
Burmannic Acid Inhibits Proliferation and Induces Oxidative Stress Response of Oral Cancer Cells


 , and
, and 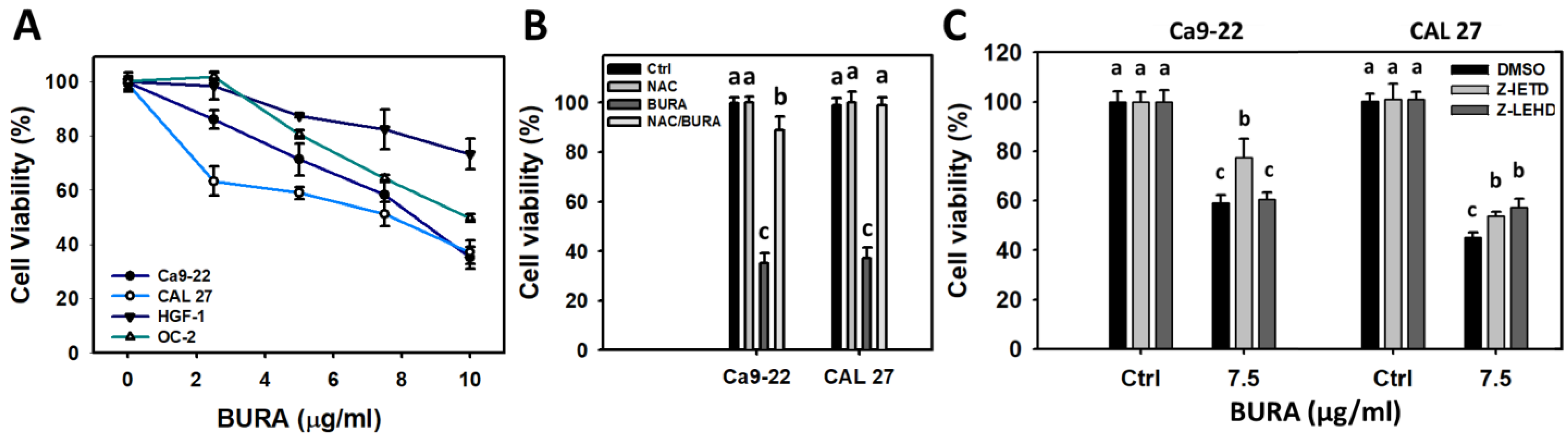{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. BURA Preparation
2.2. Reagents
2.3. Cell Cultures
2.4. Cell Viability Assay
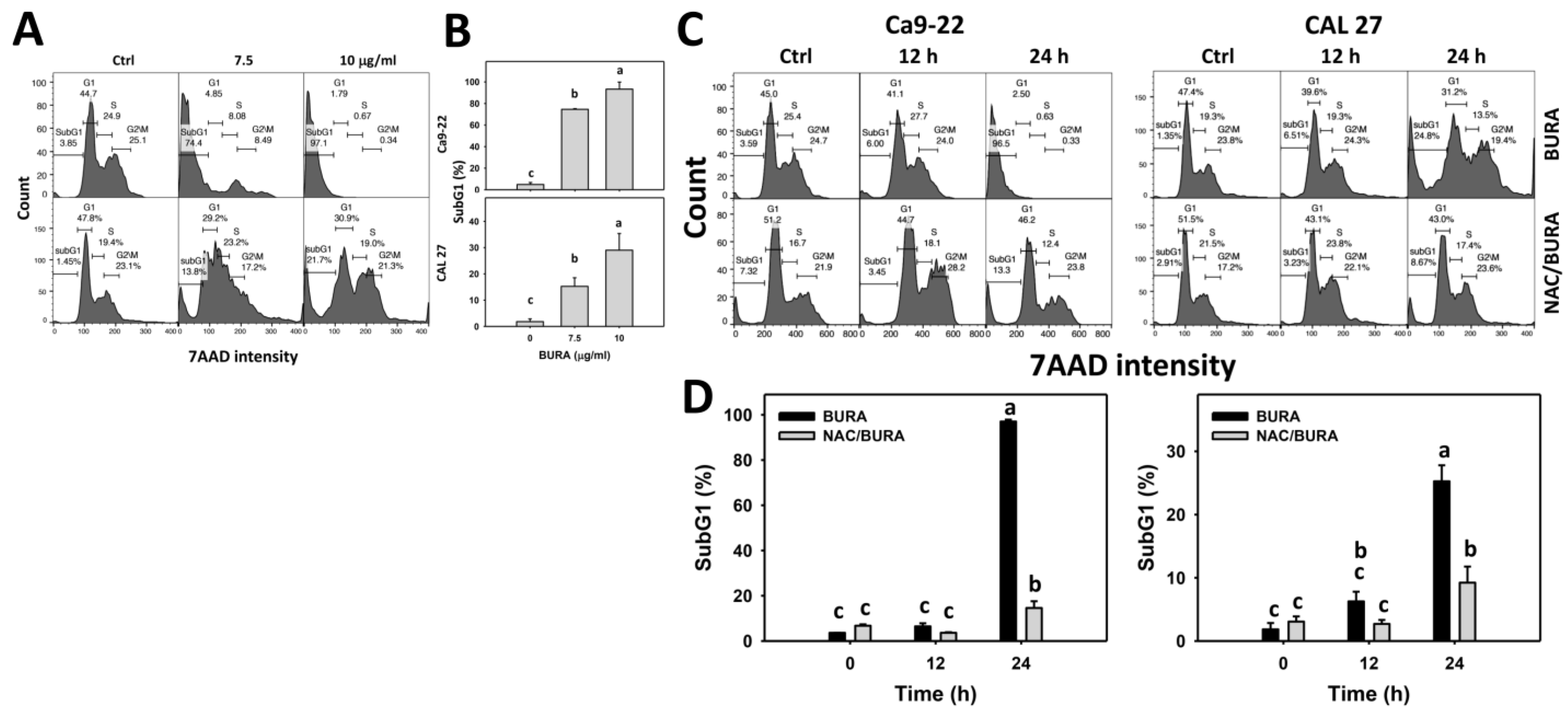
2.5. Cell Cycle Determination
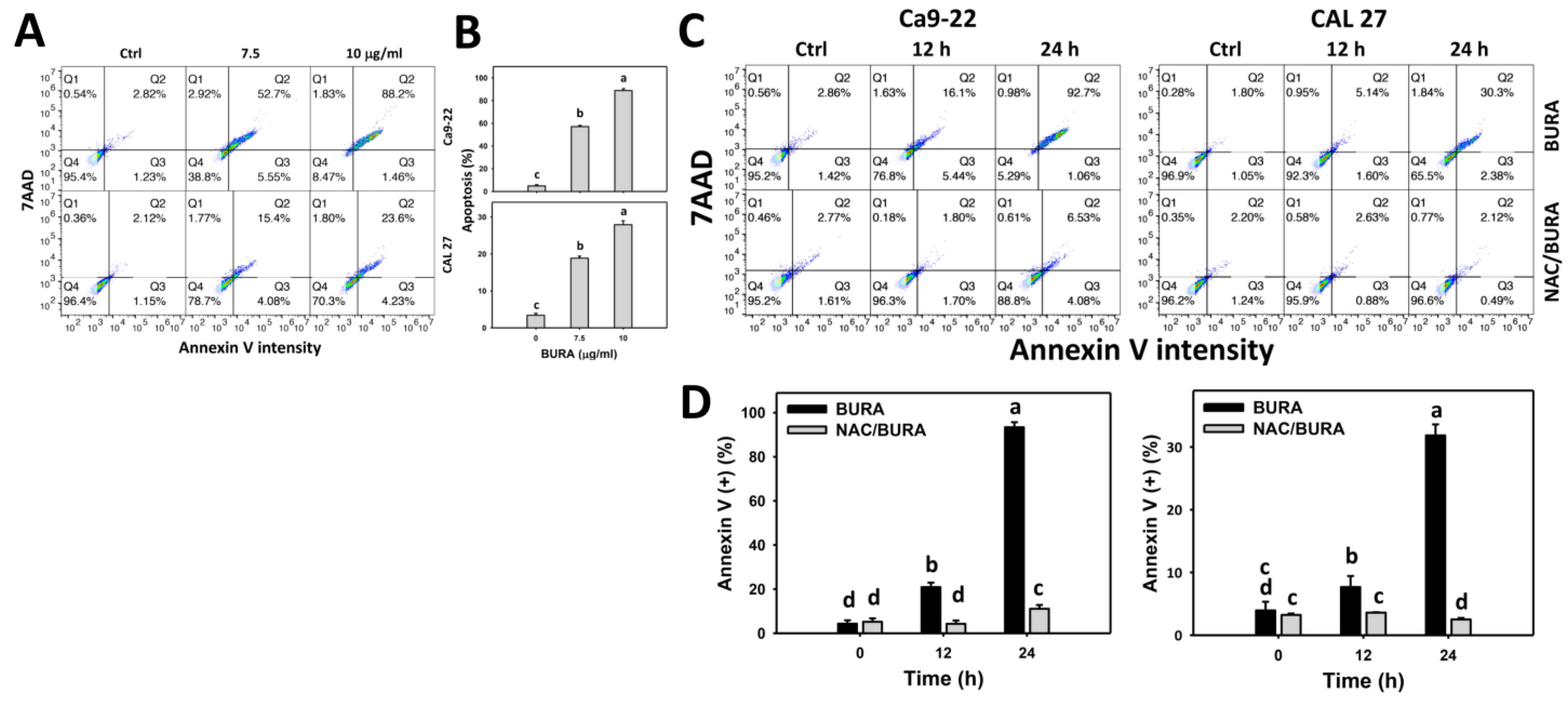
2.6. Apoptosis Determination by Annexin V/7AAD Method
2.7. Apoptosis Determination by Pancaspase and Caspase 3/8/9 (Cas 3/8/9) Assays
2.8. Oxidative Stress Determination by Mitochondrial Superoxide (MitoSOX), and Mitochondrial Membrane Potential (MMP) Assays
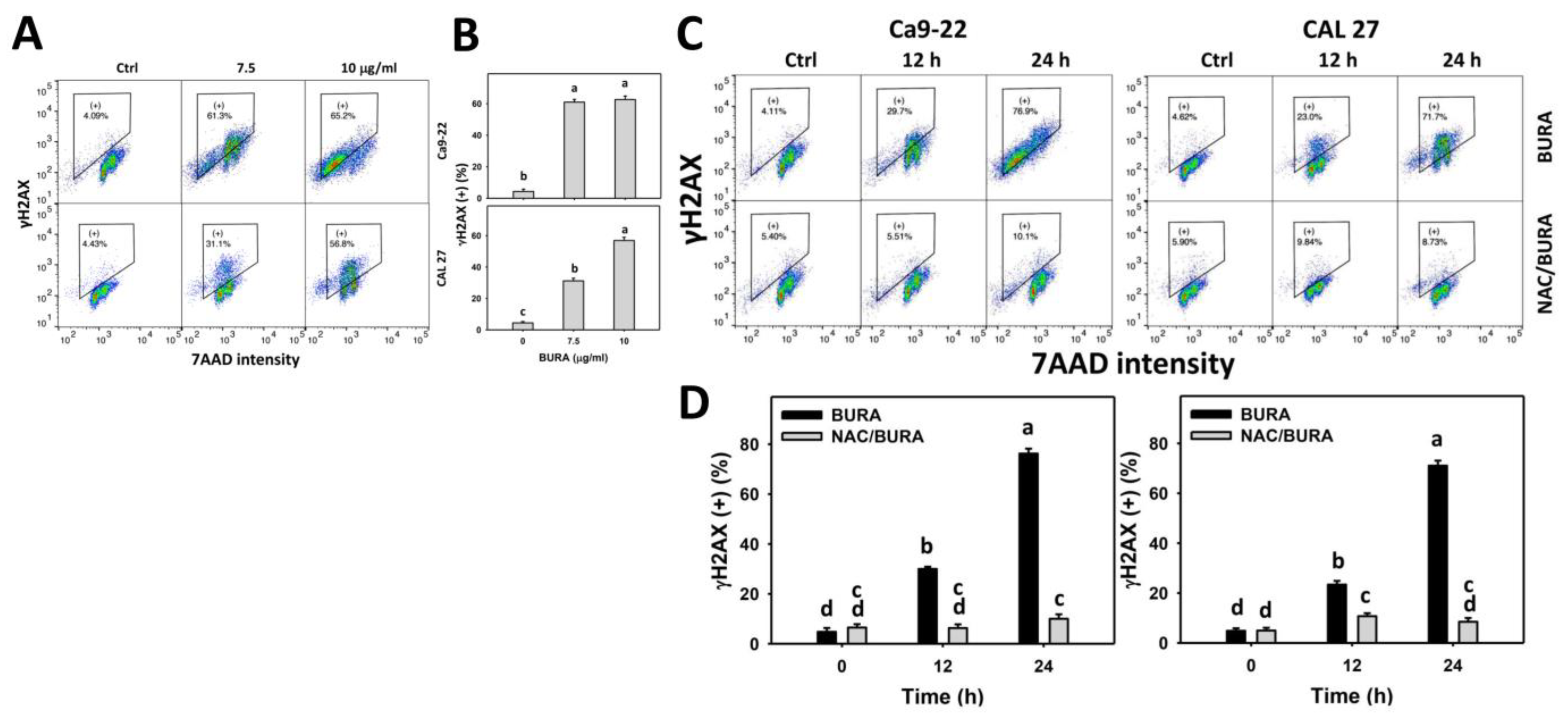
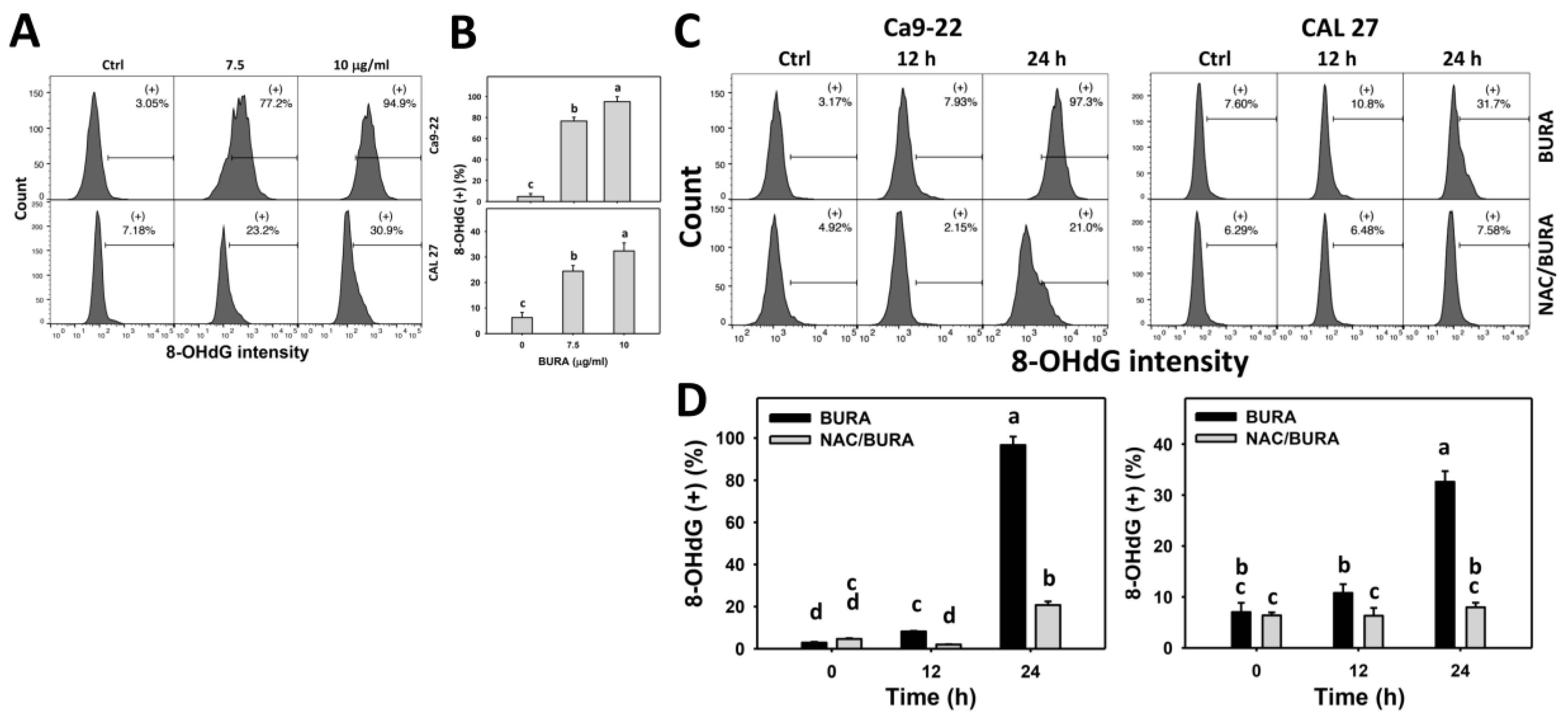
2.9. DNA Damage Determination by γH2AX and 8-Hydroxy-2-Deoxyguanosine (8-OHdG) Assays
2.10. Statistical Analysis
3. Results
3.1. BURA Shows Selective Antiproliferation in Oral Cancer Cells
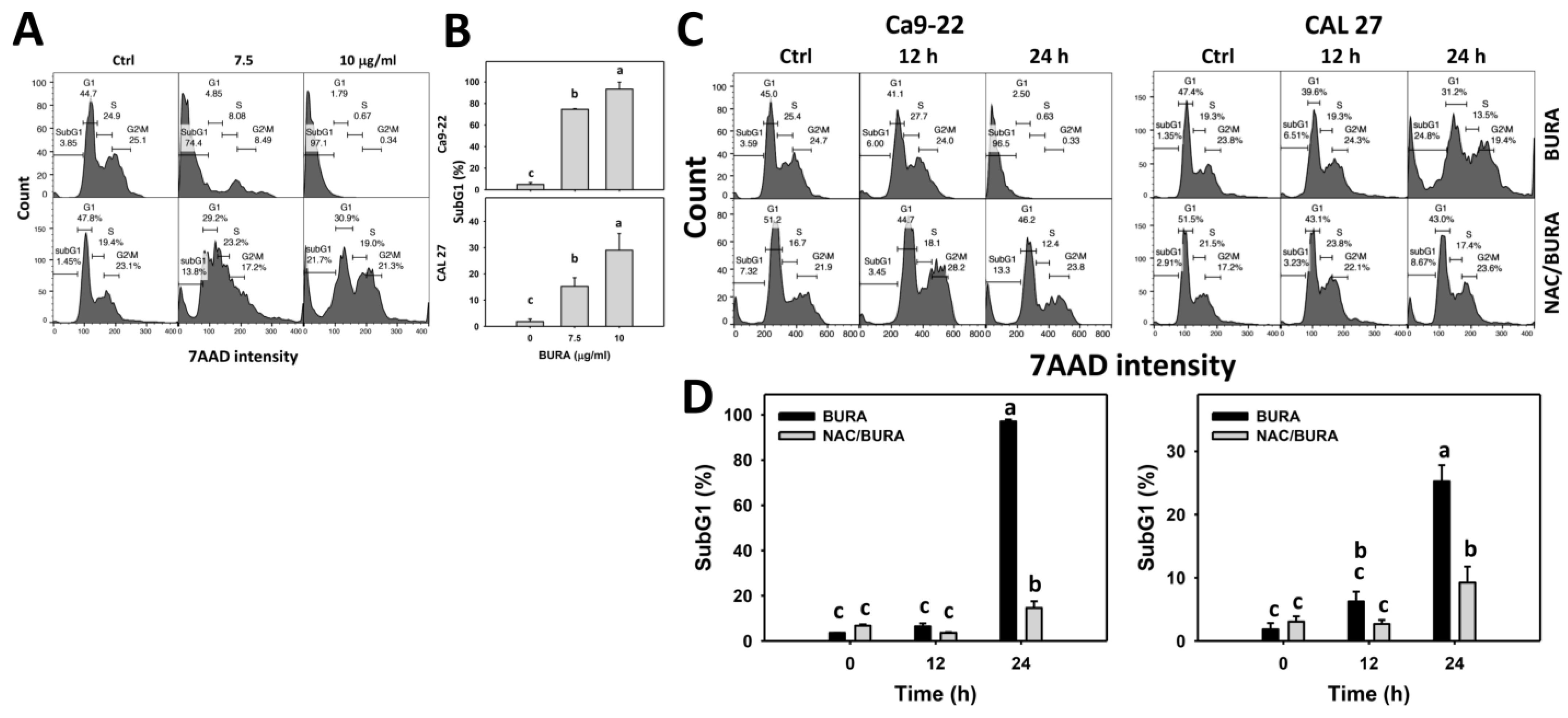
3.2. BURA Induces Cell Cycle Redistribution in Oral Cancer Cells
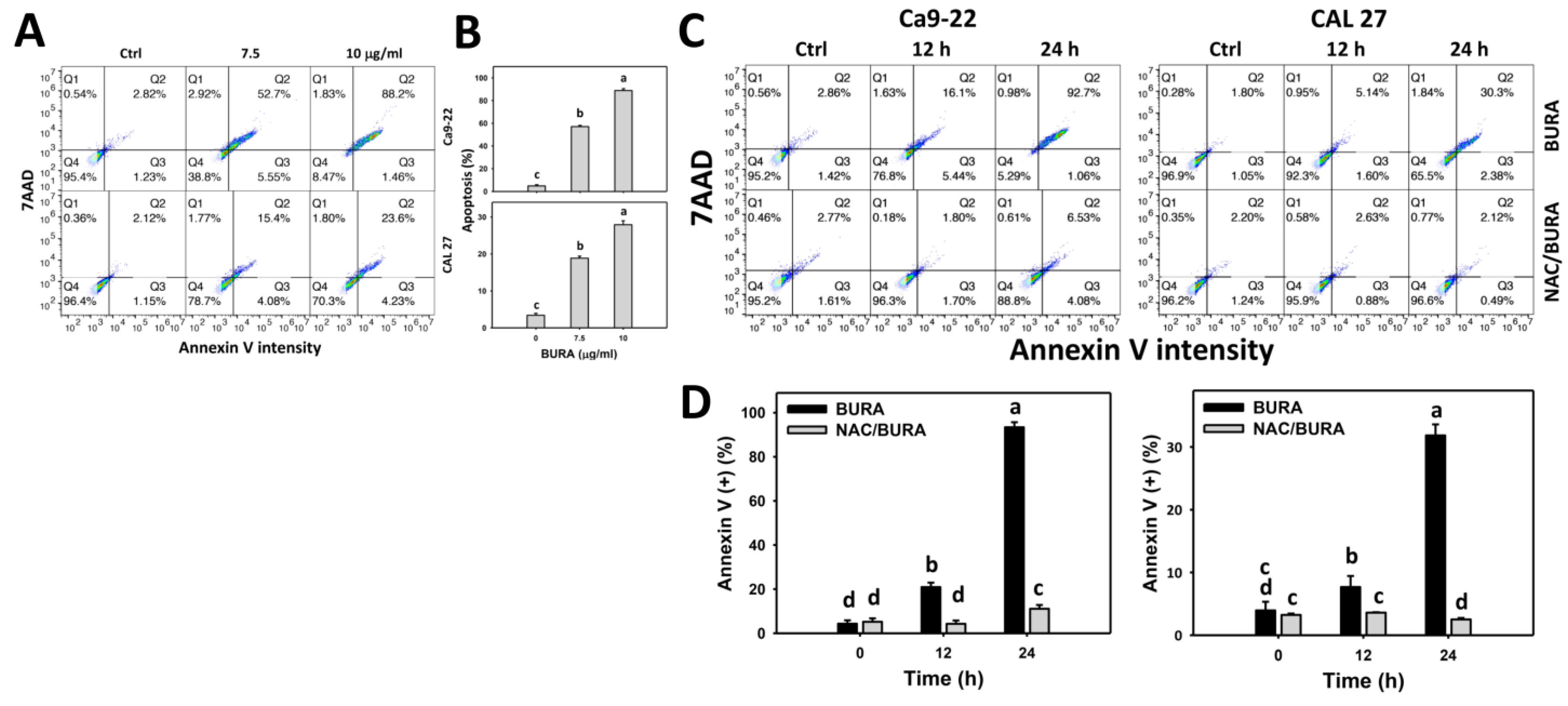
3.3. BURA Increases Annexin V-Positive Population in Oral Cancer Cells
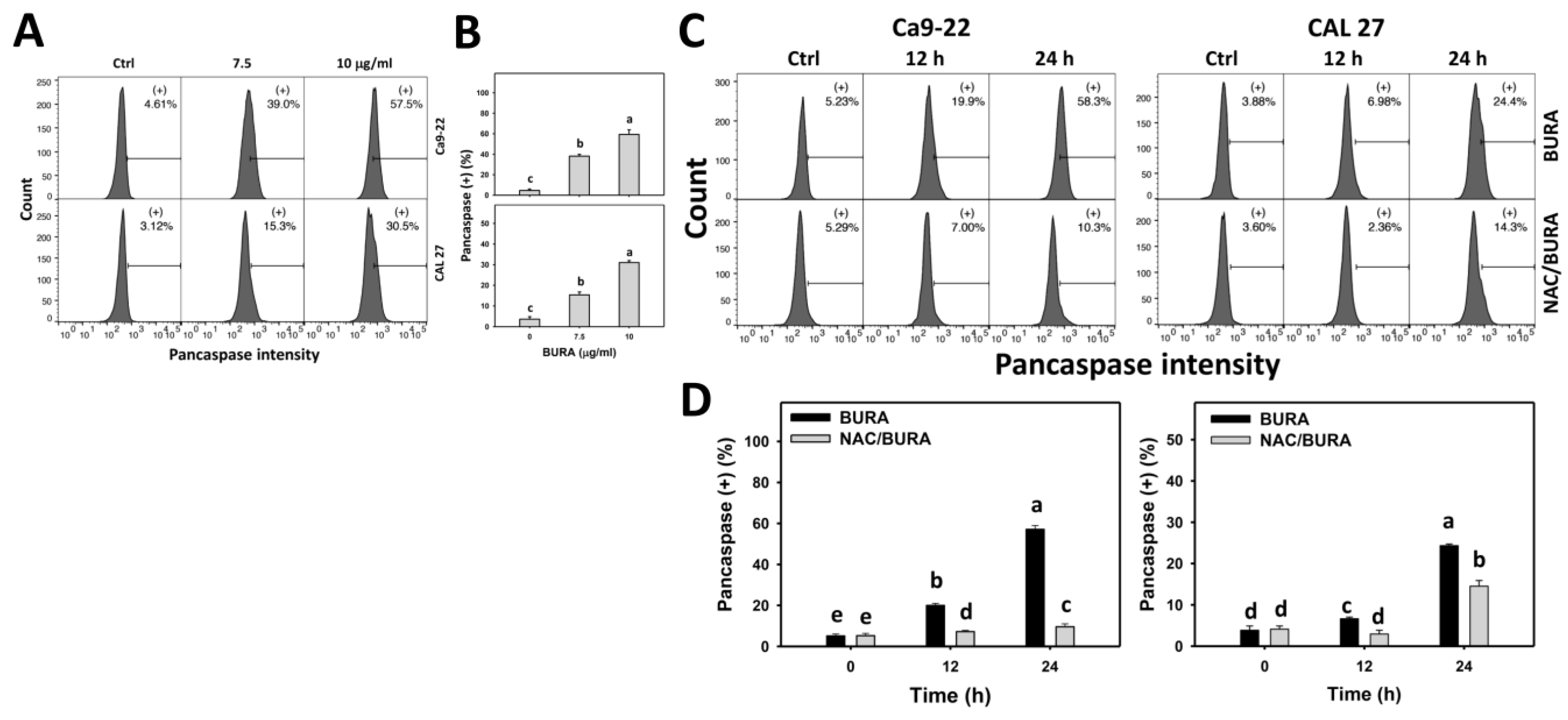
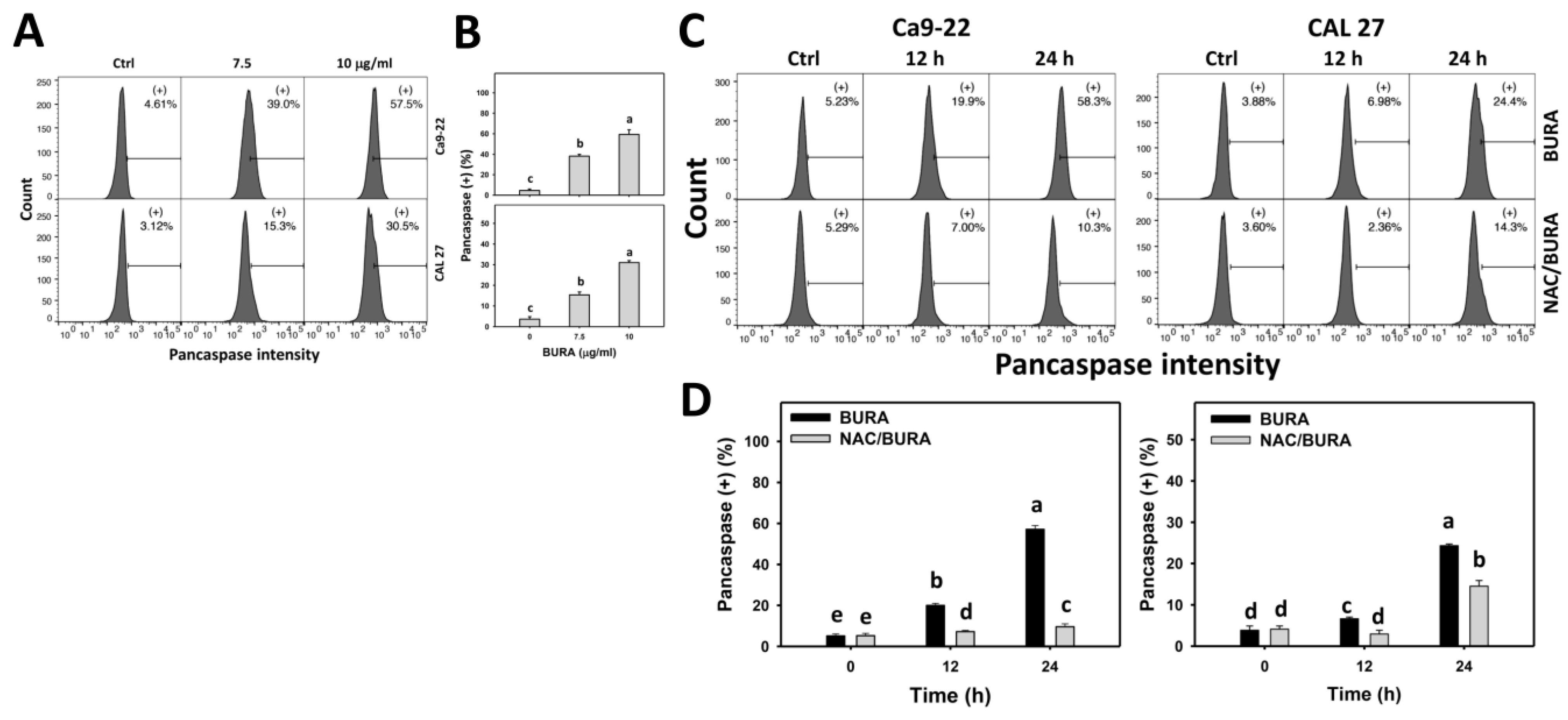
3.4. BURA Promotes Pancaspase Activation in Oral Cancer Cells
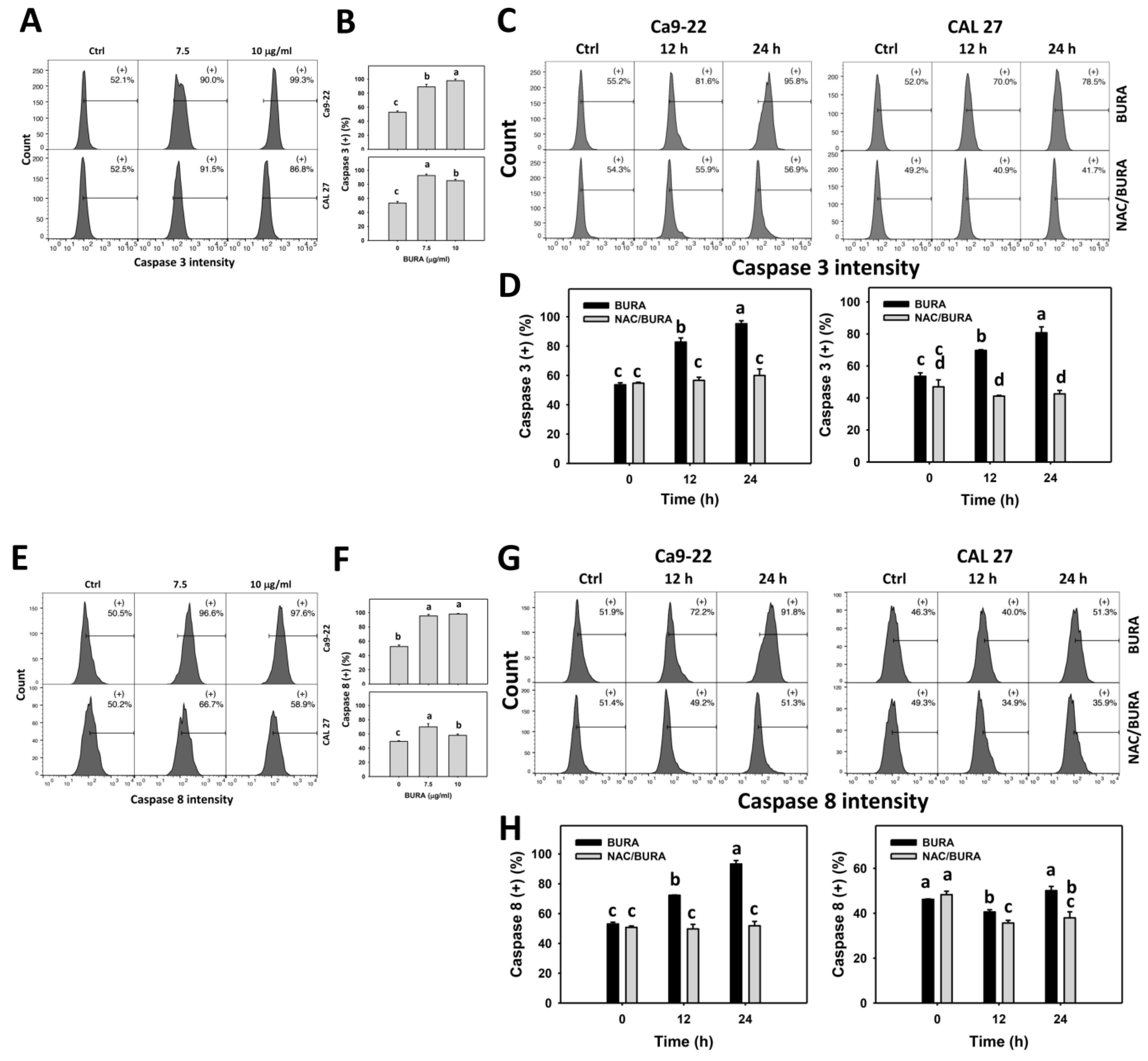
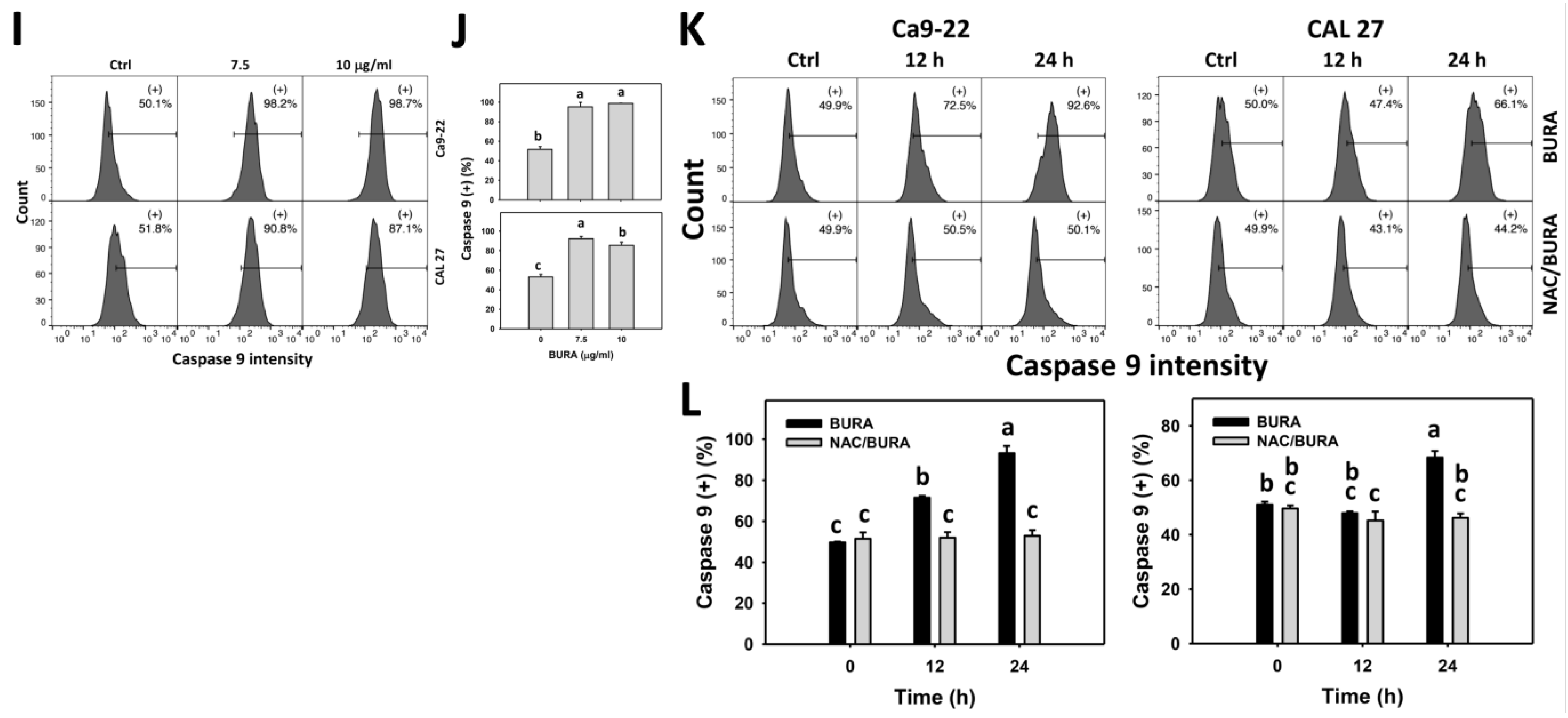
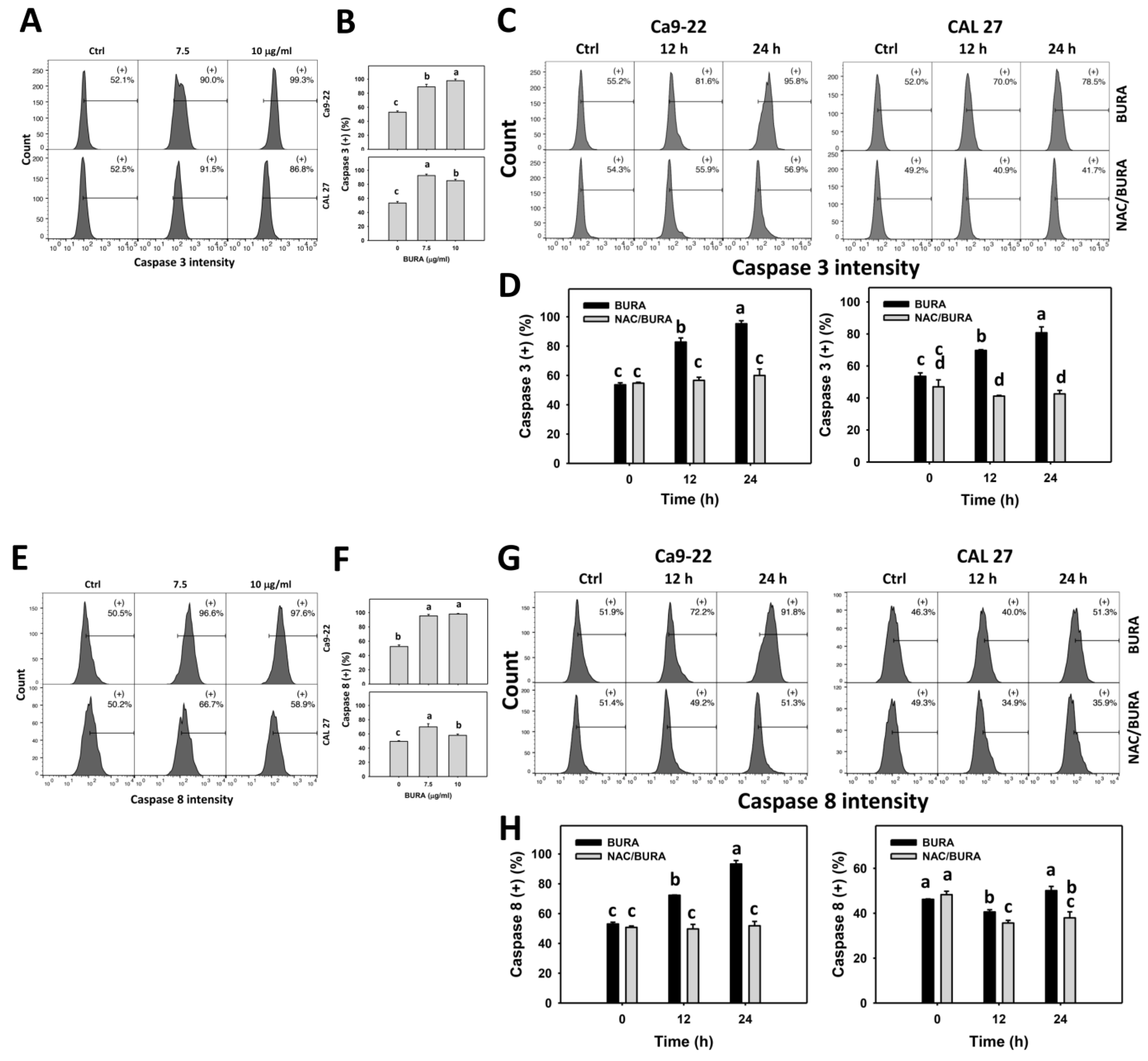
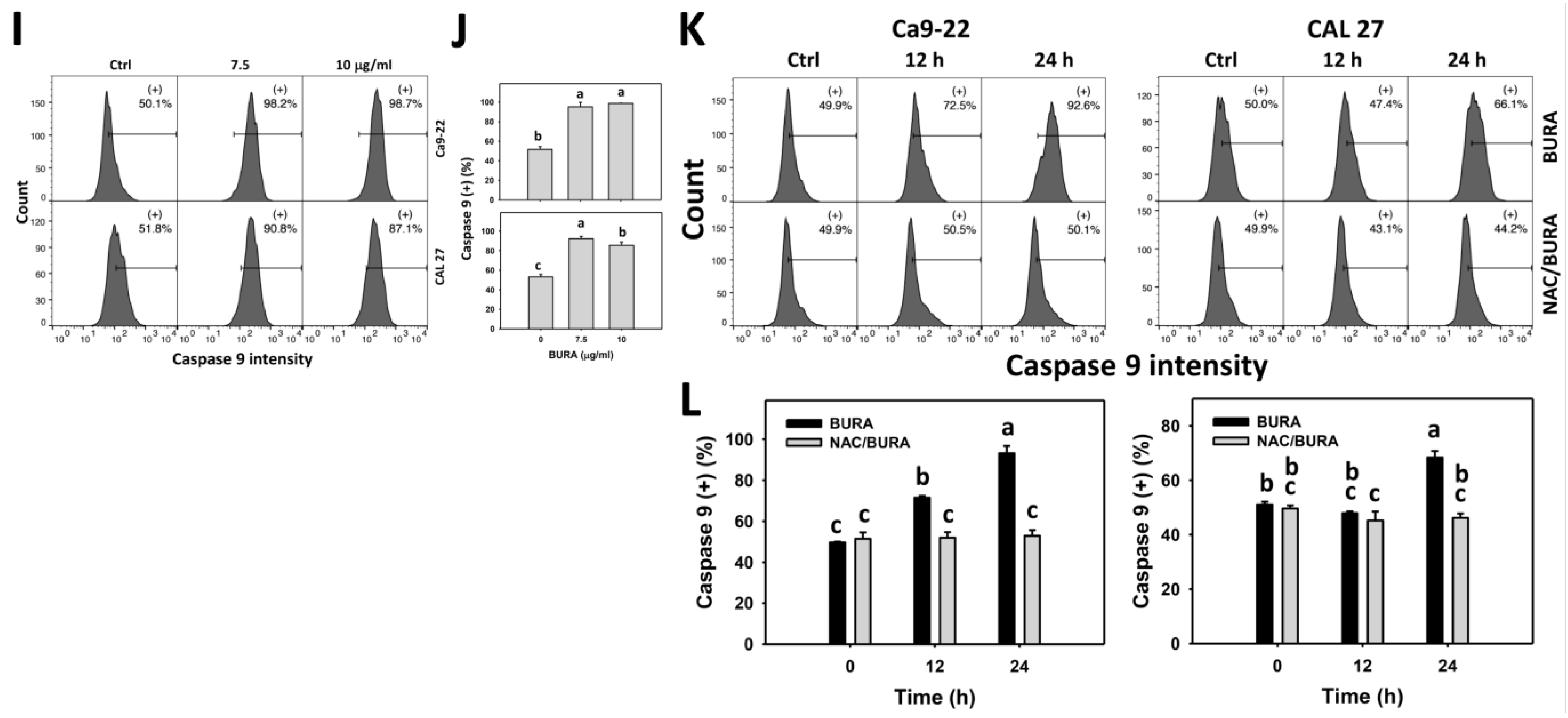
3.5. BURA Promotes Caspase Signaling Activation in Oral Cancer Cells
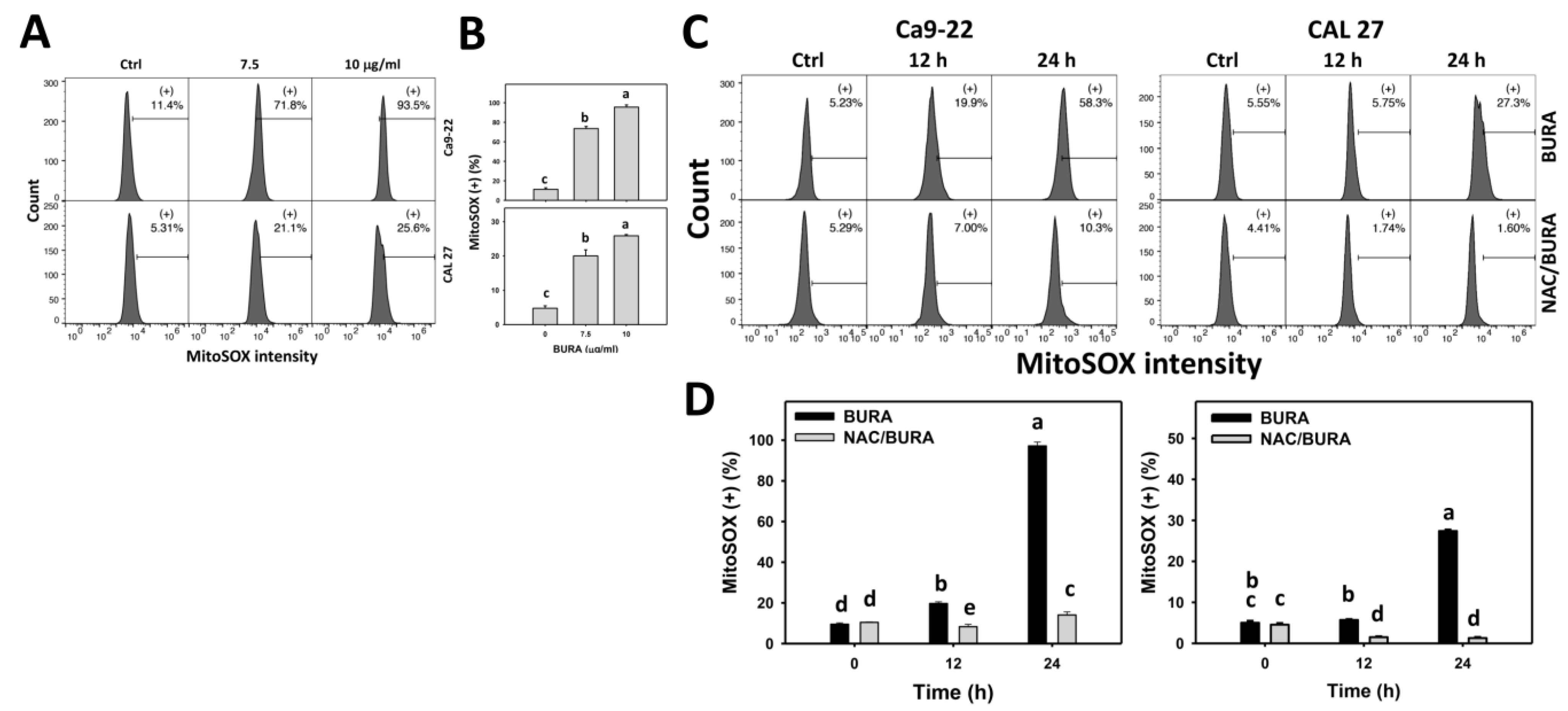
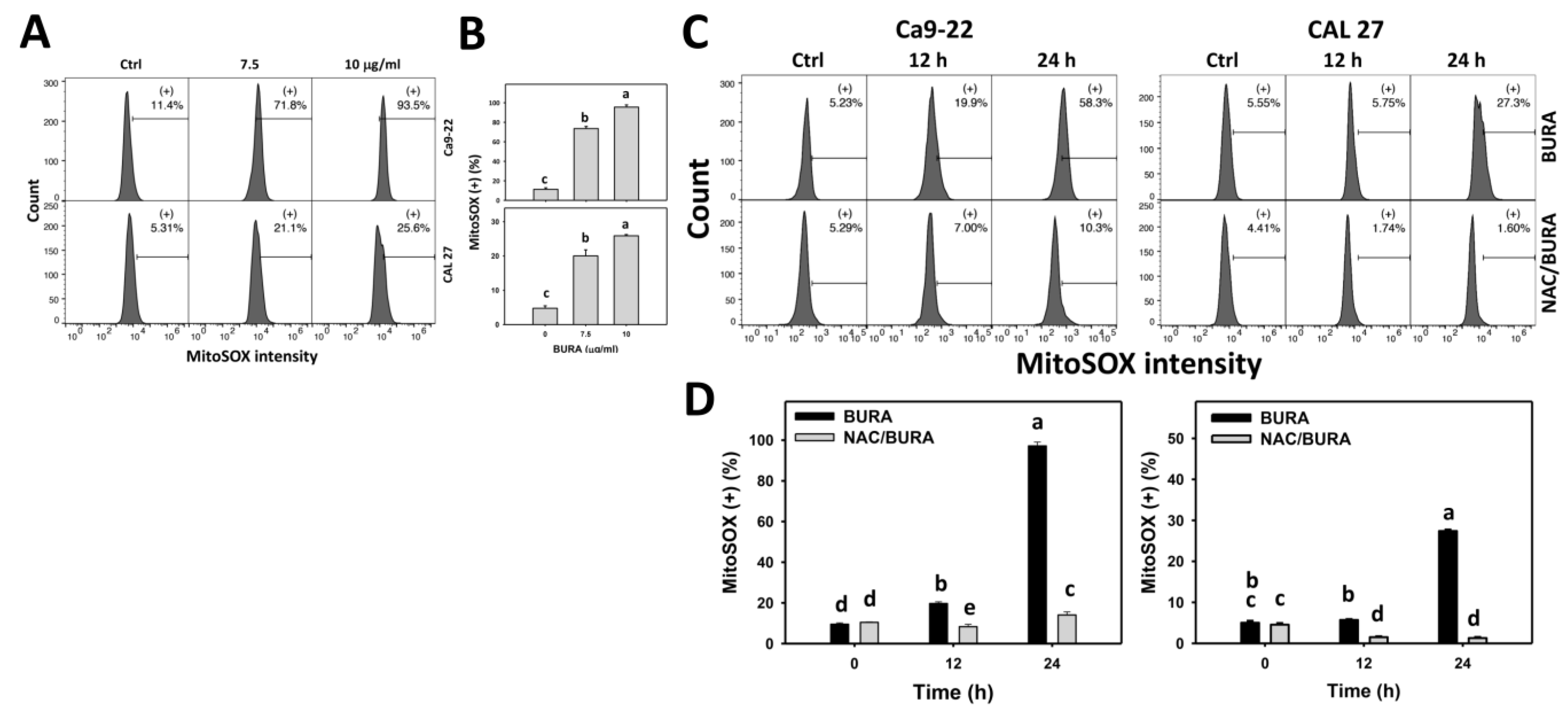
3.6. BURA Generates MitoSOX in Oral Cancer Cells
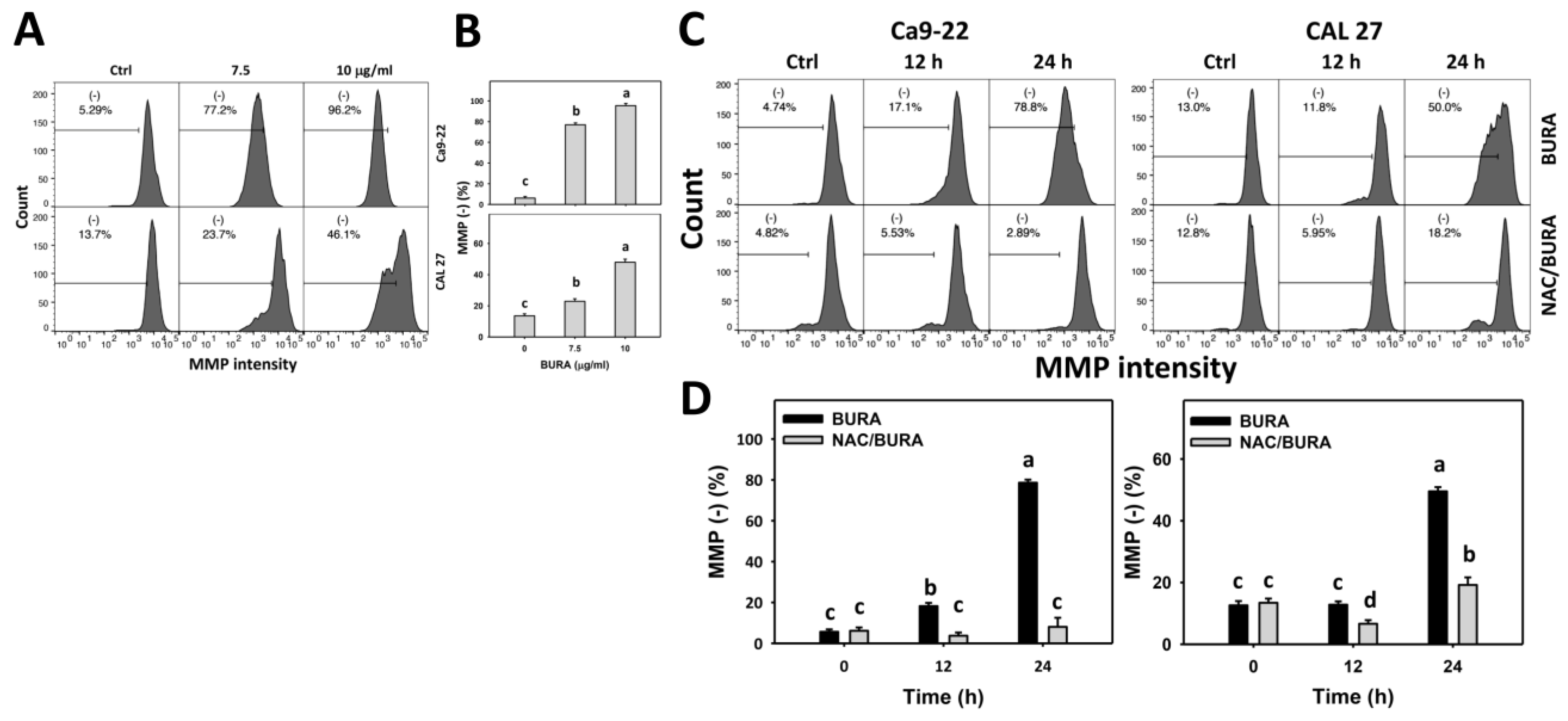
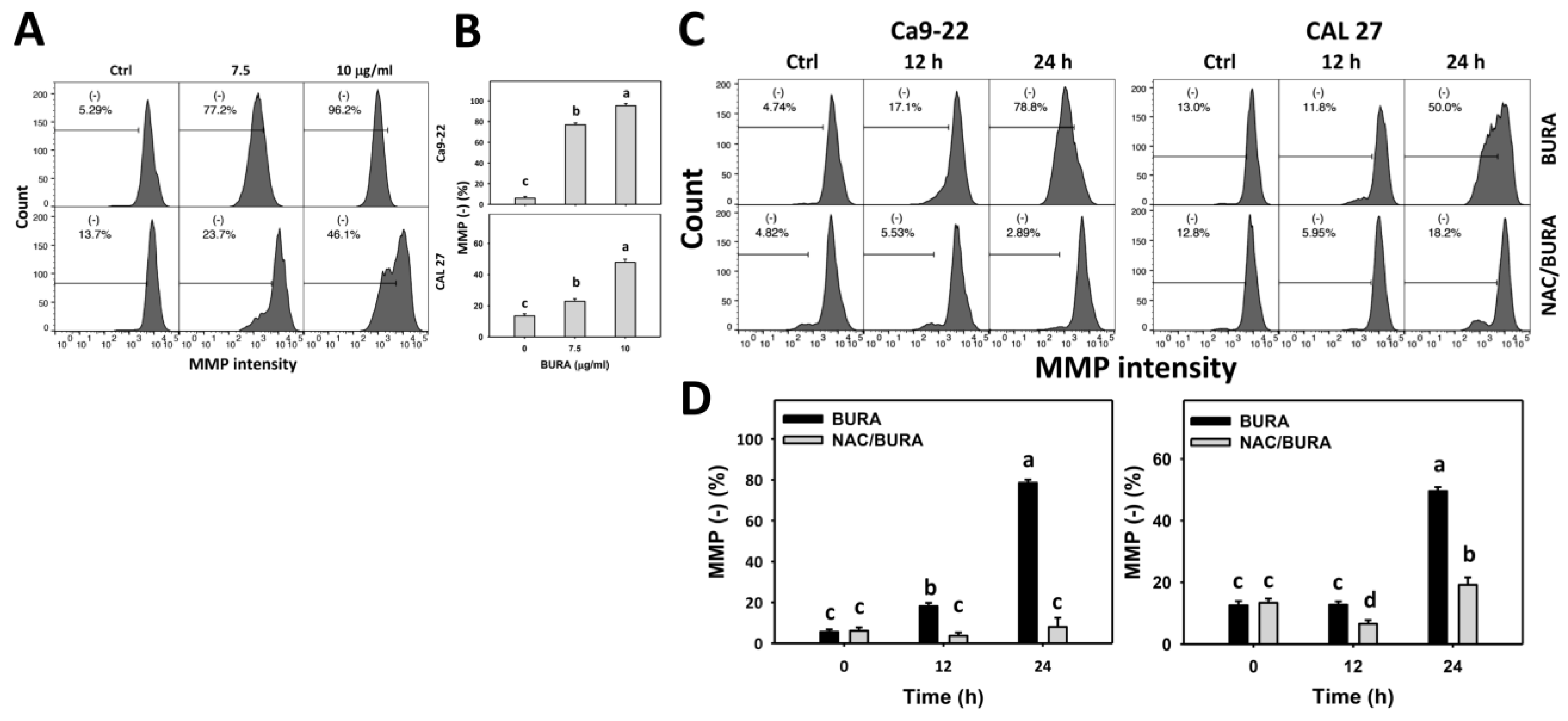
3.7. BURA Causes MMP Reduction in Oral Cancer Cells
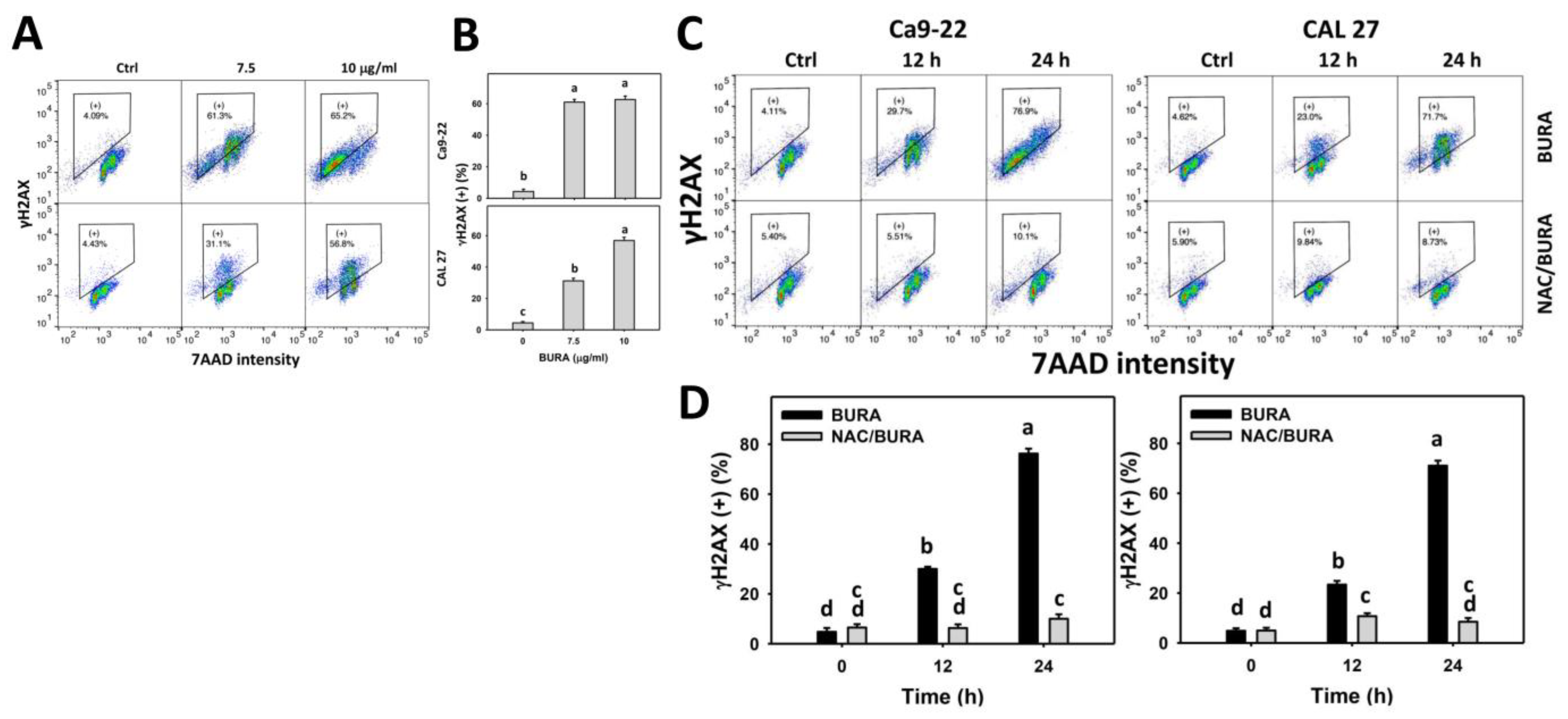
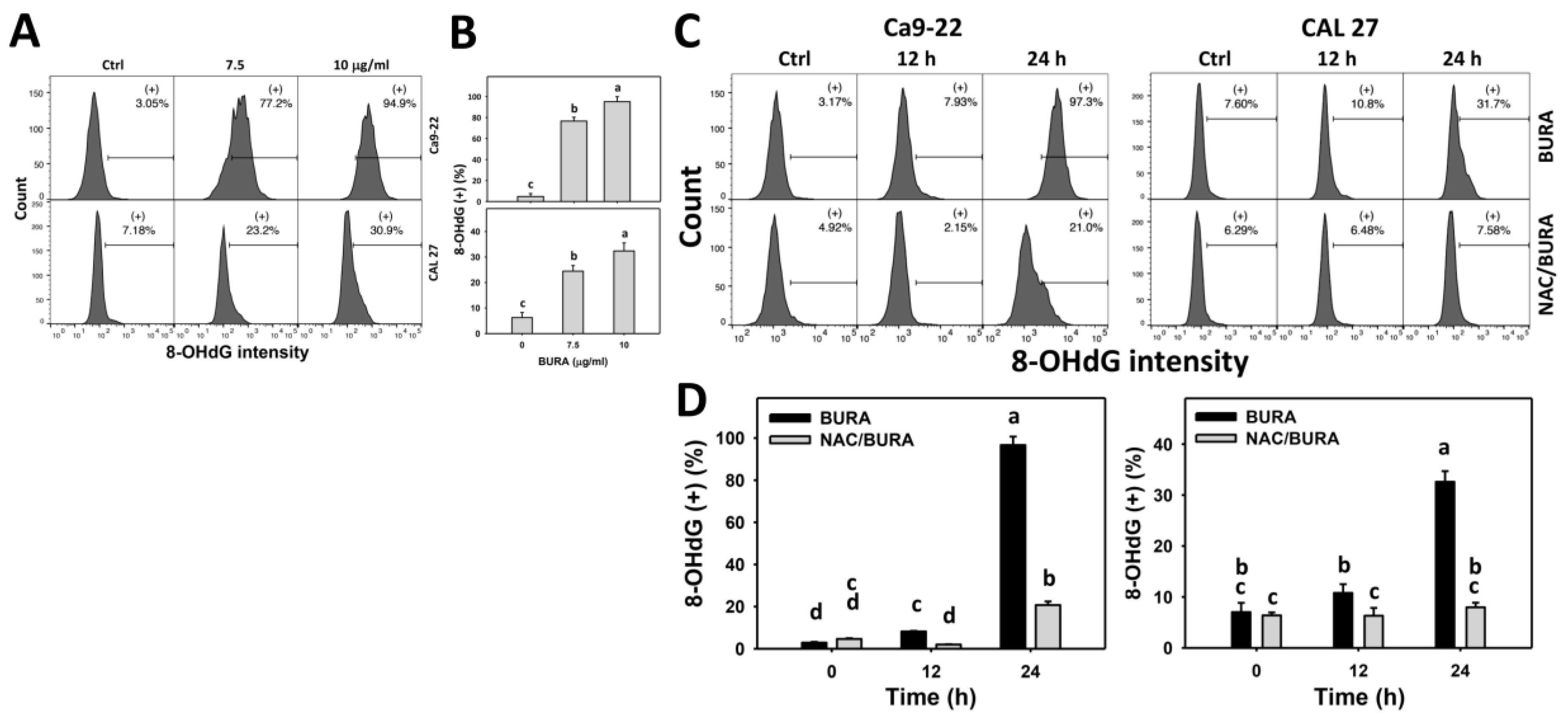
3.8. BURA Causes DNA Damage in Oral Cancer Cells
