholar] [CrossRef]
- Li, S.; Pu, X.P. Neuroprotective effect of kaempferol against a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Biol. Pharm. Bull. 2011, 34, 1291–1296. [Google Scholar] [CrossRef]
- Beg, T.; Jyoti, S.; Naz, F.; Rahul; Ali, F.; Ali, S.K.; Reyad, A.M.; Siddique, Y.H. Protective effect of kaempferol on the transgenic Drosophila model of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2018, 17, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Chen, J.; Zhang, W.; Fu, W.; Wu, G.; Wei, H.; Wang, Q.; Ruan, J. In vivo investigation on the potential of galangin, kaempferol and myricetin for protection of D-galactose-induced cognitive impairment. Food Chem. 2012, 135, 2702–2707. [Google Scholar] [CrossRef]
- BinMowyna, M.N.; AlFaris, N.A. Kaempferol suppresses acetaminophen-induced liver damage by upregulation/activation of SIRT1. Pharm. Biol. 2021, 59, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Al Sabaani, N. Kaempferol protects against hydrogen peroxide-induced retinal pigment epithelium cell inflammation and apoptosis by activation of SIRT1 and inhibition of PARP1. J. Ocul. Pharmacol. Ther. 2020, 36, 563–577. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Cao, X.B.; Xiong, N.; Wang, H.C.; Huang, J.S.; Sun, S.G.; Wang, T. Morin exerts neuroprotective actions in Parkinson disease models in vitro and in vivo. Acta Pharmacol. Sin. 2010, 31, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Sánchez-Gómez, M.V.; Ruiz, A.; Cavaliere, F.; Ortiz-Sanz, C.; Quintela-López, T.; Capetillo-Zarate, E.; Solé-Domènech, S.; Matute, C. Mangiferin and morin attenuate oxidative stress, mitochondrial dysfunction, and neurocytotoxicity, induced by amyloid beta oligomers. Oxid. Med. Cell. Longev. 2018, 2018, 2856063. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Qu, J.; Zhang, W.; Bai, M.; Zhou, Q.; Zhang, Z.; Li, Z.; Miao, J. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology 2016, 108, 1–13. [Google Scholar] [CrossRef]
- Gong, E.J.; Park, H.R.; Kim, M.E.; Piao, S.; Lee, E.; Jo, D.G.; Chung, H.Y.; Ha, N.C.; Mattson, M.P.; Lee, J. Morin attenuates tau hyperphosphorylation by inhibiting GSK3β. Neurobiol. Dis. 2011, 44, 223–230. [Google Scholar] [CrossRef]
- Lee, K.M.; Lee, Y.; Chun, H.J.; Kim, A.H.; Kim, J.Y.; Lee, J.Y.; Ishigami, A.; Lee, J. Neuroprotective and anti-inflammatory effects of morin in a murine model of Parkinson’s disease. J. Neurosci. Res. 2016, 94, 865–878. [Google Scholar] [CrossRef]
- Çelik, H.; Kucukler, S.; Çomaklı, S.; Özdemir, S.; Caglayan, C.; Yardım, A.; Kandemir, F.M. Morin attenuates ifosfamide-induced neurotoxicity in rats via suppression of oxidative stress, neuroinflammation and neuronal apoptosis. Neurotoxicology 2020, 76, 126–137. [Google Scholar] [CrossRef]
- Kuzu, M.; Kandemir, F.M.; Yildirim, S.; Kucukler, S.; Caglayan, C.; Turk, E. Morin attenuates doxorubicin-induced heart and brain damage by reducing oxidative stress, inflammation and apoptosis. Biomed. Pharmacother. 2018, 106, 443–453. [Google Scholar] [CrossRef]
- Ola, M.S.; Aleisa, A.M.; Al-Rejaie, S.S.; Abuohashish, H.M.; Parmar, M.Y.; Alhomida, A.S.; Ahmed, M.M. Flavonoid, morin inhibits oxidative stress, inflammation and enhances neurotrophic support in the brain of streptozotocin-induced diabetic rats. Neurol. Sci. 2014, 35, 1003–1008. [Google Scholar] [CrossRef]
- Bachewal, P.; Gundu, C.; Yerra, V.G.; Kalvala, A.K.; Areti, A.; Kumar, A. Morin exerts neuroprotection via attenuation of ROS induced oxidative damage and neuroinflammation in experimental diabetic neuropathy. BioFactors 2018, 44, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Shin, B.Y.; Han, J.Y.; Kim, M.G.; Wi, J.E.; Kim, Y.W.; Cho, I.J.; Kim, S.C.; Shin, S.M.; Ki, S.H. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol. Appl. Pharmacol. 2014, 274, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Guan, Y.Y.; Zhang, Z.L.; Rahman, K.; Wang, S.J.; Zhou, S.; Luan, X.; Zhang, H. Isorhamnetin: A review of pharmacological effects. Biomed. Pharmacother. 2020, 128, 110301. [Google Scholar] [CrossRef]
- Hwang, S.L.; Yen, G.C. Modulation of Akt, JNK, and p38 activation is involved in citrus flavonoid-mediated cytoprotection of PC12 cells challenged by hydrogen peroxide. J. Agric. Food Chem. 2009, 57, 2576–2582. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Ma, Y.; Luo, Y.; Cao, Z.; Lu, H. Protective effects of isorhamnetin on N2a cell against endoplasmic reticulum stress-induced injury is mediated by PKCε. Biomed Pharmacother. 2017, 93, 830–836. [Google Scholar] [CrossRef]
- Wu, Y.; Fan, L.; Wang, Y.; Ding, J.; Wang, R. Isorhamnetin alleviates high glucose-aggravated inflammatory response and apoptosis in oxygen-glucose deprivation and reoxygenation-induced HT22 hippocampal neurons through Akt/SIRT1/Nrf2/HO-1 signaling pathway. Inflammation 2021, 44, 1993–2005. [Google Scholar] [CrossRef]
- Chen, F.; Hu, M.; Shen, Y.; Zhu, W.; Cao, A.; Ni, B.; Qian, J.; Yang, J. Isorhamnetin promotes functional recovery in rats with spinal cord injury by abating oxidative stress and modulating M2 macrophages/microglia polarization. Eur. J. Pharmacol. 2021, 895, 173878. [Google Scholar] [CrossRef]