4. Discussion
4.1. BURA Shows Antiproliferation Effect in Oral Cancer Cells
4.2. BURA Shows Oxidative Stress in Oral Cancer Cells
4.3. BURA Shows Apoptosis and DNA Damage in Oral Cancer Cells
4.4. Antiproliferation Mechanisms of BURA in Oral Cancer Cells Depend on Oxidative Stress
4.5. Potential Targets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sarode, G.S.; Sarode, S.C.; Maniyar, N.; Anand, R.; Patil, S. Oral cancer databases: A comprehensive review. J. Oral Pathol. Med. 2018, 47, 547–556. [Google Scholar] [CrossRef]
- Rajguru, J.P.; Mouneshkumar, C.D.; Radhakrishnan, I.C.; Negi, B.S.; Maya, D.; Hajibabaei, S.; Rana, V. Tumor markers in oral cancer: A review. J. Family Med. Prim. Care 2020, 9, 492–496. [Google Scholar] [PubMed]
- Yen, C.Y.; Chen, C.H.; Chang, C.H.; Tseng, H.F.; Liu, S.Y.; Chuang, L.Y.; Wen, C.H.; Chang, H.W. Matrix metalloproteinases (MMP) 1 and MMP10 but not MMP12 are potential oral cancer markers. Biomarkers 2009, 14, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S., Jr. Oral cancer: Complications of therapy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1999, 88, 122–126. [Google Scholar] [CrossRef]
- Ahmad, R.; Khan, M.A.; Srivastava, A.N.; Gupta, A.; Srivastava, A.; Jafri, T.R.; Siddiqui, Z.; Chaubey, S.; Khan, T.; Srivastava, A.K. Anticancer potential of dietary natural products: A comprehensive review. Anticancer. Agents Med. Chem. 2020, 20, 122–236. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.Y.; Lin, L.C.; Chen, S.R.; Farooqi, A.A.; Cheng, Y.B.; Tang, J.Y.; Chang, H.W. Pomegranate extract (POMx) induces mitochondrial dysfunction and apoptosis of oral cancer cells. Antioxidants 2021, 10, 1117. [Google Scholar] [CrossRef]
- Das, G.; Patra, J.K.; Goncalves, S.; Romano, A.; Gutierrez-Grijalva, E.P.; Heredia, J.B.; Das Talukdar, A.; Shome, S.; Shin, H.S. Galangal, the multipotent super spices: A comprehensive review. Trends Food Sci. Technol. 2020, 101, 50–62. [Google Scholar] [CrossRef]