- Zhao, J.J.; Song, J.Q.; Pan, S.Y.; Wang, K. Treatment with isorhamnetin protects the brain against ischemic injury in mice. Neurochem. Res. 2016, 41, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.L.; Choi, R.C.; Zhu, K.Y.; Leung, K.W.; Guo, A.J.; Bi, D.; Xu, H.; Lau, D.T.; Dong, T.T.; Tsim, K.W. Isorhamnetin, a flavonol aglycone from Ginkgo biloba L.; induces neuronal differentiation of cultured PC12 cells: Potentiating the effect of nerve growth factor. Evid. Based Complement. Alternat. Med. 2012, 2012, 278273. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Usui, T.; Zar Kalai, F.; Han, J.; Isoda, H.; Nagumo, Y. Protective effects of Nitraria retusa extract and its constituent isorhamnetin against amyloid β-induced cytotoxicity and amyloid β aggregation. Biosci. Biotechnol. Biochem. 2015, 79, 1548–1551. [Google Scholar] [CrossRef]
- Asha, D.; Sumathi, T. Nootropic activity of isorhamnetin in amyloid β 25–35 induced cognitive dysfunction and its related mRNA expressions in Alzheimer’s disease. Int. J. Pharm. Sci. Res. 2016, 7, 3233–3242. [Google Scholar]
- Ishola, I.O.; Osele, M.O.; Chijioke, M.C.; Adeyemi, O.O. Isorhamnetin enhanced cortico-hippocampal learning and memory capability in mice with scopolamine-induced amnesia: Role of antioxidant defense, cholinergic and BDNF signaling. Brain Res. 2019, 1712, 188–196. [Google Scholar] [CrossRef]
- Olennikov, D.N.; Kashchenko, N.I.; Chirikova, N.K.; Akobirshoeva, A.; Zilfikarov, I.N.; Vennos, C. Isorhamnetin and quercetin derivatives as anti-acetylcholinesterase principles of marigold (Calendula officinalis) flowers and preparations. Int. J. Mol. Sci. 2017, 18, 1685. [Google Scholar] [CrossRef]
- Pérez-González, A.; Rebollar-Zepeda, A.M.; León-Carmona, J.R.; Galano, A. Reactivity indexes and O-H bond dissociation energies of a large series of polyphenols: Implications for their free radical scavenging activity. J. Mex. Chem. Soc. 2012, 56, 241–249. [Google Scholar] [CrossRef]
- Moses, G.S.; Jensen, M.D.; Lue, L.F.; Walker, D.G.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Secretory PLA2-IIA: A new inflammatory factor for Alzheimer’s disease. J. Neuroinflamm. 2006, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Novo Belchor, M.; Hessel Gaeta, H.; Fabri Bittencourt Rodrigues, C.; Ramos da Cruz Costa, C.; De Oliveira Toyama, D.; Domingues Passero, L.F.; Dalastra Laurenti, M.; Hikari Toyama, M. Evaluation of rhamnetin as an inhibitor of the pharmacological effect of secretory phospholipase A2. Molecules 2017, 22, 1441. [Google Scholar] [CrossRef] [PubMed]