- Dwivedi, V.; Shrivastava, R.; Hussain, S.; Ganguly, C.; Bharadwaj, M. Cytotoxic potential of Indian spices (extracts) against esophageal squamous carcinoma cells. Asian Pac. J. Cancer Prev. 2011, 12, 2069–2073. [Google Scholar]
- Bhagat, N.; Chaturvedi, A. Spices as an alternative therapy for cancer treatment. Sys. Rev. Pharm. 2016, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Turrini, E.; Sestili, P.; Fimognari, C. Overview of the anticancer potential of the “king of spices” piper nigrum and its main constituent piperine. Toxins 2020, 12, 747. [Google Scholar] [CrossRef]
- Mughal, M.H. Spices; a mechanistic anticancer treatise. J. Nutr. Food Res. Technol. 2019, 2, 14–19. [Google Scholar] [CrossRef]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. ROS-mediated cancer cell killing through dietary phytochemicals. Oxid. Med. Cell Longev. 2019, 2019, 9051542. [Google Scholar] [CrossRef] [PubMed]
- Singletary, K. Diet, natural products and cancer chemoprevention. J. Nutr. 2000, 130, 465S–466S. [Google Scholar] [CrossRef]
- Mo, H.; Elson, C.E. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. 2004, 229, 567–585. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Emami, S. beta-Ionone and its analogs as promising anticancer agents. Eur. J. Med. Chem. 2016, 123, 141–154. [Google Scholar] [CrossRef]
- Aloum, L.; Alefishat, E.; Adem, A.; Petroianu, G. Ionone is more than a violet’s fragrance: A review. Molecules 2020, 25, 5822. [Google Scholar] [CrossRef]
- Wertz, P.W.; Kensler, T.W.; Mueller, G.C. Inhibition of phorbol ester action in lymphocytes by 5,6-epoxy-beta-ionone. Biochem. Biophys. Res. Commun. 1978, 83, 138–143. [Google Scholar] [CrossRef]
- Subehan, S.; Kadota, S.; Tezuka, Y. In vitro mechanism-based inactivation of cytochrome P450 3A4 by a new constituent of Cinnamomum burmani. Planta Med. 2008, 74, 1474–1480. [Google Scholar] [CrossRef]