- Remya, C.; Dileep, K.V.; Tintu, I.; Variyar, E.J.; Sadasivan, C. Design of potent inhibitors of acetylcholinesterase using morin as the starting compound. Front. Life Sci. 2012, 6, 107–117. [Google Scholar] [CrossRef]
- Mohammadi, S.; Golshani, Y. Neuroprotective effects of rhamnazin as a flavonoid on chronic stress-induced cognitive impairment. J. Adv. Neurosci. Res. 2017, 4, 30–37. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, R.; Sa, Q.; Du, Y. Rhamnazin ameliorates traumatic brain injury in mice via reduction in apoptosis, oxidative stress, and inflammation. Neuroimmunomodulation 2021, 1–8. [Google Scholar] [CrossRef]
- Dai, L.; He, J.; Miao, X.; Guo, X.; Shang, X.; Wang, W.; Li, B.; Wang, Y.; Pan, H.; Zhang, J. Multiple biological activities of Rhododendron przewalskii Maxim. Extracts and UPLC-ESI-Q-TOF/MS characterization of their phytochemical composition. Front. Pharmacol. 2021, 12, 599778. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef]
- Khan, A.; Manna, K.; Das, D.K.; Kesh, S.B.; Sinha, M.; Das, U.; Biswas, S.; Sengupta, A.; Sikder, K.; Datta, S.; et al. Gossypetin ameliorates ionizing radiation-induced oxidative stress in mice liver-a molecular approach. Free Radic Res. 2015, 49, 1173–1186. [Google Scholar] [CrossRef]
- Lin, H.-H.; Hsieh, M.-C.; Wang, C.-P.; Yu, P.-R.; Lee, M.-S.; Chen, J.-H. Anti-atherosclerotic effect of gossypetin on abnormal vascular smooth muscle cell proliferation and migration. Antioxidants 2021, 10, 1357. [Google Scholar] [CrossRef]








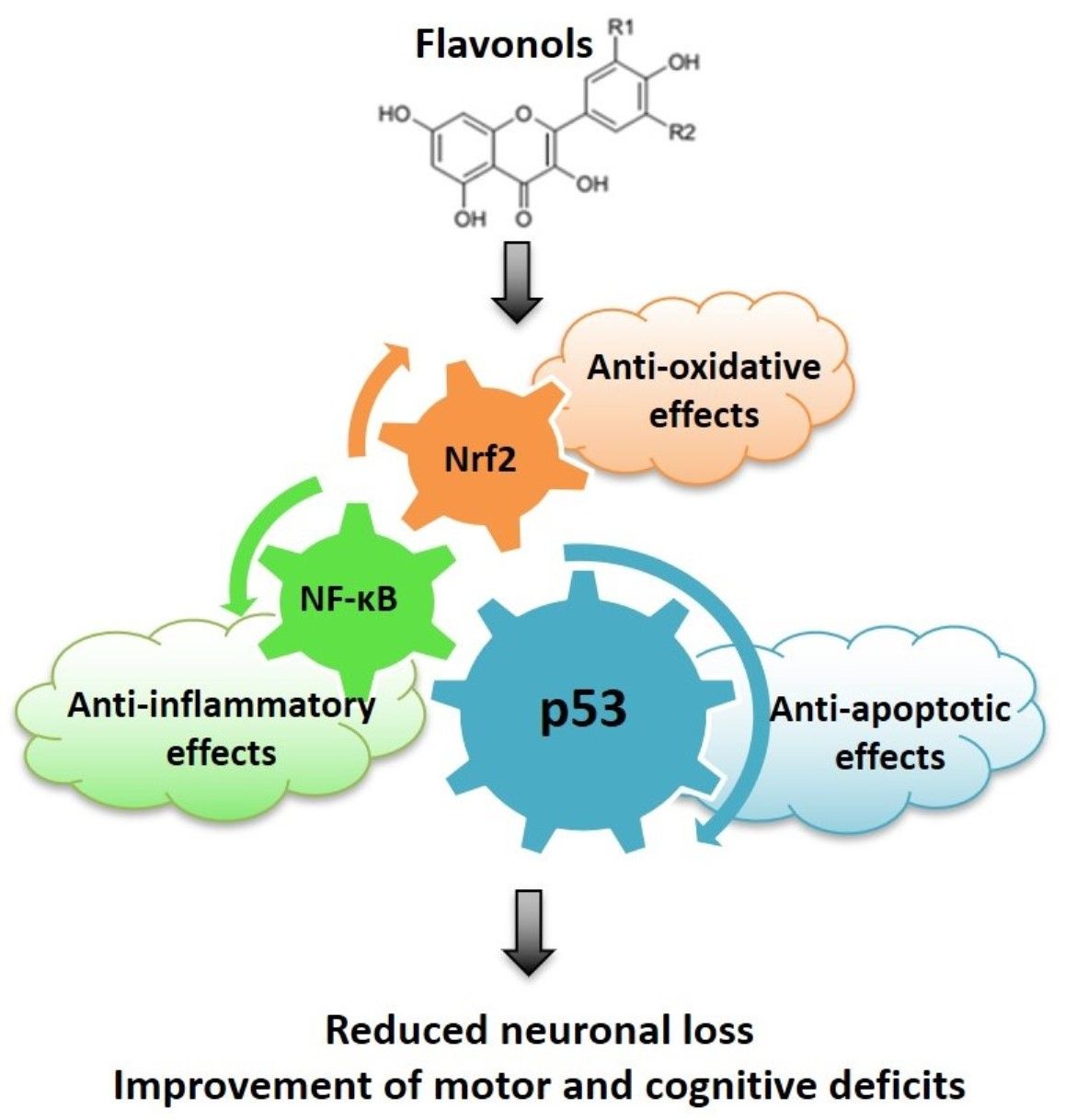{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
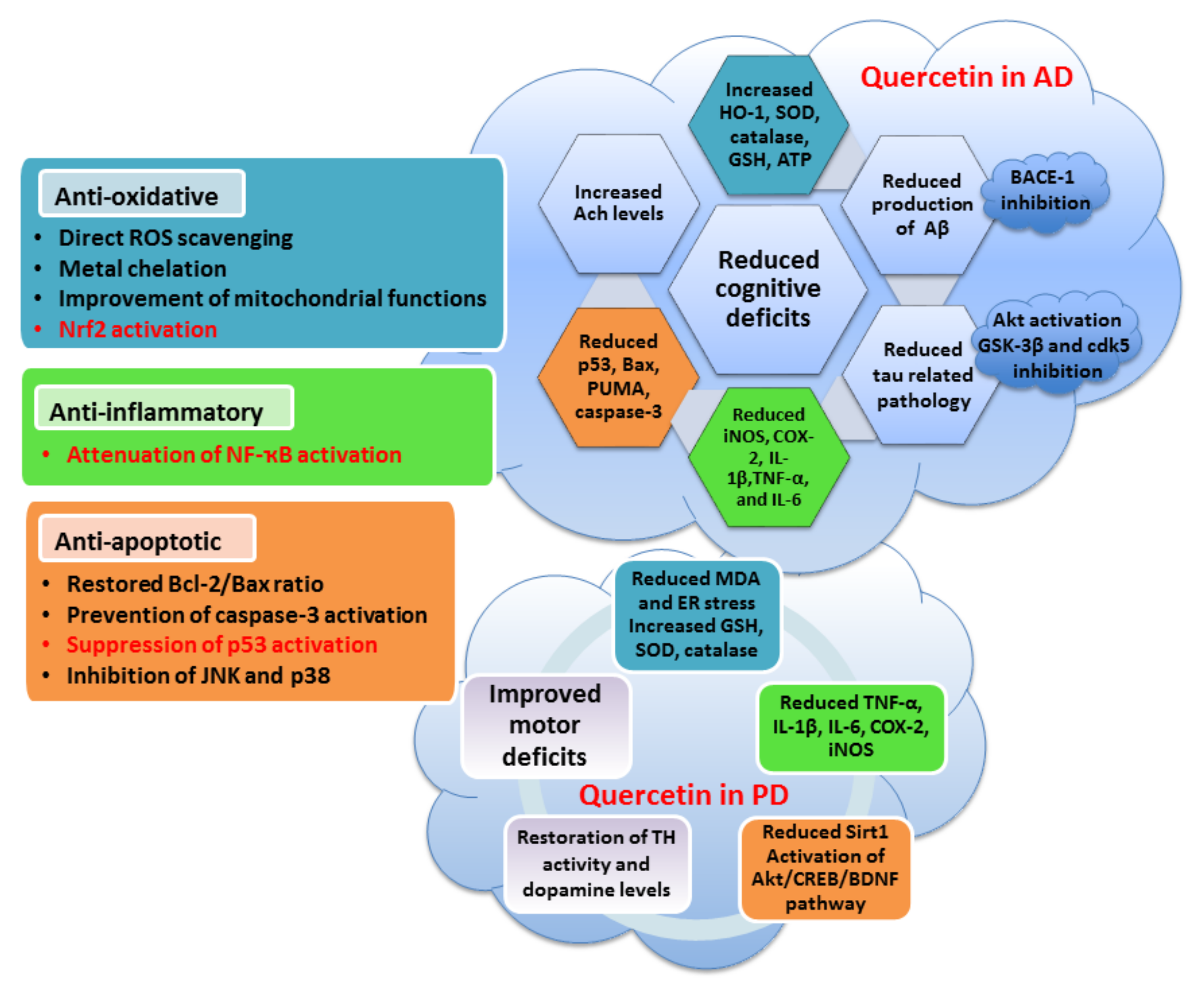{kind=link}
{kind=link}
| Model System | Treatment | Anti-Oxidative Effects | Anti-Inflammatory Effects | Anti-Apoptotic Effects | Reference |
|---|---|---|---|---|---|
| Quercetin | |||||
| PC12 cells | H2O2 | ↓ ROS production, ↓ MDA levels, ↑ GSH content, ↑ catalase, SOD and GSH-Px expression | ↓ Bax and ↑ Bcl-2 expression, ↓ cleaved caspase-3 and -9, ↓ p53 expression, ↑ activation of PI3K/Akt | [203,204,206] | |
| PC12 cells | Aβ25-35 | ↓ levels of MDA, and HO-1 protein, ↑ levels of SOD, GSH-Px, catalase, Sirt1 and Nrf2 protein | [212] | ||
| P19 neurons | H2O2 | ↓ ROS production | ↓ caspase-3/7 activation, ↓ nuclear condensation, ↓ p53 and ↑ Bcl-2 expression, ↑ activation of PI3K/Akt and ERK1/2 | [186,188] | |