- Chen, C.Y.; Hong, Z.L.; Yang, W.L.; Wu, M.H.; Huang, J.C.; Lee, J.Y. A novel homosesquiterpenoid from the stems of Cinnamomum burmanii. Nat. Prod. Res. 2012, 26, 1218–1223. [Google Scholar] [CrossRef]
- Lin, R.; Kao, C.; Liu, S.; Yeh, H.; Song, P.; Li, H.; Wang, H.; Chang, H.; Chen, C. A new apocarotenoid of Cinnamomum burmannii. Chem. Nat. Compd. 2020, 56, 604–606. [Google Scholar] [CrossRef]
- Huang, C.H.; Yeh, J.M.; Chan, W.H. Hazardous impacts of silver nanoparticles on mouse oocyte maturation and fertilization and fetal development through induction of apoptotic processes. Environ. Toxicol. 2018, 33, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.H.; Chen, C.Y.; Omar, H.A.; Huang, K.Y.; Tsao, C.C.; Chiu, C.C.; Chen, Y.L.; Chen, P.H.; Teng, Y.N. Reactive oxygen species mediate Terbufos-induced apoptosis in mouse testicular cell lines via the modulation of cell cycle and pro-apoptotic proteins. Environ. Toxicol. 2016, 31, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.S.; Lin, C.P.; Chen, Y.P.; Chao, M.R.; Li, C.C.; Liu, K.L. CYP450-mediated mitochondrial ROS production involved in arecoline N-oxide-induced oxidative damage in liver cell lines. Environ. Toxicol. 2018, 33, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.Y.; Chang, K.W.; Chen, C.F.; Chang, R.C. Characterization of two new cell lines derived from oral cavity human squamous cell carcinomas—OC1 and OC2. J. Oral Maxillofac. Surg. 1990, 48, 385–390. [Google Scholar] [CrossRef]
- Wang, H.R.; Tang, J.Y.; Wang, Y.Y.; Farooqi, A.A.; Yen, C.Y.; Yuan, S.F.; Huang, H.W.; Chang, H.W. Manoalide preferentially provides antiproliferation of oral cancer cells by oxidative stress-mediated apoptosis and DNA damage. Cancers 2019, 11, 1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.C.; Tseng, C.N.; Yang, J.I.; Huang, H.W.; Fang, Y.; Tang, J.Y.; Chang, F.R.; Chang, H.W. Antiproliferation and induction of apoptosis in Ca9-22 oral cancer cells by ethanolic extract of Gracilaria tenuistipitata. Molecules 2012, 17, 10916–10927. [Google Scholar] [CrossRef] [Green Version]