| Cerebellar granule neurons | H2O2 | ↑ GSH levels, Nrf2 translocation, ↑ expression of GCLC mRNA | [205] | ||
| P19 neurons | CuSO4 | ↓ ROS production in moderate OS, pro-oxidative activity in severe OS | ↓ caspase-3/7 activation, ↓ chromatin condensation, ↓ PUMA expression, ↑ activation of PI3K/Akt and ERK1/2 | [123] | |
| HT22 cells | Okadaic acid | ↓ levels of ROS and MDA, ↑ levels of SOD and GSH-Px, ↑ mitochondrial membrane potential | ↓ expression of NF-κB p-p65 | ↓ Bax levels, ↓ cleaved caspase-3, activation of PI3K/Akt pathway, inhibition of p38 and JNK, reduced intracellular Ca2+ levels | [213,215] |
| Drosophila AD model | Restored expression of genes involved in p53 signaling | [219] | |||
| Wild-type C57BL/6N mice | LPS | ↓ gliosis, ↓ expression of p-NF-κB, ↓ levels of TNF-α, IL-1β, COX-2 and iNOS | ↓ cytochrome c release and Bax/Bcl-2 ratio, ↓ caspase-3 activity, ↓ synaptic loss | [200] | |
| Wistar albino rats (hippocampus) | Al-lactate | ↓ ROS production, ↑ MnSOD activity, preserved mitochondrial morphology | ↓ Bax and ↑ Bcl-2 expression, ↓ cytochrome c expression, ↓ caspase-3 activation, ↓ DNA fragmentation, ↓ p53 expression | [207] | |
| APPswe/ PS1dE9 mice (C57/BL) (cortex, hippocampus) | Restoration of mitochondrial membrane potential, ROS levels and ATP production | [209] | |||
| 3xTg-AD mice (hippocampus) | ↓ activation of microglia, ↓ iNOS and COX-2 immunoreactivity, ↓ IL-1β | [210] | |||
| Sprague Dawley rats (brain homogenate) | Aβ | ↑ SOD, catalase and GSH, ↓ MDA, activation of Nrf2/HO-1 pathway | [220] | ||
| MN9D dopaminergic cells | ↑ mitochondrial biogenesis and mtDNA content | ↑ pAkt expression, ↑ BDNF levels | [225] | ||
| SK-N-MC neuroblastoma cells | Mn | ↑ SOD and catalase activity, ↑ GSH levels, ↓ MDA, ↑ levels of Nrf2 and HO-1 | ↓ level of P-IκBα and NF-κB p65 | ↓ staining with Hoechst 33342, ↓ number of apoptotic cells | [226] |
| Albino rats | rotenone | ↓ MDA levels, ↑ thioredoxin reductase reactivity | ↓ DNA fragmentation | [222] | |
| Sprague Dawley rats (brain homogenate) | Mn | ↓ ROS and protein carbonyl levels, ↑ Cu/Zn-SOD activity, ↑ Nrf2 and HO-1 mRNAs | ↓ TNF-α, IL-1β, IL-6, COX-2 and iNOS protein expression, ↓ NF-κB and iNOS mRNAs | ↓ Bax expression, ↓ cytochrome c expression, ↓ cleaved caspase-3 expression, ↓ PARP-1 levels | [226] |
| 3-NP-induced model of HD in Wistar rats (striatum) | Restored ATP levels, ↑ mitochondrial SOD and catalase activity, ↓ MDA levels and ↑ thiol content | ↓ number of pyknotic and condensed nuclei | [227] | ||