- Vignon, C.; Debeissat, C.; Georget, M.T.; Bouscary, D.; Gyan, E.; Rosset, P.; Herault, O. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS ONE 2013, 8, e68425. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.W.; Tang, J.Y.; Ou-Yang, F.; Wang, H.R.; Guan, P.Y.; Huang, C.Y.; Chen, C.Y.; Hou, M.F.; Sheu, J.H.; Chang, H.W. Sinularin selectively kills breast cancer cells showing G2/M arrest, apoptosis, and oxidative DNA damage. Molecules 2018, 23, 849. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Shih, Y.L.; Lee, M.H.; Au, M.K.; Chen, Y.L.; Lu, H.F.; Chung, J.G. Bufalin induces apoptosis of human osteosarcoma U-2 OS cells through endoplasmic reticulum stress, caspase- and mitochondria-dependent signaling pathways. Molecules 2017, 22, 437. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Huang, C.Y.; Tang, J.Y.; Liaw, C.C.; Li, R.N.; Liu, J.R.; Sheu, J.H.; Chang, H.W. Reactive oxygen species mediate soft corals-derived sinuleptolide-induced antiproliferation and DNA damage in oral cancer cells. Onco Targets Ther. 2017, 10, 3289–3297. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.S.; Tang, J.Y.; Yen, C.Y.; Huang, H.W.; Wu, C.Y.; Chung, Y.A.; Wang, H.R.; Chen, I.S.; Huang, M.Y.; Chang, H.W. Antiproliferation of Cryptocarya concinna-derived cryptocaryone against oral cancer cells involving apoptosis, oxidative stress, and DNA damage. BMC Complement. Altern. Med. 2016, 16, 94. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Huang, J.W.; Chang, F.R.; Huang, K.J.; Huang, H.M.; Huang, H.W.; Chou, C.K.; Wu, Y.C.; Chang, H.W. Golden berry-derived 4beta-hydroxywithanolide E for selectively killing oral cancer cells by generating ROS, DNA damage, and apoptotic pathways. PLoS ONE 2013, 8, e64739. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.Y.; Wang, Y.Y.; Lan, T.H.; Lin, L.C.; Yuan, S.F.; Tang, J.Y.; Chang, H.W. Low dose combined treatment with ultraviolet-C and withaferin a enhances selective killing of oral cancer cells. Antioxidants 2020, 9, 1120. [Google Scholar] [CrossRef]