| Rutin | |||||
| Sprague Dawley rats (hippocampus) | TMT | ↓ mRNA markers of reactive microglia and pro-inflammatory cytokines | [234] | ||
| Wistar rats (hippocampus) | STZ | ↓ TBARS content and nitrite levels, ↑ GSH, ↑ GPx, GR and catalase activity | ↓ GFAP, COX-2, iNOS, IL-8 and p65 NF-κB expression | Restored PARP-1 activity, prevented histological abnormalities | [235] |
| PP/PS1 (APPswe/ PSEN1dE9) mice (brain homogenate) | ↓ microglial activation, ↓ TNF-α, IL-1β, IL-6, ↑ IL-4 and IL-10 | Reversed synaptic dysfunction, enhanced microglial Aβ phagocytosis | [236] | ||
| Sprague Dawley rats (brain homogenate) | Colistin | ↑ SOD, GSH-Px and catalase content, ↑ GSH levels, ↓ MDA, ↑ levels of Nrf2 | ↓ GFAP expression, ↓ expression of NF-κB, TNF-α, ↓ nNOS activity | ↑ Bcl-2 and ↓ Bax expression, ↓ caspase-3 activity, ↓ p53 level, modulation of ERK1/2 pathway | [238] |
| Myricetin | |||||
| Primary neuronal cultures | Glutamate | ↓ ROS production | ↓ calcium overload, ↓ activation of caspase-3, ↓ nuclear fragmentation | [241] | |
| Primary neuronal cultures | Aβ | ↓ nuclear fragmentation, ↓ caspase-3 activation | [242] | ||
| MES23.5 cells | MPP+ | ↓ ROS production, restored mitochondrial membrane potential | ↓ nuclear condensation, ↓ Bax/Bcl-2 ratio, ↓ caspase-3 activation, ↓ pJNK expression | [245] | |
| Myricitrin | |||||
| C57BL/6 mice (substantia nigra) | 6-OHDA | ↓ number of Iba1-positive cells, ↓ expression TNF-α | [261] | ||
| Sprague Dawley rats (brain homogenate) | TBI of the spinal cord | ↑ SOD, GSH-Px and catalase content, ↓ MDA levels | ↓ TNF-α, IL-1β, IL-6 and NF-κB p65 subunit content, ↓ COX-2 and TGF-β1 mRNA expression | ↓ Bax/Bcl-2 ratio, ↓ p53 levels | [262] |
| Fisetin | |||||
| HT22 cells | Glutamate | ↓ ROS production, ↑ GSH levels | [265] | ||
| APPswe/ PS1dE9 mice (hippocampus) | ↓ protein carbonylation | ↓ cPLA2 levels, ↓ COX-2, ↓ astrocytic reactivity | ↑ pERK, preserved expression of synaptic markers | [268] | |
| C57BL/6N mice (hippocampus) | Aβ | ↓ expression of p-IKKβ, p-NF-ҡB, TNFα and IL-1β, ↓ number of activated Iba-1- and GFAP-positive cells | ↓ Bax expression, ↑ Bcl-2 expression, ↓ cytochrome c activation, ↓ cleaved caspase-3 and -9, ↓ expression of p53 and PARP-1, ↑ activation of PI3K/Akt, restored synaptic function | [271] | |
| C57BL/6N mice (hippocampus) | LPS | ↓ ROS production and lipid peroxidation, ↑ GSH levels | ↓ reactivity of TLR4 and pNF-ҡB, ↓ TNFα, IL-1β and COX-2, ↓ activation of Iba-1- and GFAP-positive cells | ↓ expression of cytochrome c, Apaf-1, caspase-3 and -9 and PARP-1, ↓ activation of JNK pathway, prevented morphological changes | [272] |