- Moosavi, F.; Ebadi, A.; Mohabbati, M.; Damghani, T.; Mortazavi, M.; Miri, R.; Firuzi, O. Antiproliferative effect, alteration of cancer cell cycle progression and potential MET kinase inhibition induced by 3,4-dihydropyrimidin-2(1H)-one C5 amide derivatives. Eur. J. Pharmacol. 2021, 894, 173850. [Google Scholar] [CrossRef]
- Li, Y.; Pan, J.; Gou, M. The anti-proliferation, cycle arrest and apoptotic inducing activity of peperomin E on prostate cancer PC-3 cell line. Molecules 2019, 24, 1472. [Google Scholar] [CrossRef] [Green Version]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzilai, A.; Yamamoto, K. DNA damage responses to oxidative stress. DNA Repair. 2004, 3, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Amrithaa, B.; Priya, V.V.; Gayathri, R.; Dhanasekaran, G.; Muthu, K.; Sundaram, R. Antiproliferative effect of β-sitosterol on human lung adenocarcinoma cell line A549—In vitro. Drug Invent. Today 2019, 11, 1818–1822. [Google Scholar]
- Baskar, A.A.; Ignacimuthu, S.; Paulraj, G.M.; Al Numair, K.S. Chemopreventive potential of beta-Sitosterol in experimental colon cancer model—An in vitro and In vivo study. BMC Complement. Altern. Med. 2010, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Aminuddin, A.; Ng, P.Y.; Leong, C.O.; Chua, E.W. Mitochondrial DNA alterations may influence the cisplatin responsiveness of oral squamous cell carcinoma. Sci. Rep. 2020, 10, 7885. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-inducing strategy in anticancer therapy. Oxid. Med. Cell Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ou-Yang, F.; Hou, M.F.; Huang, H.W.; Wang, H.R.; Li, K.T.; Fayyaz, S.; Shu, C.W.; Chang, H.W. Oxidative stress-modulating drugs have preferential anticancer effects—Involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin. Cancer Biol. 2019, 58, 109–117. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yen, C.Y.; Wang, H.R.; Yang, H.P.; Tang, J.Y.; Huang, H.W.; Hsu, S.H.; Chang, H.W. Tenuifolide B from Cinnamomum tenuifolium stem selectively inhibits proliferation of oral cancer cells via apoptosis, ROS generation, mitochondrial depolarization, and DNA damage. Toxins 2016, 8, 319. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, J.; Choi, C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE 2011, 6, e23211. [Google Scholar] [CrossRef]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. Regulated necrotic cell death in alternative tumor therapeutic strategies. Cells 2020, 9, 2709. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, W.; Hou, H.; Li, J.; Li, H.; Sun, X.; Zhao, L.; Li, X. Ginsenoside 20(S)-Rg3 induced autophagy to inhibit migration and invasion of ovarian cancer. Biomed. Pharmacother. 2017, 85, 620–626. [Google Scholar] [CrossRef]
- Sithara, T.; Arun, K.B.; Syama, H.P.; Reshmitha, T.R.; Nisha, P. Morin inhibits proliferation of SW480 colorectal cancer cells by inducing apoptosis mediated by reactive oxygen species formation and uncoupling of Warburg effect. Front. Pharmacol. 2017, 8, 640. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- McAdam, E.; Brem, R.; Karran, P. Oxidative stress-induced protein damage inhibits DNA repair and determines mutation risk and therapeutic efficacy. Mol. Cancer Res. 2016, 14, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Fernandes, N.V.; Yeganehjoo, H.; Katuru, R.; Qu, H.; Yu, Z.; Mo, H. beta-ionone induces cell cycle arrest and apoptosis in human prostate tumor cells. Nutr. Cancer 2013, 65, 600–610. [Google Scholar] [CrossRef]