| Swiss albino mice (cortex and hippocampus) | AlCl3 | ↓ Bax and ↑ Bcl-2 mRNA expression, ↓ expression of cytochrome c, caspase-3 and -9, ↓ expression of p53, ↓ number of TUNEL-positive neurons, ↓ JNK activation | [273] | ||
| C57BL/6N mice (striatum) | MPTP | ↓ number of TUNEL-positive neurons | [274] | ||
| Wistar rats (brain homogenate) | D-galactose | ↓ production of ROS and NO, ↓ protein carbonylation and lipid hydroperoxidation, ↑ SOD and catalase levels, ↑ total thiol content | ↓ expression of TNF-α and IL-1β | [277] | |
| C57BL/6N mice | D-galactose | ↓ ROS and MDA levels, ↑ levels of Sirt1, Nrf2 and HO-1 | ↓ pNF-ҡB, NF-ҡB, Iba-1, GFAP, IL-1β, TNF-α and iNOS | ↓ Bax and ↑ Bcl-2 expression, ↓ expression of caspase-3 and PARP-1 | [278] |
| Kaempferol | |||||
| PC12 | HNE | ↑ Bcl-2 levels, ↓ caspase-3 activation, ↓ nuclear condensation and PARP-1 cleavage, inhibited activation of NADPH oxidase and JNK phosphorylation | [283] | ||
| SH-SY5Y cells | Rotenone | ↓ production of ROS, ↓ mitochondrial carbonyls, restored mitochondrial function | Prevented round-shape phenotype, preserved nuclear morphology, ↓ reduced caspase-9/3 cleavage, ↓ activation of JNK and p38 | [168] | |
| SH-SY5Y cells | Rotenone | ↓ production of ROS, ↓ MDA content, ↑ GSH, SOD, GSH-Px and catalase levels | ↑ expression of TH | [282] | |
| Albino rats | Rotenone | ↓ production of ROS, ↓ MDA levels, ↑ GSH, SOD, GSH-Px and catalase levels | ↓ levels of TNF-α and IL-6 | [282] | |
| Drosophila model of PD | ↑ free radical scavenging, ↓ TBARS and protein carbonyl content, ↑ SOD, GSH-S-transferase and catalase activity | ↓ reduced caspase-3 and -9 activity, ↑ TH staining and L-DOPA levels | [283] | ||
| Drosophila model of AD | ↑ GSH content, ↓ TBARS and protein carbonyl content | ↓ caspase-3 and -9 activity | [286] | ||
| C57BL/6 mice (substantia nigra) | MPTP | ↑ SOD and GSH-Px activity, ↓ MDA content | ↑ TH staining | [285] | |
| Kunming mice (hippocampus) | D-galactose | ↓ TBARS, ↑ SOD | ↑ activation of ERK/CREB pathway | [287] | |
| Morin | |||||
| SH-SY5Y cells | Aβ | ↓ production of ROS | ↓ staining with Hoechst 33342 and PI | [293] | |
| Differentiated PC12 cells | MPP+ | ↓ production of ROS | ↓ caspase-3 activity | [290] | |
| Primary cultured neurons | MPP+ | ↓ ROS production, prevented mitochondrial membrane disruption | ↓ staining with Hoechst 33342 and PI | [294] | |