- Dong, H.W.; Zhang, S.; Sun, W.G.; Liu, Q.; Ibla, J.C.; Soriano, S.G.; Han, X.H.; Liu, L.X.; Li, M.S.; Liu, J.R. beta-Ionone arrests cell cycle of gastric carcinoma cancer cells by a MAPK pathway. Arch. Toxicol. 2013, 87, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Dong, H.W.; Sun, W.G.; Liu, M.; Ibla, J.C.; Liu, L.X.; Parry, J.W.; Han, X.H.; Li, M.S.; Liu, J.R. Apoptosis initiation of beta-ionone in SGC-7901 gastric carcinoma cancer cells via a PI3K-AKT pathway. Arch. Toxicol. 2013, 87, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Liu, T.; Chen, J.; Yang, Z.; Xu, S.; Fan, Y.; Zeng, J.; Chen, Y.; Ma, Z.; Gao, Y.; et al. Activation of PSGR with beta-ionone suppresses prostate cancer progression by blocking androgen receptor nuclear translocation. Cancer Lett. 2019, 453, 193–205. [Google Scholar] [CrossRef]
- Dong, H.W.; Wang, K.; Chang, X.X.; Jin, F.F.; Wang, Q.; Jiang, X.F.; Liu, J.R.; Wu, Y.H.; Yang, C. Beta-ionone-inhibited proliferation of breast cancer cells by inhibited COX-2 activity. Arch. Toxicol. 2019, 93, 2993–3003. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.-L.; Yang, K.-H.; Yang, C.-W.; Lee, M.-Y.; Chuang, Y.-T.; Chen, Y.-N.; Chang, F.-R.; Chen, C.-Y.; Chang, H.-W. Burmannic Acid Inhibits Proliferation and Induces Oxidative Stress Response of Oral Cancer Cells. Antioxidants 2021, 10, 1588. https://doi.org/10.3390/antiox10101588
Liu S-L, Yang K-H, Yang C-W, Lee M-Y, Chuang Y-T, Chen Y-N, Chang F-R, Chen C-Y, Chang H-W. Burmannic Acid Inhibits Proliferation and Induces Oxidative Stress Response of Oral Cancer Cells. Antioxidants. 2021; 10(10):1588. https://doi.org/10.3390/antiox10101588
Chicago/Turabian StyleLiu, Su-Ling, Kun-Han Yang, Che-Wei Yang, Min-Yu Lee, Ya-Ting Chuang, Yan-Ning Chen, Fang-Rong Chang, Chung-Yi Chen, and Hsueh-Wei Chang. 2021. "Burmannic Acid Inhibits Proliferation and Induces Oxidative Stress Response of Oral Cancer Cells" Antioxidants 10, no. 10: 1588. https://doi.org/10.3390/antiox10101588