| Primary cultured astrocytes | MPP+ | ↓ astrocyte activation, ↓ nuclear translocation of p65 NF-ҡB | [294] | ||
| Cultured cortical neurons | Aβ | ↓ ROS production and protein oxidation, ↑ SOD and catalase activity, ↓ calcium uptake and mitochondrial membrane depolarization, restored respiratory capacity | ↓ PI staining, ↓ cytochrome c release | [291] | |
| Sprague Dawley rats (brain homogenate) | Ifosfamide | ↓ MDA levels, ↑ GSH, SOD, GSH-Px and catalase activities, ↑ Nrf2 levels | ↓ GFAP levels, ↓ NF-ҡB and TNF-α, ↓ nNOS activity | ↓ caspase-3 and p53 levels, ↓ JNK activation | [295] |
| APPswe/ PS1dE9 mice (cortex, hippocampus) | ↓ activation of glial cells | ↓ cerebral Aβ production, ↑ expression of synaptic markers | [291] | ||
| C57BL/6 mice (striatum and substantia nigra) | MPTP | ↓ astrocyte activation, ↓ levels of TNF-α | ↓ loss of dopaminergic cells | [294] | |
| B57/BL mice (substantia nigra) | MPTP | ↑ number of TH-positive cells | [290] | ||
| Isorhamnetin | |||||
| PC12 cells | H2O2 | ↓ formation of ROS, ↑ catalase activity | ↓ calcium levels, ↓ caspase-3 activation, ↑ Akt signaling and ↓ activation of JNK pathway | [301] | |
| N2a cells | TG | ↓ formation of ROS | ↓ calcium overload, ↓ PI fluorescence, ↑ phosphorylation of PKCε | [302] | |
| HT22 neurons | OGD/R | ↑ Akt/Sirt1/Nrf2/HO-1 pathway | [303] | ||
| Albino rats (hippocampus) | Aβ25-35 | ↓ formation of H2O2, ↓ MAO activity | ↓ expression of iNOS and IL-1β mRNA | [308] | |
| Albino mice (prefrontal cortex and hippocampus) | SCP | ↓ MDA and nitrite production, ↑ GSH levels, ↑ SOD and catalase activities | [309] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jazvinšćak Jembrek, M.; Oršolić, N.; Mandić, L.; Sadžak, A.; Šegota, S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration. Antioxidants 2021, 10, 1628. https://doi.org/10.3390/antiox10101628
Jazvinšćak Jembrek M, Oršolić N, Mandić L, Sadžak A, Šegota S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration. Antioxidants. 2021; 10(10):1628. https://doi.org/10.3390/antiox10101628
Chicago/Turabian StyleJazvinšćak Jembrek, Maja, Nada Oršolić, Lucija Mandić, Anja Sadžak, and Suzana Šegota. 2021. "Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration" Antioxidants 10, no. 10: 1628. https://doi.org/10.3390/antiox10101628
APA StyleJazvinšćak Jembrek, M., Oršolić, N., Mandić, L., Sadžak, A., & Šegota, S. (2021). Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration. Antioxidants, 10(10), 1628. https://doi.org/10.3390/antiox10101628